
Abstract
Stromal cell-derived factor-1 alpha (SDF-1a) or CXCL12 is an important cytokine with multiple functions in the brain during development and in adulthood. The inflammatory response initiated by spinal cord injury (SCI) involves the processing of interleukin-1beta (IL-1ß) and IL-18 mediated by caspase-1 which is under the control of an intracellular multiprotein complex termed inflammasome. Using an SCI rat model, we found improved functional long-term recovery which is paralleled by a reduction of apoptosis after intrathecal treatment with SDF-1a. An intriguing aspect is that SDF-1a changed the number of neuroinflammatory cells in the damaged area. We further examined the cellular localization and sequential expression of several inflammasomes during SCI at 6 h, 24 h, 3 days, and 7 days as well as the role of SDF-1a as a regulatory factor for inflammasomes. Using 14-week old male Wistar rats, spinal cord contusion was applied at the thoracic segment 9, and animals were subsequently treated with SDF-1a via intrathecal application through an osmotic pump. SCI temporally increased the expression of the inflammasomes NLRP3, ASC, the inflammatory marker tumor necrosis factor-a (TNF-a), interleukin-1ß (IL-1β) and IL-18. SDF-1a significantly reduced the levels of IL-18, IL-1b, TNF-a, NLRP3, ASC, and caspase-1. Immunofluorescence double-labeling demonstrated that microglia and neurons are major sources of the ASC and NLRP3 respectivley. Our data provide clear evidence that SCI stimulates a complex scenario of inflammasome activation at the injured site and that SDF-1a-mediated neuroprotection presumably depends on the attenuation of the inflammasome complex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury (SCI) is a fatal neuro-destructing event resulting from a complete or incomplete insult which initiates a progressive state of degeneration including structural, biochemical, and physiological post injury tissue changes and reorganization [1]. The pathophysiology of SCI is characterized by two major mechanisms, a primary mechanical injury and a secondary process orchestrated by diverse pathophysiological mechanisms, including inflammation, excitotoxicity, apoptosis, and demyelination [2]. Neuroinflammatory responses involve the activation of the pro-inflammatory interleukin (IL) family such as IL-1b and IL-18 that contribute to cell death. Both ILs are closely related, possess a similar three-dimensional structure, and are synthesized as inactive cytoplasmic precursor forms [3]. Processing and activation of pro-IL-1b and IL-18 require the activation of a multiprotein caspase-1-activating complex termed inflammasome [3] which is a key signaling platform that detects pathogenic stressors. The activation of inflammasome components in innate brain immune cells typically reflects one of the primary and critical steps of neuroinflammation sequentially triggered by harmful stimuli and traumatic challenges through pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP) molecules. The NACHT, LRR, and PYD domains-containing protein (NLRP) inflammasomes NLRP1 and NLRP3 consist of three main components: a nucleotide oligomerization domain-like receptor (NLR), caspase-1 and an adaptor called apoptosis-associated speck-like protein (ASC) containing a caspase recruitment domain [3]. The NLR family contains a leucine rich repeat domain (LRR), a central NACHT domain, and a variable amino-terminal domain which in the NLRP subfamily is an N-terminal pyrin domain (PYD) [4]. The activation of NLRPs leads to the recruitment of ASC which contains a caspase activation and recruitment domain (CARD). ASC then interacts with the CARD of pro-caspase-1. The interaction with pro-caspase-1 leads to its conversion to active caspase-1 which then converts pro forms of IL-1β and IL-18 into their active forms, thereby initiating an inflammatory response [5]. Only a few studies showed inflammasome components to be differentially regulated after SCI. De Rivero and coworkers have demonstrated that NLRP1 is activated after SCI and that the neutralization of ASC after SCI reduces caspase-1 activation and decreases processing of IL-1β and IL-18 [6]. In consequence, the administration of anti-ASC neutralizing antibodies to injured rats resulted in significant tissue sparing and functional improvement [6]. Most recently, it has been found that the expression levels of NALP, ASC, and caspase-1 are increased after SCI in rats and that hyperbaric oxygen treatment mitigated this effect [7].
Stromal derived factor-1a (SDF-1a) is a potent chemo-attractant cytokine with various biological functions such as stem cell mobilization, proliferation, inflammatory cell infiltration, and angiogenesis in the central nervous system (CNS) [8]. SDF-1a is up-regulated under pathological conditions such as ischemic stroke, promotes the migration of neural progenitor cells, and participates in the regulation of the neurovascular unit [9]. The administration of SDF-1a ameliorates cell death of cortical neurons induced after stroke most likely by stimulating angiogenesis and neurogenesis and through the modulation of inflammatory responses [10]. We and other groups have recently demonstrated that the protective actions of SDF-1a finally rescuing neurons from apoptosis and improving behavioral functions [10, 11]. In SCI models, infusion of SDF-1a into the lesion site also promotes neurite growth and axonal sprouting [12, 13] which might represent an additional mode of SDF-1a action.
In a previous study, we were able to demonstrate that SDF-1a exerts neuroprotective effects after SCI [14]. SDF-1a significantly alleviated neuronal damage and stimulated functional recovery in a dose-dependent way. Furthermore, we observed reduced numbers of active caspase-3 expressing cells in the injured spinal cord after SDF-1a application. On the other hand, microgliosis was observed by SDF-1a. After SCI, microglia which are decisive regulators of tissue damage after SCI accumulate within the epicenter and the hematoma of the injured spinal cord and are a putative source of IL-1b [15].
Based on the above conceptual studies and our findings regarding the bimodal role of SDF-1a during inflammatory responses, we aimed at analyzing the role of intrathecal application of SDF-1a after SCI in an early injury phase on NLRP-inflammasome activation.
Material and Methods
Animals and Surgery
Research and animal care were approved by the Review Board for the Care of Animal Subjects of the district government (Tehran, Iran). In vivo experiments were performed in 14-week old male Wistar rats (350–400 g, Pasteur, Iran). Animals were housed under temperature-controlled conditions with alternate light and dark cycles, 12 h each and ad libitum access to water and food. For the continuous intrathecal administration of SDF-1a, catheters were placed in all rats 1 week before the induction of SCI according to the procedure outlined by Yaksh and Rudy [16] (Fig. 1a). Under anesthesia with 2.0 % isoflurane in O2 (Abbott, Germany), a sterile rat intrathecal catheter (10 cm, 10 μL void volume, ALZET, Cupertino, CA, USA) was inserted and advanced caudally to the rostral edge of the lumbar enlargement through an incision in the atlantooccipital membrane. Afterwards, 20 μL of sterile saline was flushed through the catheter to clear the tip. The catheter was fixed and the incision was subsequently sutured. The exact location of the catheter was confirmed by hind limb paralysis approx. 15 min post catheterization by injecting 10 μL 2 % lidocaine. Rats were allowed to recover for 7 days and housed individually. Spinal contusion lesions were made in accordance with the weight compression method [17]. This procedure has been previously described in detail [14]. Briefly, rats were anesthetized with 2.5 % isoflurane in O2 (Abbott, Germany), and the appropriate narcosis was controlled by reflex testing such as the absence of a pedal withdrawal response to a hard pinch. After shaving the surgical area, rats were placed in a modified stereotactic frame. Skin incision and blunt dissection of the muscle layers over the area of the vertebral T10 level (spinal T9) were performed. Afterwards, an adjustable forceps were applied to the spinous processes of the second vertebra rostral the Th8 vertebra to stabilize the spinal cord. In addition, the vertebra columns, transverse processes of Th7 and Th9, were secured by transverse process clamps of the Spinal Cord Adaptor (Stoelting Co, USA). Under a surgical microscope, dorsal laminectomy of T8 vertebra was performed using a fine rongeur. After laminectomy, the spinal cord was compressed by placing a 50 g weight on the exposed spinal cord column for 5 min using a rectangular plate which is longitudinally oriented over the spinal cord. The plate had an area of 11.0 mm2 (2.2 × 5.0 mm) and a concave shape that ensured equal distribution of the pressure on the spinal cord tissue. Immediately after injury, an infusion pump (Model 2001, ALZET, Cupertino, CA, USA) with a flow rate of 1 μL/h was filled with vehicle solution (PBS/1 % BSA) or SDF-1a (500 ng/mL dissolved in 1 % BSA in PBS), connected to the catheter and subcutaneously implanted in to the neck region. During the surgery, body temperature was maintained at 36–37 °C. Postoperative aftercare included saline rehydration (2 mL) and Baytril (0.3 mL, subcutaneously, twice daily) to prevent urinary tract infection. Additionally, surgery-induced bladder dysfunction was compensated by manual emptying twice a day.

Time line of experiments (a). Behavioral assessment (BBB) during the first 7 days after SCI (b). Time course of BBB recovery in the SCI placebo group and the effect of SDF-1a administration on BBB scoring. # p ≤ 0.05 SCI/SDF-1a vs. SCI placebo. ## p ≤ 0.001 SCI/SDF-1a vs. SCI placebo. Data represent means ± SEM and from n = 9 per group and treatment is given in arbitrary units
Locomotion Deficit
Locomotion deficits of rats after SCI were scored in an open field according to Basso, Beattie, and Bresnahan (BBB) locomotion rating scale of 0 (complete paralysis) to 21 (normal) as previously described [18]. The scale assesses hind limb movements, body weight support, forelimb to hind limb coordination, and whole body movements. Two experienced raters, blinded to experimental treatment, observed open-field locomotion during a 5 min testing period after SCI and then daily until sacrifice.
RNA Extraction and Real-Time PCR
Gene expression studies were performed with tissues corresponding to the epicenter of injury. Total RNA was extracted using peqGold RNA TriFast (PeqLab, Germany) as previously described [19]. Total RNA amount and purity were determined using 260/280 ratios of optical density from each sample (Nanodrop 1000, PeqLap, Germany). Complementary DNA of 1 μg of total RNA was synthesized using the M-MLV reverse transcription (RT)-kit and random hexanucleotide primers (Invitrogen, Germany). Measurement of expression levels of target genes was performed with a mixture consisting of 2 μL reversed-transcribed cDNA, 2 μL RNAse-free water (Invitrogen, Germany), 5 μL 2× SensiMix SYBR and Fluorescein (Bioline, Germany), and 0.5 μL primers (10 pmol/μL). Quantitative real-time PCR (qrtPCR) analysis was performed using the MyIQ detection system (Biorad, Germany). Relative quantification was calculated by the ΔΔCt-method using the qbase + software (Biogazelle, Belgium). Data were expressed as relative amount of the target gene to the amount of a reference gene (CycloA). The values of sham animals were set to 100 %. Data of interest were given as relative expression. A list of used primers and analyzed genes is given in Table 1.
Tissue Preparation and Immunohistochemistry
Rats were deeply anesthetized with ketamine/xylazine (RaziCo, Iran) and transcardially perfused with PBS followed by 4 % paraformaldehyde (Sigma, Germany). The spinal cord was carefully removed and placed in the same fixative overnight for post fixation. Formalin-fixed spinal cord samples (T9, epicenter of injury) were harvested for subsequent histological processing. To this end, tissue specimens were embedded in paraffin (Merck, Germany) and 5 μm paraffin sections were transversely cut. For each animal, four slices were analyzed with a distance of 50 μm in between.
For immunohistochemistry, a Vectastain-DAB Kit (Vector Laboratories, Burlingame, CA) was used. After deparaffinizing, sections were used for heat-induced epitope retrieval (HIER) using Tris/EDTA buffer (pH 9, 20 min). Then slices were exposed to 10 % goat serum for blocking of non-specific reaction (Sigma, Germany) for 30 min and afterwards incubated overnight at 4 °C with the respective primary antibodies (for details of antisera see Table 2). The next day, slices were washed and treated with H2O2/PBS (0.3 %, 30 min) (Roth, Germany) to block endogenous peroxidase. Afterwards, sections were incubated with the appropriate secondary antibodies followed by the ABC complex. Diaminobenzidine (DAB) was used as chromogen. Finally, sections were counterstained with hematoxylin, dehydrated in graded alcohols, and mounted.
Immunofluorescence Double-Labeling
Immunofluorescence double-labeling was performed as previously described [20]. In brief, formalin-fixed and paraffin-embedded sections (5 μm) were rehydrated and unmasked by Tris/EDTA (pH 9.0) buffer, blocked in PBS containing 2 % FCS and 1 % BSA, and incubated overnight with the primary antibodies diluted in blocking solution. A list of used antibodies is given in Table 2. The following antibodies, anti-glial fibrillary acidic protein antibody (GFAP), anti-neuronal nuclear antigen (NeuN), and ionized calcium-binding adapter molecule 1 (Iba1), were combined either with anti-ASC or anti-NLRP3. Fluorescent anti-rabbit antibody (1:500, Alexa Fluor 488, Invitrogen, Germany) and anti-mouse/anti-goat antibodies (1:500, Alexa Fluor 598, Invitrogen, Germany) were used as secondary antibodies.
ELISA
Concentrations of mature IL-1b, mature Il-18, and TNF-a were measured with enzyme-linked immunosorbent assay (ELISA) kits following the manufacturer’s protocol. Briefly, spinal cord tissue was dissected, homogenized in PBS (0.02 mol/L, pH 7.0–7.2) and centrifuged at 5000×g for 10 min at 4 °C. Supernatants were collected and analyzed by ELISA. IL-1b, TNF-a, and IL-18 protein concentrations were determined using a rat IL-1b ELISA kit (900-K91, Peprotech, Rocky Hill, USA), rat TNF-a ELISA kit (900-K73, Peprotech, Rocky Hill, USA), and a rat IL-18 ELISA kit (ABIN416245, antibodies-online.com). According to the manufacturer’s instructions, samples were assayed in duplicate at absorbance rates for TNF-a, IL-1b at 405 nm, and IL-18 at 450 nm, respectively. Concentrations were calculated from respective standard curves and expressed as pictogram per milligram entire protein.
SDS PAGE and Western Blot
NLRP3, ASC, TLR-2, and caspase-1 protein were analyzed using Western blotting. The spinal cord tissue was lysed in ice-cold RIPA buffer consisting of 50 mM Tris–HCl, pH 8.0, 1 % (v/v) Nonidet P-40 (Sigma, Igepal, CA), 0.1 % SDS (sodium dodecyl sulfate), and 0.5 % sodium deoxycholate and protease inhibitor cocktail (Complete Mini, Roche, Mannheim, Germany). Protein concentration was determined using the BCATM Protein Assay Kit (Pierce, Bonn, Germany) according to the manufacturer’s protocol. Same amounts of protein samples (approx. 30 μg per lane) were loaded, separated by 8–12 % (v/v) discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a PVDF membranes (Roche, Manheim, Germany). After blocking with 5 % skimmed milk in Tris-buffered saline containing 0.05 % Tween 20 (TBS-T) for 1 h at room temperature, PVDF membranes were incubated with primary antibodies (Table 2) overnight at 4 °C. After washing with TBS-T, the membranes were incubated with a peroxidase-conjugated goat anti-rabbit (Bio-Rad, USA) secondary antibody for 2 h at room temperature. Visualization was achieved using the enhanced chemiluminescence method (ECL plus, Pierce Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. For densitometric quantification, the intensities of the specific bands were normalized to GAPDH in the same blot using ImageJ software (Free Java software provided by the National Institute of Health, Bethesda, Maryland, USA).
Data Analysis
All data are given as means ± SEM. Statistical differences between various groups were analyzed by Student’s t test and one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test using GraphPad Prism 5 (GraphPad Software Inc., USA). The BBB test was evaluated using two-way ANOVA.
Results
The male Wistar rats (350–400 g) at the age of 14 weeks were used for these experiments. After laminectomy, contusion lesions were made at the T9 spinal segment by placing a 50 g weight on the intact dura mater for 5 min. After experimental SCI, rats were intrathecally treated with SDF-1a or PBS/BSA.
In preliminary experiments, we have evaluated the influence of SDF-1a alone without any injury. SDF-1a did not change the behavioral performance per se and did not affect any of the inflammatory cascades or inflammasome factors as reported here after SCI (data not shown).
Locomotion Deficits after SCI and SDF-1a Supplementation
The BBB locomotion rating scale was developed based on the natural progression of locomotion recovery in rats with thoracic SCI. As expected, BBB scoring reached maximum values of 21 in all animals in the sham group. After injury, BBB score dropped within 6 h to 0 (Fig. 1b). This confirms a successful and correct SCI procedure. During the first 24 h after SCI, scores remained at this low level reflecting the typical spinal shock phase. After that, BBB scores gradually but slowly increased until 7 days resulting in a maximum recovery of 2–3 scores/animal after 7 days in placebo animals. The administration of SDF-1a substitution did not significantly affect the initial motor scoring depletion during the first 24 h. Thereafter, we observed a faster and significantly better recovery at 72 h and 7 days after the insult with a maximum value of 5–6 scores/animal at 7 days (Fig. 1b).
Time Course of Cytokine and Inflammasome Expression after SCI
In the next step, we have analyzed the mRNA and protein expression profiles of key inflammatory markers during the initial 7 days after SCI and the effect of SDF-1a supplementation on these parameters. IL-1b and TNF-a mRNA and protein values immediately rose within the first 6 h, remained at this high level until 24 h, and then rapidly declined but were still significantly elevated after 72 h and 7 days compared to sham controls (Fig. 2a, b). IL-18 mRNA and protein levels stepwise increased reaching significant differences compared to sham group only at 72 h. IL-18 concentrations were at all investigated time points lower compared to the other two cytokines. Since CXCR4 is the main receptor for SDF-1a [21], we have analyzed the expression profile of the CXCR4 during the first 7 days post SCI. These data clearly show a massive upregulation of the CXCR4 receptor at day 3 and 7 post injury (Fig. 2c).

Cytokine expression in the spinal cord after SCI. a Time course of IL-1b, IL-18, and TNF-a mRNA expression during 7 days post SCI analyzed by qrtPCR. b Protein amounts at the same time points of IL-1b, IL-18, and TNF-a in the spinal cord after SCI measured by ELISA. c Time course of CXCR4 mRNA expression during 7 days after SCI.*p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 all SCI vs. Sham. Data represent means ± SEM. n = 6
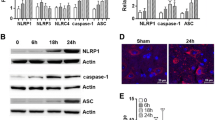
In a further attempt, we have determined the expression pattern and profile of distinct inflammasome components during the given time points and the influence of SDF-1a on inflammasome expression. As shown in Fig. 3a, NLRP1b and NLRP3 mRNA levels also rapidly increased until 6 h, whereas ASC remained low. Then, NLRP3 and ASC further rose until 72 h and dropped at 7 days. In contrast, NLRP1b already decreased after 24 h and reached basal values at 72 h post injury. The analysis of the corresponding protein values by Western blot showed that NLRP1b is already constitutively expressed in sham animals at considerable levels, then decreased being lowest at 6 and 24 h and slightly returned to basal values at 72 h (Fig. 3b). NLRP3 is only marginally found under control conditions but then increases until 72 h. A similar expression profile was seen with ASC which was already high at 24 h. Figure 3c, d give the corresponding quantitative densitometric evaluation. The analysis of cleaved active-caspase-1 protein and pro-IL-1b protein profiles revealed that pro-caspase-1 decelerates until 72 h and active-caspase-1 is strongly increased at 72 h (Figs. 3b, e). Pro-IL-1b is also clearly upregulated at 24 and 72 h.

Expression of inflammasome mRNA and protein levels and of pro-/activate-caspases-1 proteins in the spinal cord at different time points after SCI. a Time course of mRNA transcription rates. b Western blot analysis showing three representative samples per treatment and time point. c–e Quantitative assessment of protein values by densitometric evaluation. *p ≤ 0.05 and **p ≤ 0.01 SCI vs. Sham. Data represent means ± SEM. Q-PCR, n = 6; WB, n = 4
Effect of SDF-1a Treatment on Cytokine and Inflammasome Expression
Since all parameters were significantly upregulated in SCI animals after 72 h (see Fig. 2b), the influence of SDF-1a on mRNA and protein expression after SCI was determined at this time point. We observed that the administration of SDF-1a significantly reduced the SCI-dependent increase of IL-18 mRNA levels, whereas SCI-induced TNF-a and IL-1b mRNA expression was not significantly affected by SDF-1a (Fig. 4a–c). In contrast to the protein level, SDF-1a significantly attenuated the SCI-mediated upregulation of all three cytokines (Fig. 4d–f). Importantly, SDF-1a treatment did not reduce the mRNA level and protein level (data are not shown) of pro-IL1b but significantly attenuated SCI-mediated upregulation of mature IL-1b. The production of pro-IL-1b is necessary before multiprotein caspase-1-activating complex inflammasome is able to create mature IL-1b. Since SDF-1a dampened active-caspase 1, we might argue that SDF-1a diminished SCI-mediated induction of active IL-1b by preventing the increase of active-caspase-1.

Effect of SDF-1a treatment on cytokine IL-1b, IL-18, and TNF-a mRNA (a–c) and protein (d–f) expression in the spinal cord 72 h after SCI. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 all SCI vs. Sham, # p ≤ 0.05 and ## p ≤ 0.01SCI/SDF-1a vs. SCI placebo. Data represent means ± SEM. n = 6
With respect to inflammasomes, we have analyzed NLRP3 and ASC at 72 h but left out NLRP1b at that time point, since this inflammasome has already reached basal values as shown in Fig. 3a. SDF-1a significantly dampened the SCI-mediated induction of NLRP3 and ASC transcription (Fig. 5a, b) and significantly reduced NLRP3 and ASC mRNA and protein levels (Figs. 5a–d, g). In addition, SDF-1a significantly prevented the increase of active-caspase-1 without affecting pro-caspase-1 protein (Fig. 5e–g). In additional experiments, we did not find any obvious regulation of CXCR4 expression by SDF-1a (data not shown).

Effect of SDF-1a treatment on inflammasome NLRP3 and ASC mRNA and protein levels (a–d, g) and pro-/active-caspases-1 protein amounts (e–g) in the spinal cord 72 h after SCI. (g) Western blot analysis shows three representative samples per treatment and time point. **p ≤ 0.01 SCI vs. Sham, # p ≤ 0.05, ## p ≤ 0.01 and ### p ≤ 0.001 SCI/SDF-1a vs. SCI placebo. Data represent means ± SEM n = 6
Effect of SDF-1a Application on ASC+ Cell Numbers and Coocalization of ASC, and NLRP3with Iba+ Microglia, GFAP+ Astroglia, and NeuN+ Neurons, Respectively
The distribution of ASC+ cells, the effect of SDF-1a on ASC+ cell numbers, and the colocalization of ASC+ and NLRP3+ cells were investigated by immunohistochemistry and double-immunofluorescence staining in the epicenter of SCI lesion. ASC+ cells were counted in the total gray matter within the extension of the T9 segment. For each animal, a total of four slices were analyzed with a distance of 50 μm in between. Compared to sham animals, the number of ASC+ cells significantly increased after SCI 72 h. SDF-1a clearly reduced the number of ASC+ cells (Fig. 6g, h). Double-immunofluorescence staining revealed that ASC is predominantly associated with GFAP+ astroglia and Iba1+ microglia/macrophages (Fig. 6c, d). Iba1+ cells also showed considerable co-labeling with NLRP3 (Fig. 6e). GFAP+ astroglia only marginally contained NLRP3 (Fig. 6b). NeuN+ neurons occasionally co-expressed ASC (not shown) but as shown in Fig. 3a, NeuN+ were more frequently positive for NLRP3 (Fig. 6a, f).

Quantitative (cell numbers) colocalization of NLRP3 with NeuN (neuronal), Iba1 (microglia/macrophage), and GFAP (astrocyte) (a). Most of the cells expressing NLRP3 were neurons (a, f). Also a considerable number of Iba1+ cells stained for NLRP3 (a, e), whereas only few astrocytes were co-labeled (a, b). ASC is mainly expressed by microglia (d) and astrocytes (c), but neuron did not reveal ASC (not shown). g, h show the quantitative evaluation of ASC+ cell numbers in the ventral horn of the spinal cord 72 h after SCI and SDF-1a treatment and representative microphotographs of the different experimental groups. ***p ≤ 0.001 Sham vs. SCI placebo; ## p ≤ 0.01 SCI/SDF-1a vs. SCI placebo, scale bars in H (50 μm), in C-F (100 μm), and in D, E (200 μm). Data represent means ± SEM from n = 5
SDF-1a Affects Microgliosis
We reported in a previous study that microglial cells are targets for SDF-1a [14]. The numbers of Iba-1 cells were significantly higher in SDF-1a-treated compared to placebo-treated animals 4 weeks after SCI which represents a chronic stage after neurotrauma. Here, we have evaluated the effect of SDF-1a on this cell population in an acute phase after SCI (Fig. 7). We observed a slight but significant microgliosis in the SDF-1a-treated compare to placebo animals. Interestingly, microglial cells showed a different morphology after SDF-1a exposure with a more ramified phenotype (Fig. 7b) in comparison to the placebo group where Iba1+ cells were round without or with small branches. We also evaluated mRNA expression profiles of inducible nitric oxide synthase (iNOS) and arginase 1 (Arg1) which are characteristic for the pro-inflammatory M1 microglia phenotype (iNOS) and presumably more protective M2, respectively [22]. SDF-1a increased this ratio toward Arg1 and the M2 phenotype (Fig. 7c). We propose that the neuroprotective potential of SDF-1a can be ascribed to the fact that microglial function is shifted from a pro-inflammatory status toward a more protective condition. This would allow damaged neurons to better survive the critical early period after SCI onset.

Number of Iba-1+ microglia/macrophages in the gray matter of the spinal cord presented in the different treatment groups. Note the significant increase of Iba-1+ cells after SDF-1a treatment (a–b). SDF-1a up-regulates Arg1 and increases Arg1/iNOS ratios. Therefore, SDF-1a shifts microglia cells toward a M2 phenotype (c)
Discussion
SDF-1a is an interesting compound for regenerative medicine due to its ability to attract different types of stem/progenitor cells to the defect site [23]. Several studies have demonstrated that the SDF-1a/CXCR4 pathway regulates homing of engrafted stem cells to sites of tissue damage after SCI within the CNS. In particular, this treatment and stem cell migration improved recovery processes in the spinal cord [24]. Supporting this treatment with anti-SDF-1 blocking serum resulted in a marked impairment in migration and proliferation of engrafted neural stem cells [25]. Besides stimulation of cell migration, SDF-1a is able to promote neurite outgrowth of cultured neurons [12] and to protect neurons from apoptosis after ß-amyloid-induced neurodegeneration [26].
Our study clearly demonstrates that the chemokine SDF-1a reduces inflammatory responses in the spinal cord after SCI and might thereby ameliorate the measure of damages to the spinal cord tissue including functional deficits. Precisely, we found that the induction and activation of distinct key control switches centers of inflammation, the inflammasome complexes, are dampened and subsequently that the formation of pro-inflammatory cytokines related to inflammasome activity is inhibited. Further, we report that different neural cell types including neurons, astroglia, and microglia show inflammasome activation after injury, suggesting a multiplex cell-cell network implicated in local inflammation and protection by SDF-1a. Together with the previous findings which showed that chronic intrathecal delivery of SDF-1a into the damaged area after SCI protected from neuronal cell death and functional deficiency [14], we assume that the anti-inflammatory potency of SDF-1a is a key function for providing neuroprotection and functional recovery after SCI.
Acute and ongoing chronic local inflammation after SCI appears to be a key element determining the functional outcome and recovery after damage. Thus, the brain innate immune system represented by microglial cells interacts with other CNS cells primarily astroglia, the endothelium, oligodendrocytes, and hampered neurons to provoke inflammatory reactions and the stimulation of pro-inflammatory cytokine networks after SCI [27, 28]. Despite many similarities with inflammatory episodes occurring in the brain after neurotrauma, the injured spinal cord reveals distinct anatomical and molecular differences [29]. The migrating and invading macrophages represent an integral and specific feature, since the sequential activation and composition of pro-inflammatory M1 macrophages and alternatively stimulated M2a, M2b, and M2c macrophage subtypes occur during wound healing and facilitate the transition between inflammation, proliferation, and tissue remodeling [22]. Generally, SCI induces a low-grade inflammatory state which is characterized by bidirectional communication between the nervous, endocrine, and immune system [30]. Microglia accumulation peaks in the lesion on 3–7 days post SCI throughout the gray and white matter at the epicenter of the injury site [31]. These cells typically produce, after a delay of several hours, pro-inflammatory cytokines such as IL-1b and TNF-a, and recruit other cells to the damaged site [27]. Microglial cells are the primary mediators of the CNS immune defense system and crucial for shaping inflammatory responses. They represent a highly dynamic cell population which is constantly moving and surveying their environment. Acute brain damage such as SCI causes a local attraction and activation of this immune cell type which involves neuron-to-glia and glia-to-glia interactions [32]. Microglial cells can be classified into at least two subsets of phenotypes with distinct molecular and effector functions. The pro-inflammatory M1 type which produces high amounts of ROS, proteases, and pro-inflammatory cytokines and an alternatively activated M2 microglial cell subtype which promotes tissue remodeling/repair and angiogenesis [33]. It is currently believed that the dual properties (protective-destructive) of microglia cells correlate with a permanent switching of this cell type between different activity states depending on cell-cell interactions and regulatory diffusible molecules present in the near proximity. The prevailing view attributes microglia a “negative” role such as defense and debris elimination. More topical studies also suggest a protective and “positive” regulatory function. After SCI, microglia plays a decisive role in secondary injury, and the activation of a single macrophage phenotype can facilitate proper wound healing following SCI [15]. Successful orchestration of macrophage-mediated tissue repair requires fine tuning different macrophage phenotypes to harmoniously facilitate transitions among different wound healing phases [22]. Considering our data that SDF-1a treatment shifts the microglia phenotype within the spinal cord lesion site from a M1 to a M2 protective microglia cell, we also propose that SDF-1a protect neurons from cell death by interfering with microglia function. Nevertheless, further studies are required to emphasis this effect of SDF-1a on microglia.
Early after injury onset, mainly neurons and astroglia synthesize chemokines and cytokines [34, 35]. Besides orchestrating the inflammatory reaction, these pro-inflammatory factors are implicated in neuronal survival, wound healing, and myelin and debris phagocytosis. Within the first 24 h after injury, neutrophils are among the first immigrating cells occupying the injury site followed by monocytes and macrophages [35, 36]. Membrane spanning Toll-like receptors (TLRs) and the cytoplasmic sensors Nod-like receptors (NLRs), which recognize pathogens and initiate innate immune responses to protect the host also sense damaged tissue/cells and released stressor ligands, are functionally implicated in the propagation of SCI-related inflammatory cascade [37–39]. These receptors are typically expressed by a variety of spinal cord cells under regular and pathological conditions [40]. TLR and NLR are integrated in the signaling process of initiation of inflammation and form the intracellular platform responsible for activating the intracellular inflammasomes [41].
TLRs play an important role in mediating the inflammatory responses in general and in the spinal cord. It has shown that the expression of TLRs and their downstream effectors is increased after SCI [42]. TLR4 expression was primarily localized to CNS macrophages, whereas TLR2 was found in CNS macrophages and astrocytes in equivalent amounts [38]. It has been shown that TLRs are key modulators of sterile inflammation and glial activation associated with SCI, although it remains to be determined whether they mediate neuroprotection or neurodegeneration via direct effects on spinal neurons [42]. Despite considerable progress over the past decade, the role of TLRs in the physiological and pathological function of the spinal cord remains inadequately defined. As well, no data are available regarding the regulation of TLR and NLR under SDF-1a treatment protocols. Additional studies are required to define the effectiveness of SDF-1a on TLR ligands as therapeutic agents in SCI and disease. A well-characterized outcome of TLR/NLR activation and cooperation with the inflammasome complexes is the activation of cytokine processing and in particular of IL-1β and IL-18 synthesis and secretion [39, 41]. The stimulation of the cytosolic multiprotein inflammasome complexes such as NLRP3 generally requires two signals. First, a priming signal to increase the transcription of inflammasome components and a second signal that leads to the assembly and activation of the inflammasome and interleukin processing [36, 39]. In a series of studies, de Rivero Vaccari and colleagues have shown that NALP1 is expressed in neurons of the intact rat spinal cord. Further, SCI promoted the assembly and activity of this inflammasome complex, and neuronal purinergic P2X receptor subtypes are involved in SCI-mediated NALP1 activation [6]. Another report showed that the inhibition of Rho kinase signaling pathways after SCI reduced the secondary neural injury in conjunction with attenuated inflammasome activation [43]. In a recent study, we could demonstrate that polyunsaturated fatty acids such as omega-3 fish oil components are neuroprotective agents in another acute brain damaging model, i.e., ischemic stroke in rats [19]. The observed reduction of the infarct area was clearly associated with a prevention of NLRP3 activation in the brain. The present study now demonstrates that different inflammasome complex compounds are stimulated and activated in a coordinated fashion and time setting during the first 3 days and then drop until 7 days with ASC and NLRP3 remaining elevated compared to basal activities. We also show that, besides neurons, astroglia and microglia express subtypes of inflammasome devices to a substantial extent. This clearly emphasizes the importance of the local cellular inflammatory network in the initiation and maintenance of local inflammatory responses after SCI.
In the adult and healthy CNS, SDF-1a is constitutively expressed, although at relatively low levels [45] and is typically upregulated during neuroinflammatory challenges, particularly by endothelial cells of micro blood vessels and astrocytes in multiple sclerosis [46]. SDF-1a fulfills the classical role of a chemokine in the local immune responses and is implicated in the attraction of CXCR4+ T cells and monocytes to the sites of inflammation [47]. It is believed that innate immune cells migrate along a concentration gradient of chemokine (s) across the blood–brain barrier (BBB) to their target [48]. In the immune system, the binding of SDF-1a to CXCR4 induces intracellular signaling through several divergent pathways, such as upregulating the binding activity of LFA-1 and VLA-4 on T cells, thereby initiating and controlling chemotaxis [47]. SDF-1a and/or its receptors are also associated with the pathogenesis of autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus [47]. In multiple sclerosis and related animal models mimicking aspects of multiple sclerosis, SDF-1a appears to be one of the major regulators of neuroinflammation and interference with SDF-1a signaling suggests a protective role for this chemokine [49] by inducing CD4+ cell apoptosis [50] and switching antigen-specific effector T cells from a destructive to a regulatory phenotype [51]. Irrespective of the anti-inflammatory potency, SDF-1a might in addition help to overcome tissue damage within the CNS by affecting the migration and survival of oligodendrocyte precursors and facilitating remyelination [52].
In conclusion, we have demonstrated in our study the involvement of different inflammasome complexes and their modules to be sequentially activated in the zone of tissue injury after SCI. The administration of SDF-1a reduced tissue damage in the spinal cord and improved functional recovery. Our data let us reason that the SDF-1a-mediated attenuation of inflammasome activation and local inflammatory processes are crucial for the observed neuroprotective action.
References
Thuret S, Moon LD, Gage FH (2006) Therapeutic interventions after spinal cord injury. Nat Rev Neurosci 7(8):628–643
Schwab ME, Bartholdi D (1996) Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev 76(2):319–370
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140(6):821–832
de Zoete MR, Palm NW, Zhu S, Flavell RA (2014) Inflammasomes. Cold Spring Harbor perspectives in biology:a016287
Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14(9):1583–1589
de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW (2008) A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 28(13):3404–3414
Liang F, Li C, Gao C, Li Z, Yang J, Liu X, Wang Y (2015) Effects of hyperbaric oxygen therapy on NACHT domain-leucine-rich-repeat-and pyrin domain-containing protein 3 inflammasome expression in rats following spinal cord injury. Mol Medi Rep 11(6):4650–4656
Aiuti A, Webb I, Bleul C, Springer T, Gutierrez-Ramos J (1997) The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med 185(1):111–120
Cui L, Qu H, Xiao T, Zhao M, Jolkkonen J, Zhao C (2013) Stromal cell-derived factor-1 and its receptor CXCR4 in adult neurogenesis after cerebral ischemia. Restor Neurol Neurosci 31(3):239–251
Shyu W-C, Lin S-Z, Yen P-S, Su C-Y, Chen D-C, Wang H-J, Li H (2008) Stromal cell-derived factor-1α promotes neuroprotection, angiogenesis, and mobilization/homing of bone marrow-derived cells in stroke rats. J Pharmacol Exp Ther 324(2):834–849
Jaerve A, Bosse F, Müller HW (2012) SDF-1/CXCL12: its role in spinal cord injury. Int J Biochem Cell Biol 44(3):452–456
Opatz J, Küry P, Schiwy N, Järve A, Estrada V, Brazda N, Bosse F, Müller HW (2009) SDF-1 stimulates neurite growth on inhibitory CNS myelin. Mol Cell Neurosci 40(2):293–300
Jaerve A, Schiwy N, Schmitz C, Mueller HW (2011) Differential effect of aging on axon sprouting and regenerative growth in spinal cord injury. Exp Neurol 231(2):284–294
Zendedel A, Nobakht M, Bakhtiyari M, Beyer C, Kipp M, Baazm M, Joghataie MT (2012) Stromal cell-derived factor-1 alpha (SDF-1α) improves neural recovery after spinal cord contusion in rats. Brain Res 1473:214–226
Zhang Y-K, Liu J-T, Peng Z-W, Fan H, Yao A-H, Peng C, Liu L, Ju G et al (2013) Different TLR4 expression and microglia/macrophage activation induced by hemorrhage in the rat spinal cord after compressive injury. J Neuroinflammation 10:112
Yaksh TL, Rudy TA (1976) Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17(6):1031–1036
Nyström B, Berglund JE (1988) Spinal cord restitution following compression injuries in rats. Acta Neurol Scand 78(6):467–472
Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12(1):1–21
Zendedel A, Habib P, Dang J, Lammerding L, Hoffmann S, Beyer C, Slowik A (2015) Omega-3 polyunsaturated fatty acids ameliorate neuroinflammation and mitigate ischemic stroke damage through interactions with astrocytes and microglia. J Neuroimmunol 278:200–211
Clarner T, Diederichs F, Berger K, Denecke B, Gan L, Van der Valk P, Beyer C, Amor S et al (2012) Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 60(10):1468–1480
Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J et al (2004) CXCR4–SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol 35(3):233–245
Gensel JC, Zhang B (2015) Macrophage activation and its role in repair and pathology after spinal cord injury. Brain research
Nagasawa T (2014) CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J Mol Med 92(5):433–439
Takeuchi H, Natsume A, Wakabayashi T, Aoshima C, Shimato S, Ito M, Ishii J, Maeda Y et al (2007) Intravenously transplanted human neural stem cells migrate to the injured spinal cord in adult mice in an SDF-1-and HGF-dependent manner. Neurosci Lett 426(2):69–74
Carbajal KS, Schaumburg C, Strieter R, Kane J, Lane TE (2010) Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci 107(24):11068–11073
Raman D, Milatovic S-Z, Milatovic D, Splittgerber R, Fan G-H, Richmond A (2011) Chemokines, macrophage inflammatory protein-2 and stromal cell-derived factor-1α, suppress amyloid β-induced neurotoxicity. Toxicol Appl Pharmacol 256(3):300–313
Mortazavi MM, Verma K, Harmon OA, Griessenauer CJ, Adeeb N, Theodore N, Tubbs RS (2015) The microanatomy of spinal cord injury: a review. Clin Anat 28(1):27–36
Skaper S, Facci L, Giusti P (2014) Neuroinflammation, Microglia and Mast Cells in the Pathophysiology of Neurocognitive Disorders: A Review. CNS & neurological disorders drug targets
Zhang B, Gensel J (2014) Is neuroinflammation in the injured spinal cord different than in the brain? Examining intrinsic differences between the brain and spinal cord. Exp Neurol 258:112–120
Allison D, Ditor D (2014) Immune dysfunction and chronic inflammation following spinal cord injury. Spinal cord
Sroga JM, Jones T, Kigerl KA, McGaughy VM, Popovich PG (2003) Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol 462(2):223–240
Habib P, Beyer C (2015) Regulation of brain microglia by female gonadal steroids. J Steroid Biochem Mol Biol 146:3–14
Orihuela R, McPherson CA, Harry GJ (2015) Microglial M1/M2 polarization and metabolic states. British journal of pharmacology
Rice T, Larsen J, Rivest S, Yong VW (2007) Characterization of the early neuroinflammation after spinal cord injury in mice. J Neuropathol Exp Neurol 66(3):184–195
Pineau I, Sun L, Bastien D, Lacroix S (2010) Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav Immun 24(4):540–553
Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H et al (1997) Role of neutrophils in spinal cord injury in the rat. Neuroscience 79(4):1177–1182
Gaudet AD, Popovich PG (2014) Extracellular matrix regulation of inflammation in the healthy and injured spinal cord. Exp Neurol 258:24–34
Heiman A, Pallottie A, Heary RF, Elkabes S (2014) Toll-like receptors in central nervous system injury and disease: a focus on the spinal cord. Brain Behav Immun 42:232–245
Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15(2):84–97
Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW (2014) Pattern recognition receptors and central nervous system repair. Exp Neurol 258:5–16
Hanamsagar R, Hanke ML, Kielian T (2012) Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol 33(7):333–342
Kigerl KA, Popovich PG (2009) Toll-like receptors in spinal cord injury. In: Toll-like Receptors: Roles in Infection and Neuropathology. Springer, pp 121–136
Impellizzeri D, Mazzon E, Paterniti I, Esposito E, Cuzzocrea S (2012) Effect of fasudil, a selective inhibitor of Rho kinase activity, in the secondary injury associated with the experimental model of spinal cord trauma. J Pharmacol Exp Ther 343(1):21–33
Lu M, Grove EA, Miller RJ (2002) Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci 99(10):7090–7095
Li M, Ransohoff RM (2008) Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol 84(2):116–131
Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM (2005) Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol 64(8):706–715
Karin N (2010) The multiple faces of CXCL12 (SDF-1α) in the regulation of immunity during health and disease. J Leukoc Biol 88(3):463–473
Engelhardt B, Ransohoff RM (2012) Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trends Immunol 33(12):579–589
Momcilović M, Mostarica-Stojković M, Miljković D (2012) CXCL12 in control of neuroinflammation. Immunol Res 52(1–2):53–63
Colamussi ML, Secchiero P, Gonelli A, Marchisio M, Zauli G, Capitani S (2001) Stromal derived factor-1α (SDF-1α) induces CD4+ T cell apoptosis via the functional up-regulation of the Fas (CD95)/Fas ligand (CD95L) pathway. J Leukoc Biol 69(2):263–270
Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N (2008) CXCL12 (SDF-1α) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med 205(11):2643–2655
Patel JR, McCandless EE, Dorsey D, Klein RS (2010) CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc Natl Acad Sci 107(24):11062–11067
Acknowledgments
This work was supported by an internal START grant from the Medical Clinic (UKA) of the RWTH Aachen University (A. Zendedel).
Conflict of Interest
The authors declare that they have no competing interests.
Ethical Statement
All animals used in this study were housed under specific pathogen-free conditions and with access to standard rodent diet and tap water ad libitum. All experiments were conducted in conformance with the NIH Guidelines for the Care and Use of Laboratory Animals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Adib Zendedel and Sonja Johann contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zendedel, A., Johann, S., Mehrabi, S. et al. Activation and Regulation of NLRP3 Inflammasome by Intrathecal Application of SDF-1a in a Spinal Cord Injury Model. Mol Neurobiol 53, 3063–3075 (2016). https://doi.org/10.1007/s12035-015-9203-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9203-5