
Abstract
Down syndrome candidate region 1 (DSCR1) has two differentially regulated isoforms (DSCR1-1 and DSCR1-4) and is reported to play a role in a number of physiological processes, such as the inhibition of cardiac hypertrophy, attenuation of angiogenesis and carcinogenesis, and protection against neuronal death. However, the function of DSCR1 in the retina is still not clear. Therefore, we analyzed the expression and location of DSCR1 in the retina of neonatal mice with oxygen-induced retinopathy (OIR), and studied its effects on angiogenesis. The neonatal C57BL/6J mice were exposed to 75 % O2 for 5 days from postnatal day 7 (P7) to P12. At P12, the mice were returned to 21 % O2 at room air. The primary retinal ganglion cells (RGCs) were exposed to hypoxia (93 % N2, 5 % CO2, and 2 % O2) at 37 °C for 36 h. And then the mouse retinal microvascular endothelial cells (mRMECs) were treated with 25 ng/mL vascular endothelial growth factor (VEGF) or culture medium conditioned by hypoxic RGCs alone, hypoxic RGCs treated with DSCR1-4-siRNA (siDSCR1-4) or hypoxic RGCs treated with siDSCR1–4 and 200 ng/mL cyclosporin A (CsA), and then primed with VEGF (25 ng/mL). The expression of DSCR1-4 increased strongly at P16 after OIR. There was no change in messenger RNA (mRNA) expression of DSCR1-1 at P16 after OIR. The increased DSCR1 was mainly located in the RGCs of avascular retina. In addition, DSCR1-4 expression was increased in primary RGCs after hypoxia exposure. There was no change in mRNA expression of DSCR1-1 in primary RGCs after hypoxia exposure. Moreover, DSCR1-4 produced by hypoxic RGCs showed anti-angiogenic properties, with decreased cell proliferation, migration, tube formation, and inflammatory cytokines production. These properties were due to inhibited nuclear factor of activated T cell (NFATc) 1 dephosphorylation and translocation into nuclear in VEGF-treated mRMECs. Using siRNA-mediated knockdown of DSCR1-4 and NFATc1 inhibitor (Cs A) further demonstrated the inhibitory effect of DSCR1-4 on angiogenic properties in VEGF-induced mRMECs, and this effect was NFATc1-dependent. This report describes a novel effect of DSCR1-4 in the aspect of anti-angiogenesis, suggesting potential therapeutic strategies for proliferative retinopathies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proliferative retinopathies (PRs) are the major cause of vision loss. They play key roles in various retinal diseases, including diabetic retinopathy (DR) and retinopathy of prematurity (ROP) [1, 2]. Accumulating evidences suggested that vascular endothelial growth factor (VEGF) and its receptors (VEGFR-1 and VEGFR-2) could promote the occurrence of DR and ROP [1–3]. Over the past decade, anti-VEGF therapy was shown to be an effective anti-angiogenic therapy and became the major therapeutic strategy in these diseases [3]. However, anti-VEGF intravitreous injection could result in the contraction of retinal proliferative membranes, which causes the recurrence of retinal tear and macular edema [1]. Moreover, long-term anti-VEGF administration also induces tachyphylaxis [4, 5]. Therefore, exploring of other potential therapies to control pathological angiogenesis is of great necessity.
PRs develop in two phases [6]. Firstly, abnormal metabolism in DR or premature birth in ROP causes vessel regression or existing vessel loss. In the second phase, retinal vessel regression or loss induces ischemia and hypoxia of retina, which leads to the secretion of VEGF and other inflammatory cytokines, then gives rise to the pathologic retinal neovascularization (PRV) [6, 7]. Recently, more and more studies have paid attention to the cross-talk between retinal neurons and vessels, which not only shapes vascular development, but also affects pathological settings of PRs [8–11]. Understanding how retinal neurons respond to ischemic and hypoxic stimulation and its role in vessel growth regulation are important for the identification of new therapeutic targets.
Down syndrome candidate region 1 (DSCR1, also known as RCAN1, MCIP1, Adapt78, or calcipressin 1) is located in chromosome 21 and is implicated in Down syndrome [12]. DSCR1 is abundantly expressed in multiple tissues, such as brain [13–15], heart [16], tumor [12, 17, 18], and skeletal muscle [19]. DSCR1 has two differentially regulated isoforms (DSCR1-1 and DSCR1-4). Previous reports indicated that DSCR1-4 was upregulated in VEGF-induced vascular endothelial cells, and it provided a negative feedback loop that inhibiting VEGF-induced angiogenic responses and tumor growth in vivo through suppressing nuclear factor of activated T cell (NFATc) 1-dependent pathway [12, 17, 18]. However, over-expression of the isoform DSCR1-1 promoted angiogenesis [12, 17, 18]. Furthermore, numbers of studies showed that DSCR1 was upregulated in peri-infarct cortex neurons after experimental stroke [13] and protective against oxidant-induced neuronal apoptosis [14, 20]. Over-expression of DSCR1 could inhibit the neutrophil infiltration and microglia activation, and protect neurons from apoptosis during post-ischemic neuronal injury [15]. Such observations suggest that DSCR1 plays a significant role in angiogenic responses and neuroprotection. However, the expression of DSCR1 in ischemic and hypoxic retina and the regulation of PRV by DSCR1 in retina remains unknown.
Herein, our study reports for the first time the expression and distribution exchanges of DSCR1-4 in mouse retina of an oxygen-induced retinopathy (OIR) model, and further explores the anti-angiogenic properties and possible mechanisms of DSCR1-4 secreted from ischemic retinal ganglion cells (RGCs) in VEGF-induced mouse retinal microvascular endothelial cells (mRMECs). These results may gain an insight into the possible roles of DSCR1 in PRs, further making appropriate suggestions for clinical treatments.
Materials and Methods
Reagents
The Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12), fetal bovine serum (FBS), endothelial cell medium (ECM), and neurobasal cell medium (NCM) were purchased from Gibco BRL (Grand Island, NY, USA). The poly-L-lysine, glutamine, brain-derived neurotrophic factor (BDNF), bovine serum albumin (BSA), ciliary neurotrophic factor (CNTF), forskolin, papain, collagenase, DL-cysteine, ovomucoid, 0.004 % DNase I, cyclosporin A (CsA), and VEGF were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Gentamicin, B27 supplement, phosphate-buffered saline (PBS) solution and red-light-absorbing dye labeled Griffonia simplicifolia isolectin B4 (IB4) were purchased from Invitrogen Life Technologies Co. (Carlsbad, CA, USA). Antibodies against DSCR1-4, VEGF, NFATc1, HDAC1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and Thy-1.1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Proliferating cell nuclear antigen (PCNA), Ki67, OX-42, and β-actin were purchased from Cell Signaling (Beverly, CA, USA). DSCR1-4 was purchased from Sigma Chemical Co. (St. Louis, MO, USA). NeuN was purchased from Millipore (Millipore, CA, USA). βIII-tubulin was purchased from Covance (Covance, Princeton, NJ, USA). The nuclear and cytoplasmic protein extraction kit was purchased from Beyotime Institute of Biotechnology (Shanghai, China).
Mouse OIR Model
C57BL/6J mice was purchased from the Animal Laboratory of Zhongshan Ophthalmic Center (Guangzhou, China). The mouse OIR model was induced as our previously studies [21–23]. Briefly, the neonatal C57BL/J mice were exposed to 75 % O2 for 5 days from postnatal day 7 (P7) to P12. At P12, the mice were returned to 21 % O2 at room air. In the OIR and control groups, no animals were lost before the determined time points. The mouse were used for retinal flat mounting and DSCR1, βIII-tubulin, or IB4 labeling (n 4 × 6 = 24), Western blot analysis (n 10 × 6 = 60), real-time PCR (n 10 × 6 = 60).
Immunofluorescent Confocal Microscopy
After various treatments, cells were harvested and washed with PBS twice for 10 min. After being fixed in 4 % formaldehyde in PBS for 30 min, the cells were permeabilized with 0.1 % Triton-X100 in PBS for 1 h and then blocked by 1 % BSA. The primary antibodies NeuN (anti-rabbit, 1:100); βIII-tubulin (anti-mouse, 1:200); NFATc1 (anti-rabbit, 1:100); or Ki67 (anti-rabbit, 1:100) were incubated at 4 °C overnight, and then incubated with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit or anti-mouse IgG. Cells were incubated with DAPI for 10 min. Images were observed under confocal microscope (Carl Zeiss, Oberkochen, Germany).
Whole Mount Retinal Immunofluorescence
To evaluate the distribution and localization of DSCR1-4 in retina at P16 after OIR, six rats from each group were sacrificed, and 12 eyes from each group were fixed in 4 % formalin at room temperature for 2 h. The retinas were dissected and blocked in PBS containing 0.5 % Triton X-100 and 5 % BSA at 4 °C overnight. Subsequently, the retinas were incubated with IB4 (1:50, a marker for vessels), DSCR1-4 (anti-rabbit, 1: 50) and/or βIII-tubulin (anti-mouse, 1:200) at 4 °C overnight. Retinas were washed with PBS 15 min for three times, and mounted on microscope slides. Areas of avascular and neovascularized areas in retinas were examined by fluorescence microscopy (AxioCam MRC; Carl Zeiss, Thornwood, NY). The expression of DSCR1 and βIII-tubulin in the avascular and neovascularized areas of retinas were examined by confocal microscopy (Zeiss510; CarlZeiss).
The Microdissection of Avascular and Neovascularized Zones
To evaluate the distribution of DSCR1-4 in retina at P16 after OIR, rats from each group (n 4 × 6 = 24) were sacrificed, and the retinas were dissected and blocked in PBS containing IB4 (1:50, a marker for vessels) at 4 °C for 30 min and treated with RNase inhibitor at 4 °C for 10 min. Retinas were washed with PBS 10 min for three times, and mounted on microscope slides at 4 °C. The avascular and neovascularized zones of retinas from OIR mice were microdissected by ophthalmic microsurgical instruments under fluorescence microscopy (AxioCam MRC; Carl Zeiss, Thornwood, NY). The collected total protein was obtained for Western blotting analyses.
Protein Extraction and Western Blotting Analyses
For Western blot analysis, total protein was obtained by lysing in a buffer containing 1 % Triton X-100, 0.5 % sodium deoxycholate, 1 M Tris-HCl pH 7.5, 0.5 M EDTA, 1 % NP-40 (nonidet P-40), 10 μg/mL aprotinin, 10 % sodium dodecyl sulfate, 10 μg/mL leupeptin, and 1 mM phenylmethylsulfonyl fluoride. In a parallel experiment, nuclear and cytosol proteins were prepared according to the manufacturer’s instructions. 20–50 μg protein was separated with sodium dodecyl sulfate-PAGE and transferred to polyvinylidine difluoride filter (PDF) membrane (Millipore, Bedford, MA). After blocking with 5 % defatted milk in PBS-Tween-20 for 1 h at room temperature, the PDF membrane was incubated with primary antibody against DSCR1-4 (anti-rabbit, 1:400); VEGF (anti-mouse, 1:500); NFATc1 (anti-goat, 1:300); PCNA (anti-mouse, 1:500); HDAC1 (anti-mouse, 1:500); β-actin (anti-mouse, 1:800); and GAPDH (anti-rabbit, 1:800) at 4 °C overnight. After being washed with PBS-Tween-20 every 15 min for three times, the membrane was incubated with goat-anti-rabbit or goat-anti-mouse second antibody conjugated horseradish peroxidase (1:10,000; Abgent) for 2 h and then scanned with the Odyssey infrared imaging system (LI-COR Bioscience).
Real-Time Polymerase Chain Reaction
Total RNA of retinas or primary RGCs of mice, or mRMECs were extracted using TRIzol and converted to complementary DNA (cDNA) by using a cDNA first-strand synthesis system (Fermentas, Canada). The PCR primers were designed based on the NCBI messenger RNA (mRNA) and genome DNA sequence database. The primers of target genes were as follows: COX-2 forward primer: 5′-3′-ccagatgatatctttggggagac and reverse primer: 5′-3′-cttgcattgatggtggctg. iNOS forward primer: 5′-3′-acaacaggaacctaccagctca and reverse primer: 5′-3′-gatgttgtagcgctgtgtgtca. MCP-1 forward primer: 5′-3′-actgaagccagctctctcttcctc and reverse primer: 5′-3′-ttccttcttggggtcagcacagac. DSCR1-1 forward primer: 5′-3′-gccaccatcgcctgtcacctg and reverse primer: 5′-3′- gctcttaaaatactgaaaggtg. DSCR1-4 forward primer: 5′-3′- tgggtctgtagcgctttcactg and reverse primer: 5′-3′- gaaagtgaaaccagggccaaatt. VEGF forward primer: 5′-3′-gcacataggagagatgagcttcc and reverse primer: 5′-3′-ctccgctctgaacaaggct. β-actin forward primer: 5′-3′-ggcggactatgacttagttg and reverse primer: 5′-3′-aaacaacaatgtgcaatcaa. DNA fragments of PCR products were designed to amplify within 200-bp length. Results were normalized from β-actin of respective samples. Briefly, each reaction contained 0.8 μL primer (containing 100 pM forward and reverse primers), 2 μL of cDNA (0.1 μL of RNA equivalent), 5 μL of Sofast EvaGreen supermix, and 2.2 μL of H2O. Quantitative real-time pCR was performed at 94 °C for 45 s and 55 °C for 45 s to denaturation, and at 72 °C for 45 s for 50 cycles to extension. All data were analyzed by CFX manager software (BioRad, Hercules, CA, USA).
Purification and Identification of Primary RGCs
The cultures and purification of primary RGCs was performed as described previously [24, 25] with minor changes. Briefly, retinas (n 12 × 2 = 24) from C57BL/6J mouse at P1 to P2 were incubated at 37 °C in a 15 U/mL papain and 70 U/mL collagenase in PBS solution for 10–15 min. To yield single-cell suspension, the tissue was triturated in a solution containing 1 mg/mL BSA, 0.004 % DNase I and 2 ng/mL ovomucoid through an arrow-bore pasteur pipette. After centrifugation at 1500 rpm for 5 min, the cells were rewashed in 10 mg/mL ovomucoid and 10 mg/mL BSA solution, and re-suspended in PBS with 0.1 % BSA.
The cell suspension was incubated in the OX-42 antibody (a specific microglia cells marker, 1:100) coated flasks at 37 °C for 30 min. The suspension was gently shaken every 20 min to remove all adherent cells. Non-adherent cells were placed in the Thy-1.1 (a specific RGCs marker, 1:100) coated flasks. The cells were incubated for 50 min and the flasks were gently washed with PBS for five times. Finally, adherent cells on Thy-1.1 antibody coated flasks were washed in 10 mg/mL ovomucoid and 10 mg/mL BSA solution. After centrifugation at 1500 rpm for 5 min, the cells were seeded on 50 mg/mL poly-L-lysine-coated flasks. The purity of the primary RGCs in cultures were determined by staining with the antibody NeuN and βIII-tubulin, two specific RGCs markers (Supplemental Fig. 1). The percentage of RGCs in the cultures was about 95 %.
Primary RGCs Cultures
Purified primary RGCs were plated at 5 × 103 cells/well and cultured in medium containing NCM with 1:50 B27 supplement, 50 ng/mL CNTF, 50 ng/mL BDNF, 10 % FBS, 1 mM glutamine, 10 mM forskolin, and 10 mg/mL gentamicin. Cultures were maintained at 37 °C in a humidified incubator containing 5 % CO2 and 95 % air.
Hypoxia Model of Primary RGCs in Vitro
To study the DSCR1-4 expression in hypoxia-induced primary RGCs, medium was removed and replaced with DMEM/F12 containing 1 % FBS and 1 % glutamine. The cells were exposed to hypoxia in a chamber at 93 % N2, 5 % CO2, and 2 % O2 at 37 °C for different time (0, 4, 8, 12, 24, 36, and 48 h). In control group, the cells were incubated at 37 °C incubator with 95 % air and 5 % CO2.
Transfection with siRNA
The primary RGCs were transfected using Oligofectamine (Life Technologies) according to the manufacturer’s protocol. The siRNA nucleotides targeting mouse DSCR1-4 were purchased from Dharmacon (Chicago, IL). The siRNA sequence was as follows: mouse DSCR1-4 siRNA, 5′-aaacgagucagaauaaacuuu-3′. Four hours after transfection, analyses were conducted at 48 h after transfection.
mRMECs Culture and Identification
Primary mRMECs were purchased from Cell Biologics (cat # CD-1065, Chicago, IL 60612) and were cultured in ECM supplemented with 10 % FBS. Routine evaluation for FITC-marked CD31 showed that cells were >99 % pure (data not shown). Cells at passages 5–8 were used in this study. All cultures were incubated at 37 °C in 5 % CO2 and 95 % air.
mRMECs Treatment
The treatments of mRMECs were divided into five groups. (1) Control group (only cultured in ECM supplemented with 10 % FBS), (2) Treated with VEGF (25 ng/mL) alone, (3) Treated with culture medium conditioned by hypoxic RGCs and then primed with VEGF (25 ng/mL), (4) Treated with culture medium conditioned by hypoxic RGCs treated with siRNA to DSCR1-4 (siDSCR1-4) and then primed with VEGF (25 ng/mL), and (5) Treated with culture medium conditioned by hypoxic RGCs treated with siDSCR1-4 and CsA (200 ng/mL), and then primed with VEGF (25 ng/mL).
Cell Viability Assay
Cell viability was measured by MTT assay. mRMECs were seeded into 96-well plates at a density of 1 × 10 cells/well. After various treatments, the cells were incubated with 10-μL MTT solution (0.25 mg/mL), and incubated at 37 °C for 4 h in a humidified 5 % CO2 atmosphere. After dimethyl sulfoxide (100 μl/well) was added to each well, absorbance was read at 540 nm using a microplate reader (BioRad, Hercules, CA, USA). Cell viability was expressed as the ratio of clustered mRMECs to control group.
Enzyme-Linked Immunosorbent Assay
mRMECs were plated into 96-well plates (1 × 105 cells/well). After various treatments, a 100-μL aliquot of each the mRMECs culture medium supernatant was collected for determination of the IL-8, ICMA-1, and VCAM-1 concentrations by enzyme-linked immunosorbent assay (ELISA). The levels of IL-8, ICMA-1, and VCAM-1 were measured using ELISA kits (R&D Systems, Minneapolis, MN).
Migration Assay
First, 1 × 105 cells in serum-free medium were seeded in the upper compartment of a transwell chamber (Corning, Lowell, MA). After various conditioned culture medium treatment on the lower membrane for 24 h, the migrated cells on the lower membrane were stained with 0.1 % crystal violet and 20 % methanol. The positive colonies were counted from four fields of one membrane under microscope (×20 objective), and the average was calculated.
Tube Formation of mRMECs in Matrigel
The 96-well plates were precoated with a 1:1 mixture of cold Matrigel (10 mg/mL; BD Biosciences): ECM. mRMECs were plated at cells/well in ECM supplemented with or not with different treatment. After 12 h, pictures were taken for each condition. Capillary tube formation was evaluated by measuring the number of tubes per field.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism (v6.0) (GraphPad Software Inc.). In all cases, P < 0.05 was considered statistically significant. Student’s t test was used when two groups were compared. One-way ANOVA followed by Tukey multiple comparison was used when three or more groups were compared.
Results
Changes of DSCR1-4 Expression in the Retina after OIR
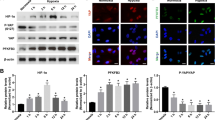
Western blotting analysis was performed to study the expression pattern of DSCR1 protein level in different time points after OIR. DSCR1 has two exons: DSCR1-1 (37 KD) and DSCR1-4 (28 KD) [17, 18, 26, 27]. According to our results and the DSCR1 antibody instructions, we found only one band and the weight was 28 KD, so we confirm that the result of western blot is DSCR1-4. As shown in Fig. 1a, b, DSCR1-4 protein level increased from P14, peaked at P16, and then gradually decreased at P18 and P20. Besides, according to previous studies [26], we designed primers of DSCR1-1 and DSCR1p4. Real-time PCR analysis for DSCR1 mRNA was performed. As shown in Fig. 1c, the DSCR1-4 mRNA level was elevated after OIR. The most robust increase was found at P14 and declined gradually. There was no change in mRNA expression of DSCR1-1 in P16 after OIR (Fig. 1d). These results demonstrated that OIR induced DSCR1-4 protein and mRNA expression in a time-dependent manner in retina.

Protein and mRNA expression of DSCR1 in retina after OIR. a, b Western blotting results showed time course of DSCR1-4 protein level in the retina after OIR. Quantification graphs (relative density) of the intensity of staining of DSCR1-4 to GAPDH at each time point. c, d Time course of DSCR1-4 (c) and DSCR1-1 (d) mRNA level in the retina after OIR. The mRNA level of Normal-P12 group set at 1. Quantification graphs (relative density) of the intensity of staining of DSCR1 to Normal-P12 group. The data are mean ± SEM (n = 6, ***P < 0.001; significantly different from Normal-P12 group)
DSCR1-4 Is Upregulated in the RGCs of Central Avascular Retina after OIR
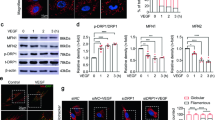
To further determine the temporal changes of DSCR1-4 in retina after OIR, microdissection was used to dissect the avascular and neovascularized zones of retinas from OIR mice (P16). Western blotting (Fig. 2e, f) and real-time PCR (Fig. 2g) analysis showed that the increased expression of DSCR1-4 was precisely located to central avascular zones in microdissected retinas. In addition, we performed immunofluorescent staining on whole mount retinas to show the double labeling of DSCR1-4 with a vascular marker IB4 (Fig. 3a), which also showed that DSCR1-4 expression was increased in avascular zones of retinas from OIR mice (P16), compared with that in neovascularized areas (Fig. 3b). But no changes were found between peripheral and central retina of normoxic controls (P16).

Distribution of DSCR1-4 in retina after OIR by Western blotting and qRT-PCR. a–d The immunostaining on whole mount retinas taken at P16 of OIR (b) demonstrating the principal characteristics of the A (yellow, c) and N (green, d) zones compared to normal retina at P16 (a). e–g Next, we micro-dissected the zones (A and B) of retinas from OIR mice at P16. The protein expression (by Western blotting, e, f) and mRNA expression (by qRT-PCR, g) of DSCR1-4 were strongly increased between A and NV zones at P16. These data are means ± SEM. (n = 6, ***P < 0.001 significantly different from the NV zones). A zones avascular zones, V zones neovascularized zones, P retina peripheral retina, C retina central retina

Distribution of DSCR1-4 in retina after OIR by immunohistochemistry. a Confocal imaging of immunohistochemistry on whole mount retinas (OIR at P16) reveals an obviously increased expression of DSCR1-4 in A zones as confirmed by merging with a vascular marker IB4. b DSCR1-4 positive cells were quantified in six randomly selected fields. DSCR1-4 expression in the P and C retina of normoxic controls are comparable as negative control. These data are means ± SEM. (n = 6, ***P < 0.001 significantly different from the NV zones). A zones avascular zones, V zones neovascularized zones, P retina peripheral retina, C retina central retina
To further address the cell types expressing DSCR1-4, double labeling immunofluorescent staining was performed on the avascular and neovascularized zones of retinas from OIR mice (P16) with RGC-specific markers: βIII-tubulin. The results of Fig. 4 revealed that increased DSCR1-4 expression on the avascular zones was almost co-localized with βIII-tubulin, indicating that the localization of DSCR1-4 appeared to be confined to the RGCs of avascular zones from OIR mice.

Localization of DSCR1-4 in retina after OIR. Confocal imaging of immunohistochemistry on whole mount retinas (OIR at P16) reveals a predominant expression of DSCR1-4 in A zones by RGCs as confirmed by merging with RGCs marker βIII-tubulin. A zones avascular zones, V zones neovascularized zones, P retina peripheral retina, C retina central retina
DSCR1-4 and VEGF Is Upregulated in Hypoxia-Induced RGCs
To further study time course of DSCR1 and VEGF expression in hypoxic RGCs after OIR, we exposed primary RGCs to hypoxia (2 % O2) for different durations (0, 4, 8, 12, 24, 36, and 48 h). Western blotting (Fig. 5a, b) and real-time PCR (Fig. 5c) analysis showed VEGF level started to increase at 4 h and reached its peak at 12 h. VEGF level gradually reduced to normal level from 36 to 48 h. DSCR1-4 protein (Fig. 5a, b) and mRNA (Fig. 5c) levels were low in control group (0 h). But after exposure to hypoxia, the expression of DSCR1-4 gradually increased and peaked at 36 h, whereas VEGF reduced to normal level at 36 h. There was no change in mRNA expression of DSCR1-1 in primary RGCs after hypoxia exposure (Fig. 5d). Our immunofluorescent staining results (Fig. 5e) were consistent with the Western blotting and real-time PCR results, and showing significantly increased VEGF at 12 h and DSCR1-4 at 36 h in primary RGCs after hypoxia exposure. Therefore, mRMECs incubated with conditioned medium from RGCs were exposed to hypoxia for 36 h (low VEGF, high DSCR1-4) in the subsequent experiments.

The expression of DSCR1-4 and VEGF in hypoxia-induced primary RGCs. a, b Western blotting results showed time course of DSCR1-4 and VEGF protein levels in hypoxia-induced primary RGCs. Quantification graphs (relative density) of the intensity of staining of DSCR1-4 and VEGF to GAPDH at each time point. c, d Real-time PCR results showed time course of DSCR1-4, VEGF, and DSCR1-1 mRNA levels in hypoxia-induced primary RGCs. The mRNA level of hypoxia-untreated group set at 1. Quantification graphs (relative density) of the intensity of staining of DSCR1 to hypoxia-untreated group. e Immunofluorescent staining showed co-localization of DSCR1-4 and VEGF in hypoxia-induced primary RGCs. Scale bar = 25 μm. The data are mean ± SEM (n = 6, **P < 0.01; ***P < 0.001; ## P < 0.01; ### P < 0.001; significantly different from the primary RGCs)
To further investigate the function of hypoxic RGCs secreted DSCR1-4 in mRMECs angiogenesis, we conducted siRNA knockdown for DSCR1-4 as shown in Supplemental Fig. 2.
DSCR1-4 Produced by Hypoxic RGCs Prevents NFATc1 Dephosphorylation and Translocation into Nuclear in VEGF-Induced mRMECs
Previous studies showed that expression of DSCR1-4 could block NFATc1 activity, which involved in mediating VEGF-induced angiogenesis in endothelial cells [28, 29]. However, whether the secretion of DSCR1-4 in hypoxic RGCs could affect NFATc1 activity in VEGF-induced mRMECs remains unclear. In the current study, western blotting analysis showed that VEGF markedly reduced phosphorylation of NFATc1 (Fig. 6a, c), and induced the total NFATc1 expression (Fig. 6a, b) in mRMECs. In addition, Western blotting (Fig. 6d–f) and immunofluorescent staining (Fig. 6g) results showed that VEGF markedly increased the nuclear translocation of NFATc1 in mRMECs. Exposure to 12 h of hypoxic RGCs medium (DSCR1-rich medium) increased the phosphorylation of NFATc1, and reduced total NFATc1 expression and nuclear translocation of NFATc1 in VEGF-induced mRMECs. However, these effects were partly abolished by DSCR1-4 knockdown in hypoxic RGCs. As a control, CsA, a specific inhibitor of NFATc1, showed the same inhibitory effect on NFATc1 activity with DSCR1-4 in VEGF-induced mRMECs. These results indicated that the secretion of DSCR1-4 in hypoxic RGCs was able to inhibit NFATc1 activity in VEGF-induced mRMECs.

Effect of DSCR1-4 produced by hypoxic RGCs on VEGF-induced NFATc1 phosphorylation and translocation into nuclear in mRMECs treated with VEGF (25 ng/mL) for 6 h or culture medium conditioned by hypoxic RGCs alone, hypoxic RGCs treated with siRNA to DSCR1-4 (siDSCR1-4) or hypoxic RGCs treated with siDSCR1-4 and CsA (200 ng/mL), and then primed with VEGF (25 ng/mL) for 6 h. a Western blotting analysis showed the protein level of p-NFATc1 and NFATc1 in different groups. b, c The bar chart showed the ratio of p-NFATc1 and NFATc1 to β-actin in different groups. d Western blotting analysis showed the levels of NFATc1 in both nuclear and cytosolic fractions in different groups. Expression of HDAC1 and β-actin (an internal control) was used for monitoring NFATc1 translocation into nuclear. e, f The bar chart showed the ratio of NFATc1 to HDAC1 in nuclear and β-actin in cytosolic fractions in different groups. g Immunofluorescent staining results showed the NFATc1 translocation into nuclear in different groups. Meanwhile, the phenotype of nuclei was also investigated via DAPI staining. Scale bar = 25 μm. All data are presented as means ± SD. (n = 3, *P < 0.05; **P < 0.01; ***P < 0.001)
DSCR1-4 Produced by Hypoxic RGCs Prevents VEGF-Induced Proliferative Responses in mRMECs
Because NFATc1 is required for VEGF-induced mRMECs proliferation, we further examined whether the secretion of DSCR1-4 in hypoxic RGCs could inhibit VEGF-induced proliferation in mRMECs. Cell viability and levels of cell proliferating markers (PCNA and Ki67) were measured as an index of cell proliferation. As shown in Fig. 7, VEGF stimulates mRMECs proliferation (increase cell viability and upregulation of PCNA and Ki67), which is partially inhibited after being exposed to 12 h of DSCR1-rich medium. However, these effects were partly abolished by DSCR1-4 knockdown in hypoxic RGCs. CsA recovered the effect of DSCR1 on VEGF-induced mRMECs proliferation. These results indicated that the secretion of DSCR1-4 in hypoxic RGCs was able to inhibit proliferation in VEGF-induced mRMECs.

Effect of DSCR1-4 produced by hypoxic RGCs on VEGF-induced proliferative responses in VEGF-induced mRMECs. mRMECs treated with VEGF (25 ng/mL) for 12 h or culture medium conditioned by hypoxic RGCs alone, hypoxic RGCs treated with siRNA to DSCR1-4 (siDSCR1-4) or hypoxic RGCs treated with siDSCR1-4 and CsA (200 ng/mL), and then primed with VEGF (25 ng/mL) for 12 h. a MTT assay showed the cell viability was assayed in different groups. The control group set at 100 %. b Western blot analysis showed the protein levels of PCNA were measured in different groups. c The bar chart showed the ratio of PCNA protein to β-actin in different groups. d–h Immunofluorescent staining results showed the Ki67 expression (i) in different groups. d Control condition. e Control condition in the presence of VEGF, f in the presence of VEGF and hypoxic RGCs culture medium, g in the presence of VEGF and hypoxic RGCs culture medium treated with siDSCR1-4, and h in the presence of VEGF, hypoxic RGCs culture medium treated with siDSCR1-4, and CsA. Meanwhile, the phenotype of nuclei was also investigated via DAPI staining (blue). Scale bar = 50 μm. i Quantitative analysis of percentages of PCNA positive cells in different groups. PCNA positive cells were quantified in six randomly selected fields. These data are means ± SEM. (n = 3, *P < 0.05; **P < 0.01; ***P < 0.001)
DSCR1-4 Produced by Hypoxic RGCs Prevents VEGF-Induced Migratory Responses in mRMECs
Transwell migration assay was also performed to test whether migratory responses were modified by DSCR1-rich medium treatment. As shown in Fig. 8, mRMECs treated for 12 h with 25 ng/mL VEGF exhibited a statistically significant increase in migratory responses compared to untreated mRMECs. DSCR1-rich medium obtained from hypoxic RGCs substantially inhibited the VEGF-induced migratory responses in mRMECs. Using the siRNA-mediated knockdown of DSCR1-4 expression and NFATc1 specific inhibitor CsA, we showed that DSCR1-4 in hypoxic RGCs could inhibit the migratory responses in VEGF-induced mRMECs, and this inhibitory effect was NFATc1-dependent.

Effect of DSCR1-4 produced by hypoxic RGCs on VEGF-induced migratory responses in mRMECs. mRMECs treated with VEGF (25 ng/mL) for 12 h or culture medium conditioned by hypoxic RGCs alone, hypoxic RGCs treated with siRNA to DSCR1-4 (siDSCR1-4), or hypoxic RGCs treated with siDSCR1-4 and CsA (200 ng/mL), and then primed with VEGF (25 ng/mL) for 12 h. a–e Transwell migration assay showed the migration of mRMECs in different groups. Representative photomicrographs of the migration chamber membranes with attached mRMECs are shown in different groups. a Control condition. b Control condition in the presence of VEGF, c in the presence of VEGF and hypoxic RGCs culture medium, d in the presence of VEGF and hypoxic RGCs culture medium treated with siDSCR1-4, and e in the presence of VEGF, hypoxic RGCs culture medium treated with siDSCR1-4 and CsA. f Following migration of the cells through the membrane pores, the number of mRMECs was counted on the lower surface of the transwell membrane in different groups. mRMECs were quantified in six randomly selected fields. Scale bar = 50 μm. These data are means ± SEM. (n = 6, **P < 0.01; ***P < 0.001)
DSCR1-4 Produced by Hypoxic RGCs Prevents VEGF-Induced Tube Formation in VEGF-Induced mRMECs
As shown in Fig. 9, the formation of capillary tubes was evaluated in Matrigel plugs in vitro. When mRMECs were seeded on these plugs, spontaneous formation of endothelial tubes occurred. The addition of DSCR1-rich medium inhibited VEGF-induced capillary-like network formation, as demonstrated by decreased tube numbers (Fig. 9f). However, these effects were partly abolished by DSCR1-4 knockdown in hypoxic RGCs. CsA recovered the effect of DSCR1-4 on VEGF-induced capillary-like network formation in mRMECs. These findings suggested that the secretion of DSCR1-4 in hypoxic RGCs could inhibit the capillary-like network formation in VEGF-induced mRMECs.

Effect of DSCR1-4 produced by hypoxic RGCs on VEGF-induced tube formation in VEGF-induced mRMECs. mRMECs treated with VEGF (25 ng/mL) for 12 h or culture medium conditioned by hypoxic RGCs alone, hypoxic RGCs treated with siRNA to DSCR1-4 (siDSCR1-4) or hypoxic RGCs treated with siDSCR1-4 and CsA (200 ng/mL), and then primed with VEGF (25 ng/mL) for 12 h. a–e Tube formation assay showed the pro-angiogenic structure formation of mRMECs in different groups. a Control condition. b Control condition in the presence of VEGF, c in the presence of VEGF and hypoxic RGCs culture medium, d in the presence of VEGF and hypoxic RGCs culture medium treated with siDSCR1, and e In the presence of VEGF, hypoxic RGCs culture medium treated with siDSCR1, and CsA. f Quantitative analysis of number of mRMECs sprouting (pro-angiogenic structure) in different groups. mRMECs were quantified in six randomly selected fields. These data are means ± SEM. (n = 6, *P < 0.05; **P < 0.01; ***P < 0.001)
DSCR1-4 Produced by Hypoxic RGCs Prevents Inflammatory Cytokines Production in VEGF-Induced mRMECs
As shown by RT-PCR analysis in Fig. 10, VEGF treatment significantly increased the COX-2, iNOS, and MCP-1 mRNA levels in mRMECs, which were markedly attenuated after exposed to DSCR1-rich medium. Our ELISA results also showed that DSCR1-rich medium attenuated the increase of IL-8, ICAM-1, and VCAM-1 production in VEGF-induced mRMECs. However, these effects were partly abolished by DSCR1-4 knockdown in hypoxic RGCs. CsA recovered the effect of DSCR1-4 on VEGF-induced inflammatory cytokines production in mRMECs. These findings suggested that the secretion of DSCR1-4 in hypoxic RGCs could inhibit the activation of the inflammatory cascade in VEGF-induced mRMECs.

Effect of DSCR1 produced by hypoxic RGCs on VEGF-induced activation of inflammatory cytokines in mRMECs. mRMECs treated with VEGF (25 ng/mL) for 12 h or culture medium conditioned by hypoxic RGCs alone, hypoxic RGCs treated with siRNA to DSCR1-4 (siDSCR1-4) or hypoxic RGCs treated with siDSCR1-4 and CsA (200 ng/mL), and then primed with VEGF (25 ng/mL) for 12 h. RT-PCR showed the mRNA levels of COX-2 (a), iNOS (b), and MCP-1 (c) in different groups. ELISA analysis showed the protein levels of IL-8 (d), ICAM-1 (e), and VCAM-1 (f) in different groups. These data are means ± SEM. (n = 6, *P < 0.05; **P < 0.01; ***P < 0.001)
Discussion
In this present study, we use a mouse OIR model to explore the potential role of DSCR1 in mediating PRV. DSCR1 has two differentially regulated isoforms (DSCR1-1 and DSCR1-4). Our data revealed that DSCR1-4 was progressively upregulated in the RGCs of central avascular retina after OIR. DSCR1-4 was also upregulated in hypoxia-induced primary RGCs in vitro. However, there was no change in mRNA expression of DSCR1-1 at P16 after OIR or in hypoxia-induced primary RGCs in vitro. In addition, we found that DSCR1-4 produced by hypoxic RGCs reduced cell proliferation, migration, tube formation, inflammatory cytokine production in VEGF-induced mRMECs, indicating its anti-angiogenic properties. Moreover, DSCR1-4 produced by hypoxic RGCs inhibited NFATc1 dephosphorylation and translocation into nuclear in VEGF-induced mRMECs. These results indicated that these anti-angiogenic properties of DSCR1-4 are NFATc1-dependent.
OIR animal model in mouse had been established in our previous studies and was widely used in ROP- or PDR-related research [22, 23, 30]. This model exhibits two stages. During the first hyperoxic phase, vascular development arrests, existing blood vessels degenerate and form the vaso-obliterated zones in central retina [6, 31]. During the second hypoxic phase, the avascular retina becomes ischemic and triggers a compensatory release of pro-angiogenic factors, leading to abnormal, and unregulated vaso-proliferation, which induces pre-retinal neovascularization [6, 31]. Previous animal studies showed that DSCR1 was detected in neuron after ischemic stroke [13] and that overexpression of DSCR1 plays anti-inflammatory and anti-apoptotic effects therefore improves the outcomes of following stroke [15]. Parallel with these studies, our data revealed that DSCR1-4 was progressively upregulated in the retina after OIR. In addition, double immunofluorescent labeling suggested that increased DSCR1-4 mainly was located in RGCs of avascular zones from OIR mice. On the other hand, in vitro studies also showed that DSCR1-4 expression was elevated in hypoxia-induced primary RGCs. According to these results, we speculated that DSCR1-4 might play an essential role in angiogenesis after OIR and be relevant to PRV.
Neurovascular cross-talk shapes vascular development in PRs [32, 33]. After blood vessel degeneration, the metabolical starvation of RGCs was induced by the retinal ischemic state, subsequently, several adaptive cellular changes occurred [32]. Ischemic RGCs secret vaso-repulsive molecule (semaphorin 3A) to inhibit VEGF-induced endothelial proliferation and migration, and to impede retinal revascularization away from the avascular neural retina [8]. In addition, ischemic RGCs induce Sirtuin 1 expression, which promotes revascularization in avascular neural retina [10]. Our findings showed that DSCR1-4 secreted by hypoxic RGCs could inhibit angiogenesis, with decrease of cell proliferation, migration, tube formation, and inflammatory cytokines production. This was proved by that the siRNA-mediated knockdown of DSCR1-4 partly abolished these inhibitory effects. DSCR-1 is a typical unstable protein and can be upregulated after cellular adaptation to oxidative stress [13, 34]. In the current study, DSCR1-4 was progressively upregulated in the RGCs of central avascular retina after OIR and in hypoxia-induced primary RGCs in vitro. In addition, the anti-angiogenic role of DSCR1 in mRMECs is only effective with immediately treatment with RGC culture medium after being induced by hypoxia for 36 h. While, the anti-angiogenic role of DSCR1 in mRMECs became weakened if treated with culture medium conditioned by hypoxic RGCs, which have been stored for 12–24 h at 4 °C or −20 °C. We speculated that the anti-angiogenic role of DSCR1 in mRMECs is transitorily effective after hypoxic stimulation in primary RGCs.
Many recent studies have demonstrated that the NFATc1-dependent pathway is an important mediator of VEGF signaling in endothelial cells [28, 29, 35]. VEGF binds to its primary receptor (VEGFR-1 and VEGFR-2) and then stimulates a variety of signaling factors, including the rapid increase of intracellular calcium level and the calcineurin activity [28, 36]. The activated calcineurin induced NFATc1 dephosphorylation and nuclear translocation in endothelial cells, resulting in the expression of angiogenesis-related genes, which promoted angiogenesis, accompanying with increase of cell proliferation, migration, tube formation, and inflammatory cytokines production [28, 29, 36]. It was reported that NFATc1 inhibitor (INCA-6 or FK-506) inhibited proliferation and tube formation in VEGF-induced mRMECs, and significantly reduced pathologic neovascularization in OIR rat model [35]. DSCR1-4 has been shown to inhibit calcium-calcineurin-mediated NFATc1 dephosphorylation, nuclear translocation, and activity level in endothelial cells [16, 37]. The biological roles of DSCR1-4 include protection against calcium-mediated oxidative stress and inhibition of VEGF-mediated signaling during angiogenesis [17]. Therefore, it is crucial to identify whether the secretion of DSCR1-4 in hypoxic RGCs inhibits angiogenesis is through NFATc1-dependent pathway. It was noteworthy that this could be proved in the current study, evidenced that DSCR1-4 produced by hypoxic RGCs prevented NFATc1 dephosphorylation and translocation into nuclear in VEGF-induced mRMECs. While, the siRNA-mediated knockdown of DSCR1-4 partly abolished the effect of DSCR1-4 on NFATc1 activity. On the other hand, CsA, an immunosuppressant drug, is a potent inhibitor of calcineurin activity [38]. CsA binds to intracellular proteins called immunophilins, and the resultant CsA-cyclophilin A complexes inhibit calcineurin activity [38]. CsA blocked VEGF-induced nuclear translocation of NFATc1 and simultaneously inhibited VEGF induced angiogenesis in microvascular endothelial cells [39, 40]. In our present study, CsA not only inhibited NFATc1 activity, but also partly prevented angiogenesis in VEGF-induced mRMECs treated with DSCR1 knockdown conditioned medium.
DSCR1 is widely expressed in the central nervous system during early embryonic development [41]. Previous studies have shown that DSCR1 is involved in cellular adaptation to oxidative stress and could regulate the neuronal apoptosis through caspase-3 [34] or activating CREB-Mediated Bcl-2 expression [14]. Previous studies have shown that the neuronal cells increased DSCR1 expression after exposure to damaging stimuli associated with calcium overloading [42, 43]. We speculated that increased expression of the DSCR1 in primary RGCs after hypoxia stimulation may be transiently affected by calcium-mediated stresses. After DSCR1 was knocked out, it may affect the specific ion such as calcium or cytokines secreted from primary RGCs, which may not only regulate the apoptosis of RGCs, but also affects the anti-angiogenesis pathway in endothelial cells following the NFAT inhibition. Our further study will focus on whether the anti-angiogenic role of DSCR1 is depend on regulating the specific ion such as calcium or cytokines secreted from RGCs.
Angiogenesis is initiated by the release of inflammatory cytokines, which subsequently promotes local endothelial cells populations, as well as increased local migration and tube formation [29, 44]. Various studies have determined that NFATc1 promoted inflammatory cytokines production, which in turn induced pro-angiogenic behaviors of tumor cells and endothelial cells [29, 44]. Our results revealed that DSCR1-4 in hypoxic RGCs inhibited NFATc1 translocation and activity, as well as influenced inflammatory cytokines production including COX-2, iNOS, MCP-1, IL-8, ICAM-1, and VCAM-1, which may in turn mediate vascular growth in VEGF-induced mRMECs. These results on DSCR1-4 dependent regulation of NFATc1 are in agreement with previous studies showing that DSCR1-4 is essential for NFATc1 translocation and activity, and production of inflammatory cytokines in tumor cells and endothelial cells in response to hypoxia [12, 37].
In our present study, preliminary test was performed to construct plasmid expression vector of RNA interference targeting DSCR1-4 via intravitreal injection. However, low transfection efficiency was detected after intravitreal injection. To our best knowledge, no successful siRNA transfection targeting on retinal DSCR1-4 was reported. Nevertheless, we consider that the present study have strongly indicated that DSCR1 produced by hypoxic RGCs displayed anti-angiogenic properties in VEGF-induced mRMECs through NFATc1-dependent pathway. Further study will be performed on DSCR1-4 knockout mice to show the anti-angiogenic role of DSCR1-4 in vivo.
Conclusion
Our study demonstrated for the first time that DSCR1-4 was significantly upregulated in the RGCs of avascular retina from OIR mouse model. Moreover, DSCR1-4 produced by hypoxic RGCs displayed anti-angiogenic properties in VEGF-induced mRMECs through NFATc1-dependent pathway (Fig. 11). Thus, besides its role as a vaso-repulsive molecule, DSCR1-4 could be used as an anti-angiogenic factor and may be a potential new target for therapeutic angiogenesis in patients with PRs.

Schematic diagram showed that ischemic RGCs secreted DSCR1-4, which displayed anti-angiogenic properties in mRMECs via NFATc1-dependent pathway
References
Li XX (2012) Intraocular antiangiogenic drugs in clinical application advantages and disadvantages. Zhonghua Yan Ke Za Zhi [Chin J Ophthalmol] 48(10):870–873
Rubio RG, Adamis AP (2016) Ocular angiogenesis: vascular endothelial growth factor and other factors. Dev Ophthalmol 55:28–37. doi:10.1159/000431129
Kwong TQ, Mohamed M (2014) Anti-vascular endothelial growth factor therapies in ophthalmology: current use, controversies and the future. Br J Clin Pharmacol 78(4):699–706. doi:10.1111/bcp.12371
Binder S (2012) Loss of reactivity in intravitreal anti-VEGF therapy: tachyphylaxis or tolerance? Br J Ophthalmol 96(1):1–2. doi:10.1136/bjophthalmol-2011-301236
Gasperini JL, Fawzi AA, Khondkaryan A, Lam L, Chong LP, Eliott D, Walsh AC, Hwang J, Sadda SR (2012) Bevacizumab and ranibizumab tachyphylaxis in the treatment of choroidal neovascularisation. Br J Ophthalmol 96(1):14–20. doi:10.1136/bjo.2011.204685
Chu ZJ, Dou GR, Wang YS, Qu XJ, Zhang Y (2013) Preliminary study of retinal pathological features in preterm birth pups exposed to an animal model of oxygen-induced retinopathy in mice. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 251(8):1937–1943. doi:10.1007/s00417-013-2366-8
Fulton AB, Akula JD, Mocko JA, Hansen RM, Benador IY, Beck SC, Fahl E, Seeliger MW, et al. (2009) Retinal degenerative and hypoxic ischemic disease. Documenta ophthalmologica Advances in ophthalmology. 118(1):55–61. doi:10.1007/s10633-008-9127-8
Joyal JS, Sitaras N, Binet F, Rivera JC, Stahl A, Zaniolo K, Shao Z, Polosa A, et al. (2011) Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A. Blood 117(22):6024–6035. doi:10.1182/blood-2010-10-311589
Michan S, Juan AM, Hurst CG, Cui Z, Evans LP, Hatton CJ, Pei DT, Ju M, et al. (2014) Sirtuin1 over-expression does not impact retinal vascular and neuronal degeneration in a mouse model of oxygen-induced retinopathy. PLoS One 9(1):e85031. doi:10.1371/journal.pone.0085031
Chen J, Michan S, Juan AM, Hurst CG, Hatton CJ, Pei DT, Joyal JS, Evans LP, et al. (2013) Neuronal sirtuin1 mediates retinal vascular regeneration in oxygen-induced ischemic retinopathy. Angiogenesis 16(4):985–992. doi:10.1007/s10456-013-9374-5
Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal JS, Cho JH, Honore JC, Kermorvant-Duchemin E, et al. (2008) The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med 14(10):1067–1076. doi:10.1038/nm.1873
Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, et al. (2009) Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 459(7250):1126–1130. doi:10.1038/nature08062
Cho KO, Kim YS, Cho YJ, Kim SY (2008) Upregulation of DSCR1 (RCAN1 or Adapt78) in the peri-infarct cortex after experimental stroke. Exp Neurol 212(1):85–92. doi:10.1016/j.expneurol.2008.03.017
Kim SS, Jang SA, Seo SR (2013) CREB-mediated Bcl-2 expression contributes to RCAN1 protection from hydrogen peroxide-induced neuronal death. J Cell Biochem 114(5):1115–1123. doi:10.1002/jcb.24452
Brait VH, Martin KR, Corlett A, Broughton BR, Kim HA, Thundyil J, Drummond GR, Arumugam TV, et al. (2012) Over-expression of DSCR1 protects against post-ischemic neuronal injury. PLoS One 7(10):e47841. doi:10.1371/journal.pone.0047841
Lange AW, Molkentin JD, Yutzey KE (2004) DSCR1 gene expression is dependent on NFATc1 during cardiac valve formation and colocalizes with anomalous organ development in trisomy 16 mice. Dev Biol 266(2):346–360
Qin L, Zhao D, Liu X, Nagy JA, Hoang MV, Brown LF, Dvorak HF, Zeng H (2006) Down syndrome candidate region 1 isoform 1 mediates angiogenesis through the calcineurin-NFAT pathway. Molecular cancer research: MCR 4(11):811–820. doi:10.1158/1541-7786.MCR-06-0126
Ryeom S, Baek KH, Rioth MJ, Lynch RC, Zaslavsky A, Birsner A, Yoon SS, McKeon F (2008) Targeted deletion of the calcineurin inhibitor DSCR1 suppresses tumor growth. Cancer Cell 13(5):420–431. doi:10.1016/j.ccr.2008.02.018
Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS (2000) A protein encoded within the down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem 275(12):8719–8725
Lee EJ, Lee JY, Seo SR, Chung KC (2007) Overexpression of DSCR1 blocks zinc-induced neuronal cell death through the formation of nuclear aggregates. Mol Cell Neurosci 35(4):585–595. doi:10.1016/j.mcn.2007.05.003
Wang W, Xia T, Yu X (2015) Wogonin suppresses inflammatory response and maintains intestinal barrier function via TLR4-MyD88-TAK1-mediated NF-kappaB pathway in vitro. Inflammation research: official journal of the European Histamine Research Society [et al] 64(6):423–431. doi:10.1007/s00011-015-0822-0
Yang L, Xu Y, Li W, Yang B, Yu S, Zhou H, Yang C, Xu F, et al. (2015) Diacylglycerol kinase (DGK) inhibitor II (R59949) could suppress retinal neovascularization and protect retinal astrocytes in an oxygen-induced retinopathy model. Journal of molecular neuroscience: MN 56(1):78–88. doi:10.1007/s12031-014-0469-2
Liang X, Zhou H, Ding Y, Li J, Yang C, Luo Y, Li S, Sun G, et al. (2012) TMP prevents retinal neovascularization and imparts neuroprotection in an oxygen-induced retinopathy model. Invest Ophthalmol Vis Sci 53(4):2157–2169. doi:10.1167/iovs.11-9315
Winzeler A, Wang JT (2013) Purification and culture of retinal ganglion cells from rodents. Cold Spring Harbor protocols 2013(7):643–652. doi:10.1101/pdb.prot074906
Xu Z, Jiang F, Zeng Y, Alkhodari HT, Chen F (2011) Culture of rat retinal ganglion cells. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban 31(3):400–403. doi:10.1007/s11596-011-0389-0
Yao YG, Duh EJ (2004) VEGF selectively induces Down syndrome critical region 1 gene expression in endothelial cells: a mechanism for feedback regulation of angiogenesis? Biochem Biophys Res Commun 321(3):648–656. doi:10.1016/j.bbrc.2004.06.176
Mitchell AN, Jayakumar L, Koleilat I, Qian J, Sheehan C, Bhoiwala D, Hushmendy SF, Heuring JM, et al. (2007) Brain expression of the calcineurin inhibitor RCAN1 (Adapt78). Arch Biochem Biophys 467(2):185–192. doi:10.1016/j.abb.2007.08.030
Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, Bischoff J, Vikkula M, et al. (2008) Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med 14(11):1236–1246. doi:10.1038/nm.1877
Johnson EN, Lee YM, Sander TL, Rabkin E, Schoen FJ, Kaushal S, Bischoff J (2003) NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells. J Biol Chem 278(3):1686–1692. doi:10.1074/jbc.M210250200
Yang B, Xu Y, Yu S, Huang Y, Lu L, Liang X (2015) Anti-angiogenic and anti-inflammatory effect of Magnolol in the oxygen-induced retinopathy model. Inflammation research: official journal of the European Histamine Research Society [et al]. doi:10.1007/s00011-015-0894-x
Kermorvant-Duchemin E, Sapieha P, Sirinyan M, Beauchamp M, Checchin D, Hardy P, Sennlaub F, Lachapelle P, et al. (2010) Understanding ischemic retinopathies: emerging concepts from oxygen-induced retinopathy. Documenta ophthalmologica Advances in ophthalmology 120(1):51–60. doi:10.1007/s10633-009-9201-x
Sapieha P, Hamel D, Shao Z, Rivera JC, Zaniolo K, Joyal JS, Chemtob S (2010) Proliferative retinopathies: angiogenesis that blinds. Int J Biochem Cell Biol 42(1):5–12. doi:10.1016/j.biocel.2009.10.006
Feng Y, Busch S, Gretz N, Hoffmann S, Hammes HP (2012) Crosstalk in the retinal neurovascular unit—lessons for the diabetic retina. Experimental and clinical endocrinology & diabetes: official journal, German Society of Endocrinology [and] German Diabetes Association 120(4):199–201. doi:10.1055/s-0032-1304571
Sun X, Wu Y, Chen B, Zhang Z, Zhou W, Tong Y, Yuan J, Xia K, et al. (2011) Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase-3 activation. J Biol Chem 286(11):9049–9062. doi:10.1074/jbc.M110.177519
Bretz CA, Savage S, Capozzi M, Penn JS (2013) The role of the NFAT signaling pathway in retinal neovascularization. Invest Ophthalmol Vis Sci 54(10):7020–7027. doi:10.1167/iovs.13-12183
Jang GH, Park IS, Yang JH, Bischoff J, Lee YM (2010) Differential functions of genes regulated by VEGF-NFATc1 signaling pathway in the migration of pulmonary valve endothelial cells. FEBS Lett 584(1):141–146. doi:10.1016/j.febslet.2009.11.031
Hesser BA, Liang XH, Camenisch G, Yang S, Lewin DA, Scheller R, Ferrara N, Gerber HP (2004) Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood 104(1):149–158. doi:10.1182/blood-2004-01-0273
Su L, Tam N, Deng R, Chen P, Li H, Wu L (2014) Everolimus-based calcineurin-inhibitor sparing regimens for kidney transplant recipients: a systematic review and meta-analysis. Int Urol Nephrol 46(10):2035–2044. doi:10.1007/s11255-014-0783-1
Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, et al. (2001) Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin a: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med 193(5):607–620
Rafiee P, Heidemann J, Ogawa H, Johnson NA, Fisher PJ, Li MS, Otterson MF, Johnson CP et al. (2004) Cyclosporin a differentially inhibits multiple steps in VEGF-induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell communication and signaling: CCS 2 (1):3. doi:10.1186/1478-811X-2-3
Harris CD, Ermak G, Davies KJ (2005) Multiple roles of the DSCR1 (Adapt78 or RCAN1) gene and its protein product calcipressin 1 (or RCAN1) in disease. Cellular and molecular life sciences: CMLS 62(21):2477–2486. doi:10.1007/s00018-005-5085-4
Davies KJ, Harris CD, Ermak G (2001) The essential role of calcium in induction of the DSCR1 (ADAPT78) gene. Biofactors 15(2–4):91–93
Porta S, Serra SA, Huch M, Valverde MA, Llorens F, Estivill X, Arbones ML, Marti E (2007) RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: a potential pathogenic process in neurodegeneration. Hum Mol Genet 16(9):1039–1050. doi:10.1093/hmg/ddm049
Suehiro J, Kanki Y, Makihara C, Schadler K, Miura M, Manabe Y, Aburatani H, Kodama T, et al. (2014) Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium. J Biol Chem 289(42):29044–29059. doi:10.1074/jbc.M114.555235
Acknowledgments
Chinese government provided financial support in the form of the National Natural Science Foundation of China (nos. 81470030, 81670850, and 81670873).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
In our study, all procedures with animals were approved by the Institutional Animal Care and Use Committee of Zhongshan Ophthamic Center, and were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Conflict of Interest
The authors declare no competing interests.
Additional information
Yue Xu, Boyu Yang, and Yaguang Hu contributed equally to this work.
Electronic supplementary material
Supplemental Figure 1
Identification of RGCs markers. The RGCs showed strong staining with antibody against NeuN (red, a) and βIII-tubulin (green, b), two RGCs markers. DAPI (blue, c). The merged images (yellow, d) represented the co-localization of NeuN, βIII-Tubulin, and DAPI. Scale bars 50 μm (GIF 52 kb)
Supplemental Figure 2
Effect of siDSCR1-4 on hypoxia-induced DSCR1-4 expression in RGCs. Western blots and real-time PCR showed that DSCR1-4 protein (a, b) and mRNA (c) levels were significantly decreased in the hypoxia-induced RGCs treated with siRNA to DSCR1-4 compared to only hypoxia-induced RGCs. These data are means ± SEM. (n = 3, ***P < 0.001) (GIF 12 kb)
Rights and permissions
About this article

Cite this article
Xu, Y., Yang, B., Hu, Y. et al. Secretion of Down Syndrome Critical Region 1 Isoform 4 in Ischemic Retinal Ganglion Cells Displays Anti-Angiogenic Properties Via NFATc1-Dependent Pathway. Mol Neurobiol 54, 6556–6571 (2017). https://doi.org/10.1007/s12035-016-0092-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0092-z