
Abstract
Despite a large amount of research which aims at defining the pathophysiology of human demyelination (i.e., multiple sclerosis), etiological bases of disease have been unknown so far. The point of intersection of all assumed etiological factors, which are mainly based upon immunological cascades, is neuroinflammation. The precise definition of the place and role of all pathogenetic factors in the occurrence and development of the disease is of crucial importance for understanding the clinical nature and for finding more effective therapeutic options. There are few studies whose results give more precise data about the role and the importance of other factors in neuroinflammation, besides immunological ones, with regard to clinical and paraclinical correlates of the disease. The review integrates results found in previously performed studies which have evaluated oxidative stress participation in early and late neuroinflammation. The largest number of studies indicates that the use of antioxidants affects the change of neuroinflammation course under experimental conditions, which is reflected in the reduction of the severity and the total reversibility in clinical presentation of the disease, the faster achieving of remission, and the delayed and slow course of neuroinflammation. Therapies based on the knowledge of redox biology targeting free radical generation hold great promise in modulation of the neuroinflammation and its clinical presentations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Neuroinflammation
The central nervous system (CNS) is a very sensitive tissue for damages which has little capacity for regeneration [1]. Due to this fact, inflammation reactions, which are the pathophysiological basis for all CNS damages regardless of etiology, have, as a consequence, nervous tissue damages of different levels of manifestation and reversibility [2].
CNS tissue is an “immunologically privileged” part of an organism because of the existence of the blood-brain barrier (BBB) and the absence of the lymphatic system. The blood-brain barrier is a physical and transport barrier for toxic matter and cells which could invade the CNS [3, 4]. It is composed of two biological barriers. The first one is formed of tight junctions of endothelial cells of brain microcirculation; it contains multiple efflux pumps, which eliminate toxic and decomposing metabolic products from the CNS into the systemic circulation. The second one is formed of a basement membrane and astrocytic extensions which form the so-called glial membrane. For these reasons, the invasion of circulating cells into the CNS is conducted through two separate processes: (1) transendothelial migration into the perivascular space and then (2) infiltration of CNS parenchyma by passing through the glial membrane [5]. These two processes are regulated independently so that circulating cells can migrate only into the perivascular space without the possibility of passing into CNS parenchyma, if the glial membrane is morphologically and functionally intact [6].
Immunologically competent cells present in the CNS under physiological conditions are microglial cells. They represent the residential cell population of the CNS. The assumption is that they are created by the transformation of circulating monocytes and the cells of myeloid lineage which colonize the CNS during its embryonic and early neonatal development [7, 8]. Under physiological conditions, microglia forms an extensive network of long cellular extensions with a relatively small cell body. Under pathological conditions, after the activation, microglia transforms into a phagocytic cell phenotype which causes the formation of many amoeboid protoplasmatic extensions like the ones formed by activated macrophages [9] (Fig. 1). Besides, activated microglia forms and secretes different proinflammatory cytokines, chemokines, reactive oxygen species (ROS), and nitric oxide (NO·) by means of which it communicates with other cells of the CNS [10]. In the perivascular space, there physiologically exists a so-called subpopulation of inactive macrophages which, in a pathological sense, have a role of antigen-presenting cells, and thus, they activate a specific immune system [11]. Besides these cells, there are also rear T lymphocytes in charge of CNS protection, which enter the CNS parenchyma through transendothelial migration, by means of adhesion molecules present on the surface of endothelial capillary cells, but, without antigens and/or autoimmune pathology, they do not initiate immunological processes [12].

The role of immune cells in the pathogenesis of neuroinflammation and demyelination
The inflammation process is initiated only in the state when there is an increased number of monocytes and T lymphocytes, delivered to CNS from the systemic circulation; when their transendothelial transport is facilitated and accelerated; as well as when there are antigens in the CNS on which T lymphocytes are sensitized by means of antigen-presenting cells [13]. Once the inflammatory process is initiated in the CNS, it is conducted like in any other organ in the further course of its propagation. There is an increased formation of molecular messengers which attract monocytes and lymphocytes, microglia multiplies, and the activation of the complement system is initiated and antibodies are created [14].
Immunological protection of the CNS is reduced in some of its parts, like the parenchyma, chambers, and meninges, so these parts could become the place of inflammatory reaction similar to the one occurring in other organs [2, 15]. On the other hand, the cortex is less suitable for infiltration compared to the white matter and the spinal cord [16]. The existence of different regional sensitivity to CNS inflammation could be a consequence of different histological structures, given that the cortex contains millions of neuronal bodies, which, in a way, achieve immunosuppressive effect by strengthening the BBB [17]. These biological features represent a contribution to understanding the existence of predilection places for the appearance of inflammation in the CNS.
BBB disruption, which exists in all the neuroinflammatory processes in varying degrees of manifestation, is a key mechanism by which a “protected” CNS tissue is exposed to the effects of activated cells of innate or acquired immunity. Therefore, determining the relation of albumin concentration in the CSF and serum, and MRI of the CNS with a paramagnetic agent such as gadolinium, chelated by diethylene triaminopentacetate (Gd-DTPA) in the form of a dimeglumine salt, which penetrates the CNS only if the BBB has been damaged, has an important role in evaluation of BBB damage severity, as well as the intensity of a neuroinflammatory process [18, 19].
Oxidative Metabolism and Its Specificities in the CNS
Free radicals are atoms, molecules, or ions with unpaired electrons. They are created in biological systems because of breaking of covalent bonds in the process of homo- or heterolysis or by means of electron transfer. Due to exceptional chemical instability, free radicals enter chemical reactions with other free radicals and nonradical molecules. This makes them the leading pathogenetic link in many pathological conditions [20]. Free radicals are created in various conditions: during oxidative phosphorylation in mitochondria, in the process of phagocytosis, in the reactions of biotransformation of exogenous and endogenous substrata in endoplasmic reticulum, in enzymatic reactions which catalyze oxidases, in the process of eicosanoid synthesis, and in oxidoreduction reactions in the presence of metal with variable valency [21–30]. Lipid peroxidation occurs as a consequence of free radical secretion, even though during the very process of lipid peroxidation free radicals are being created in increased concentration. All reactive chemical species (radical and nonradical) can be oxygen species, reactive species of nitrogen, and carbon- and sulfur-reactive species [31–33].
Oxidative modification of proteins implies a covalent protein modification which is a consequence of the effect of reactive chemical species or the products of their interaction with other biomolecules. CNS diseases, primarily the ones based on a neuroinflammatory process, are conditions where these types of changes occur a lot [34]. Advanced oxidation protein products (AOPP) and advanced glycation end products (AGE) are considered exceptionally pathogenic due to the fact that they are generators of free radicals and independent promoters of the further spreading of the process of their uncontrollable production [35–40].
Lipid peroxidation is one of the most commonly studied processes of redox cell signalization disorders where free radicals play the key role. Through the different mechanisms, lipid peroxidation disrupts membrane barrier function, inactivates membrane enzymes, and increases permeability for water, monovalent and divalent ions, and often for high molecular weight compounds. Lysosomal membrane damage enables the exit of hydrolytic enzymes, while mitochondrial membrane damage leads to Ca ion release and the activation of enzymes dependent on this ion [40–49].
Oxidative stress is a pathological condition which implies overproduction of ROS under conditions when their elimination is reduced [50–60]. Under physiological conditions, owing to the efficiency of enzymatic antioxidative potential of the organism (superoxide dismutase (SOD), catalase, etc.) (Fig. 2) and nonenzymatic molecules (GSH, vitamin C, vitamin E, bilirubin, etc.), ROS concentration is low and then ROS manifest a series of useful physiological features. They imply the cell division and growth regulation, apoptosis regulation, oxidative modifications of biomolecules in extracellular space, protection from pathogen invasion, etc. [61]. Reactive oxygen species and products of their interaction with other biomolecules cause DNA mutations, ion channel damage, intensification of the lipid peroxidation process, and oxidation of proteins and other biomolecules [62, 63]. Under the conditions of oxidative stress, ROS can directly initiate a cascade of proinflammatory actions by stimulating cytokine production and BBB damaging and by enabling cells of the immune system to penetrate into the CNS [64]. On the other hand, the most important mediator in the described oxidative processes is ONOO−, which is created as a product of NO and O2 − reaction [65, 66].

The antioxidative mechanism
CNS cells are exposed to low concentrations of ROS under physiological conditions, but due to the good antioxidative protection, their adverse effects are negligible [67]. CNS structures are particularly sensitive to ROS effects, which is a consequence of a highly activated metabolism of the CNS, when 2–5 % of the used O2 is turned into ROS in the process of cell breathing in mitochondria [1]. ROS realize their effects in their immediate environment in relation to the place of their production, diffusing in the diameter of up to 0.3 nm. By comparison, the size of the interlaminar space of the myelin sheath is 10 nm.
On the other hand, the CNS has a relatively limited potential for anaerobic metabolism which makes it especially sensitive to hypoxia [68]. Under conditions of small O2 availability, there is a dramatic increase of O2 − concentration in mitochondria [4]. Finally, some CNS cells, like oligodendrocytes, for example, have a physiologically low level of antioxidative potential, as opposed to the relatively high energy turnover [69]. A high percentage of lipids in the CNS is the basis of its tendency to initiate lipid peroxidation processes. It has been shown that the brain tissue is more sensitive to the described damages, in comparison to the tissue of the spinal cord or the peripheral nervous system [70]. This could be a consequence of the lower content of thiol redox system in the spinal cord and the peripheral nerves in comparison to the brain tissue [16]. The aforementioned differences are also related to the different GSH concentrations, taking into account that GSH participates in ROS reduction, but, on the other hand, it reactivates antioxidative enzymes in the CNS by means of independent mechanisms. GSH is mostly located in astrocytes (up to 5 mM), which is a lot more than in other CNS structures [71].
Oxidative Stress in Neuroinflammation
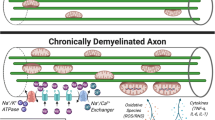
The mitochondrial respiratory chain, xanthine oxidase, NADPH oxidase, lipooxygenase, cyclooxygenase (COX), and nonenzymatic processes like dopamine and noradrenaline autooxidation appear as the main sources of ROS creation in the CNS. In oligodendrocytes, peroxisomes are an important ROS source because they are present in large numbers in the process of active myelination [72]. Microglial cells are an important source of ROS, under the conditions of neuroinflammation, as a consequence of their activation. Microglial cells are considered to be ROS depots under these pathological conditions and key initiators of the demyelination process which is ROS mediated. Peroxynitrite is important for macrophage activation due to the fact that it converts lipoproteins into forms which, independently of other factors, initiate phagocytic activity of macrophages because it binds to the so-called scavenger receptors of macrophages [9]. Neurons are also a source of ROS, given that ROS are created in the processes of electrical activity of a cell. It is assumed that transport of Ca into the axons, as well as Ca release from intracellular depots into the interior of the cell, is of greatest importance in this process. The intensive metabolism of axons under conditions of neuroinflammation is, at the same time, the explanation of the occurrence of their spontaneous, repetitive triggering in them, with the occurrence of corresponding clinical equivalents. This is followed by the increase of extracellular concentration of K, which simultaneously activates astrocytes, due to the fact that astrocytes are important for buffering extracellular changes in K ion concentration [72–77].
Some studies of demyelinating plaque have found the increased presence of LDL molecules, modified by lipid peroxidation, in the very plaques, macrophages, and astrocytes, in parallel with the increase of the concentration of lipid peroxidation end products [78–80]. This process leads to arachidonic acid release, which is enzymatically converted into prostaglandin or is converted to isoprostanes by nonenzymatic metabolism. Isoprostanes are created by free radical lipid peroxidation of arachidonic acid, and among all isoprostanes, 8-epi-PGF2α is especially used as a biomarker of oxidative stress. Prostaglandins, especially E2, independently or in conjunction with NO·, can increase BBB permeability [79]. However, prostaglandins are considered as conditional pathogens in neuroinflammation, due to the fact that their end product in the nervous tissue is dependent on the intensity of oxidative stress in the CNS [72, 81].
Oxidative stress in the CNS also causes changes in transendothelial transport which enables further evolution of the neuroinflammatory process [67, 82, 83]. The most important effect of ROS is damage of oligodendrocytes, which are the key cells in the process of myelin creation in the CNS. Even though the mechanisms of damaging of these cells are not completely clarified, it has been proven that oxidative stress and NO· have a very important role in that process [84]. There are studies which have shown that oligodendrocytes are sensitive to ROS concentrations much lower than toxic concentrations of other glial cells, astrocytes, and microglia. A protective effect on this cell line has been achieved by adding antioxidants [72, 85]. During the periods of myelin production and remyelination processes, the peroxisomal fraction of oligodendrocytes increases the volume and density, in accordance with metabolic requirements, thus representing an important independent ROS source [86–88]. Therefore, in the process of remyelination which is parallel to the process of active demyelination, oligodendrocytes are particularly predisposed to damage by means of oxidative stress [89]. In this way, altered MBP and PLP are more susceptible to trypsin degradation, which suggests that under in vitro conditions, myelin, being altered by oxidative stress, is easily subjected to degradation by the effects of extracellular protease, like the ones released by myelin [90]. Delamination of myelin sheath makes new amounts of myelin components prone to being damaged by ROS. ROS activity leads to the increase of concentration of mRNA and protein expression of matrix metalloproteinase (MMP-1), an interstitial collagenase which actively participates in the process of further demyelination [72, 91, 92].
Oligodendrocytes are especially sensitive to enormous release of glutamate, much more than astrocytes [93]. During acute attacks of the disease, concentrations of glutamate are increased which is, among other things, a consequence of inhibition of its takeover by astrocytes, resulting from the effects of proinflammatory cytokines, TNF-α, INF-γ, and IL-1β on astrocytes. Excitotoxicity of glutamate is mediated by the reduction of cystine concentration, which leads to the reduction of GSH concentration, too [94]. Oligodendrocytes in all their development forms, starting from the progenitor cell to the mature oligodendrocyte, have half the concentration of GSH in comparison to astrocytes. It is a consequence of the lower activity of glutathione synthetase, half the activity of GR compared to astrocytes, and far lower activity (15 % of astrocyte activity) of GSH-Px. Immature oligodendrocytes are sensitive to oxidative damages compared to mature forms. Lower activity of SOD has also been shown in oligodendrocyte culture compared to microglial cells and astrocytes [72, 95, 96]. Oligodendrocytes contain the biggest amount of Fe which is otherwise located in the CNS [97]. In neuroinflammation, due to microglia activation, Fe can be released from depots, ferritin, and transferrin, and, under conditions of a lower activity of SOD, it can trigger oxidative stress. Fe deposits have been found during radiological examinations of MS patients [98, 99].
Pathogenetic Importance of Oxidative Stress Modulation in Neuroinflammation
The Importance of Modulating the Process of Lipid Peroxidation
One of the leading pathogenetic mechanisms which oxidative stress use to achieve its pathogenetic effect in neuroinflammation is the process of lipid peroxidation [100]. There are some studies which have found the increased intensity of lipid peroxidation in experimental autoimmune encephalomyelitis (EAE) [101], indicating that the intensity of this process could be a direct consequence of immunoinduction toward its own antigens of the myelin sheath which has a high lipid content [17]. The association of the loss of immunological tolerance toward its own antigens MBP and MOG and the increase of the products produced by means of malondialdehyde (MDA)-mediated covalent alteration of proteins and the interaction of MDA with cysteine, lysine, and histidine were proven in this way [102]. Some results indicate the direct relation between the increase of MDA concentration and the severity of EAE neurological findings [103] and the relation between the increase of MDA concentration and the radiological findings of MS patients [104].
Some studies have shown that the modification of albumin affected by MDA, during neuroinflammation, causes the initiation of the immune response toward this protein, along with the simultaneous activation of a specific immunity, which aggravates the course of neuroinflammation and the severity of its clinical manifestation [105, 106]. Recent studies indicate that the complement factor H, as the main MDA-binding protein, can block the initiation of this type of immunostimulation. The probable mechanism in the basis of this effect is the takeover of molecules modified by MDA activity by phagocytic cells, which prevents the loss of immunotolerance and, in that way, the adverse effects in the nervous tissue which could be initiated [107]. The reduction of the intensity of oxidative and nitrosative stress directly reduces the intensity of lipid peroxidation, along with the improvement of the clinical manifestation of neuroinflammation [108]. The effect of reducing the intensity of lipid peroxidation has been proven by other studies [108, 109], indicating that MDA could be a sensitive marker of neuroinflammation intensity [110].
Some results indicate that the increase of MDA concentration in plasma and CSF has been observed in different clinical phenotypes of neuroinflammation [79, 111]. There are results which point to the close relationship between the intensity of lipid peroxidation process and the concentration of proinflammatory cytokines [41], indicating the decline of the intensity of this process along with a reduction of the intensity of the inflammatory reaction [112]. This effect has been observed after the use of corticosteroid therapy, wherein the larger decline of MDA concentration has been in the plasma than in the CSF [48]. The increase of MDA concentration in the CSF is a direct consequence of the increased intensity of lipid peroxidation process in the CNS during neuroinflammation, which obviously exists during the clinical remission of the disease. This is supported by the finding of the increased MDA concentration in the CSF after stabilization of the BBB by the use of corticosteroid therapy, which suggests that the increase of MDA concentration in the CNS is not a consequence of an “overflow” of this biomarker from the periphery through BBB damage, due to the fact that a high level of MDA is also reflected after therapeutic stabilization of the BBB [48]. This finding has been supported by the results of recent studies which indicate the larger increase of MDA concentration in the CSF than in the plasma in MS patients [104]. For this reason, the level of MDA in the CSF is considered to be the marker which directly reflects the intensity of neuroinflammation, especially due to inverse correlations of MDA level and the volume of the intact nervous tissue, which has been proven in neuroinflammation [113].
Taking into account that lipid peroxidation is a common link for the activity of nitrosative and oxidative stress [46, 114], it is considered as the crucial pathogenetic mechanism in the process of neuroinflammation and the nervous tissue damages mediated by it [115]. There are some results which indicate the absence of difference in MDA values in CSF and plasma between the patients with different clinical phenotypes of neuroinflammation [104]. The majority of end products of lipid peroxidation are taken from plasma or CSF by macrophages or they are being bound to the proteins of CSF or plasma [45]. In this way, MDA can be absorbed through the arachnoid villi into the blood, which reduces its concentration in CSF at the expense of increasing the concentration in plasma [61], which could be the explanation for the absence of any difference in MDA values in CSF and plasma between different clinical phenotypes of neuroinflammation [104].
It has been shown that erythrocytes of the patients suffering from autoimmune diseases [77, 116], including the ones suffering from MS, show greater rigidity of the cell membrane and the tendency toward spontaneous and induced cell lysis, which is in direct correlation with the intensity of inflammation [117] and its clinical presentation [118, 119]. It has been proven that the basis of these processes is the increased intensity of lipid peroxidation, taking into account that the erythrocyte membrane is a suitable substrate for initiating this process under conditions of the increased nitrosative and oxidative stress, which exist in neuroinflammation [41, 45, 100]. Similar results have been found by some of the recent studies [120, 121] which also indicate the existence of positive correlations between the process of lipid peroxidation and paraclinical findings of neuroinflammation. The authors found the explanation of the obtained correlations in the increased concentration of the products produced by the covalent modification of different biomolecules by the effect of intermediate and end products of the process of lipid peroxidation. These products, important for the further pathogenetic mechanisms of neuroinflammation, also include the advanced oxidation protein products (AOPP) [24, 122, 123].
Pathogenetic Aspects of the Role and Importance of AOPP in Neuroinflammation
Since the very discovery of AOPP, it has been defined as a stable marker of oxidative modification of protein [124], which was also confirmed by some of the recent studies [100]. The increased concentration of AOPP under conditions of oxidative and nitrosative stress is not surprising considering the fact that proteins, in accordance with their concentration which is larger than other biomolecules in the organism, are the primary target of ROS [99, 100].
The production of AOPP is explained by the chemical interaction of chlorine oxidants (chloramine and hypochlorite), which are abundantly produced in neuroinflammation, and proteins [18, 125]. However, it has been proven that AOPP are not only the end products of such chemical interaction, but, after creation, they also have a role as active mediators in the further evolution of the inflammatory process. AOPP realize their pathogenetic importance in neuroinflammation by the induction of the production of various cytokines and adhesion molecules which are important for the process of increasing BBB permeability [126]. Taking into account that AOPP are created by the effect of chlorine oxidants produced by nonspecific immune cells, which are the first line of defense in neuroinflammation, they could be considered as indicators of the intensity of the earliest inflammatory reaction [127]. This statement is supported by the finding of the positive correlation between the value of AOPP in CSF and plasma in MS patients and the parameters of the biological-biochemical syndrome of inflammation, primarily the number of leukocytes, the concentration of CRP, and fibrinogen [128].
Oxidatively modified proteins are hydrophobic and subject to denaturation, fragmentation, and aggregate formation with other oxidatively modified biomolecules [129]. In accordance with the fact that they are formed from proteins, AOPP could be considered as the direct marker of the content of albumin and the total proteins in a given tissue. However, the confirmation of such assumptions has not been obtained by the results of some recent studies because AOPP concentration in the CSF has been larger in patients with a smaller CSF content of proteins [128]. Taking into account the aforementioned facts, it seems that other proteins, not only albumins, though being the main protein fraction of plasma and CSF, participate in the formation of AOPP. In their work, Heinecke and associates [130] performed an analysis of the connection between AOPP concentration and other markers of protein damage and showed the direct connection with serum concentration of dityrosine which is considered to be a marker of oxidative stress intensity in general, while a far weaker connection was observed between AOPP concentration and other products of protein modification [131]. Similar results have been found by other studies which have analyzed the electrophoretic finding of AOPP, wherein the existence of two different peaks has been observed, of 670 and 70 kDa [130]. It has been shown that the peak of the higher molecular weight is formed by the aggregates of oxidatively modified albumins which mostly form disulfide bonds, as well as dityrosine aggregates, while the peak of the lower molecular weight contained albumins and other plasma proteins in a monomeric or structurally modified form.
Even though, on one side, AOPP are more susceptible to proteolytic degradation, and on the other side, they are taken over in slight quantities by phagocytic cells, which leads to the increase of their extracellular concentration [53, 125]. In accordance with this, higher AOPP values in patients with neuroinflammation suggest the existence of intensive inflammatory processes, which is confirmed in some studies of the parameters of the biological-biochemical syndrome of the inflammation of these patients [128].
During neuroinflammation, AOPP can directly perform the increase of the expression of transcriptional factors and cytokines, like NF-κB, TNF-α, iNOS, and some adhesion molecules like ICAM-1 and VCAM-1, which are considered to be of crucial importance in the inflammatory processes of the CNS and the induction of the immune response on the whole [132, 133]. On the other hand, preservation of defense capacities is necessary so that the nervous tissue and the whole organism could react as a response to this stimulus [134]. Over time, during the course of the disease, defense mechanisms become exhausted, which is, to some extent, present even during periods of clinical remission [83]. Therefore, the increased AOPP concentration, which indicates a more intensive inflammatory cascade, causes a higher level of nervous tissue damage and thus a more severe clinical picture of neuroinflammation only under conditions of disrupted defense potentials of the organism [135, 136]. Even though positive correlations between AOPP concentrations and the clinical finding and the length of MS were observed [137, 138], while interpreting these findings, AOPP concentration was not of crucial importance, but it was the disruption of defense mechanisms [137], because the effect shown by AOPP in relation to myelin and axon damage was potentiated in the group of patients with longer duration of the disease [128].
It is assumed that in the interaction with myelin sheath, AOPP is directly involved in the process of neoepitope creation, which leads to immunoinduction and favoring of further damaging of the nervous tissue [126]. AOPP can also have a direct immunogenic effect in the CNS by means of the products of the incomplete proteolytic degradation [129]. Starting form this statement, the existence of morphological changes in the brain tissue in MS patients, seen by radiological examination [129], could be understood as a consequence of this effect of AOPP on the nervous tissue [139].
The results of the earlier studies show the increased concentration of oxidatively modified proteins in erythrocyte membranes under conditions of oxidative stress [47]. There is some evidence of the direct link between the erythrocytic content of AOPP and MDA and the severity of the clinical manifestation and the length of neuroinflammatory condition [20, 139, 140].
The Damaging of GSH Homeostasis and the Level of SH Groups in Neuroinflammation
Considering the higher concentration, and thus the greater importance, in comparison to other redox-regulating molecules in the cell, GSH concentration changes have been studied in many prooxidative conditions [56, 141, 142]. The results of these studies agree with the conclusion that the decline of GSH concentration is responsible for the evolution of oxidative damages and the occurrence of their clinical correlates.
The decline of GSH concentration and its increase after the use of a substance which realizes the modulatory effect on the intensity of the nitrosative and oxidative stress have shown a negative correlation with the severity of EAE clinical manifestations [99]. Due to the fact that GSH synthesis in neurons is directly dependent on the concentration of the existing GSH and cysteine availability, it is not surprising that GSH declines in EAE, when GSH is spent due to the buffering of redox oxidative disbalance, and that it increases after the use of N-acetylcysteine (NAC) which is the direct donor of cysteine. Similar results have also been published in some other studies [143]. The takeover of cysteine is mediated by the cystine/glutamate transporter which is expressed on the BBB and other CNS structures [144]. Due to the fact that this process mediates the injection of cystine into the cell in contra-transport with glutamate, the activity of this transporter is reduced under conditions of disruption of the gradient of the extracellular concentration of the glutamate which initiates this process. The disruption of this gradient exists in neuroinflammation. The increased expression of this glutamatergic transporter, EAAT1, in brain tissue of EAE animals [104] confirms the existence of glutamatergic excitotoxicity. In this way, glutamatergic excitotoxicity leads to the disruption of the antioxidative capacity of the CNS [145, 146].
Special sensitivity of oligodendrocytes to the oxidative damages is caused by the physiologically lower content of GSH [147, 148]. Other glial cells activated in neuroinflammation because of high GSH concentrations show a higher level of resistance to the nitrosative and oxidative damages [149]. There are studies which indicate that the supplementation of the precursors of GSH synthesis increases the resistance and the regenerative ability of oligodendrocytes [148]. This effect is probably mediated also by the prevention of glutamatergic excitotoxicity, which is confirmed by the reduction of the expression of the EAAT1 transporter in the brain of EAE animals treated with NAC [104, 128].
There are results which indicate that the use of NAC directly reduces TNF-α expression and thus the cell damages mediated by it [148–150]. This mechanism of NAC effect lays in the basis of NO· concentration decline in EAE animals treated with NAC, due to the fact that the reduction of TNF-α also reduces its direct stimulatory effect on iNOS activity [151, 152]. Some studies indicate that thiol supplementation prevents cell death mediated by the activation of ASK1 complex, also known as apoptosome, which reduces cell damages in the CNS [153], the consequence of which is the benign clinical presentation of the disease. The increased production of NO· during inflammation is important for the process of GSH consumption in the CNS [154]. In the direct interaction of NO· and GSH and other low molecular weight antioxidants, S-nitrosoglutathione (GSNO) and S-nitrosothiols (RSNO) are created [142, 155]. In this way, the thiol content is reduced and the redox oxidation buffering capacity of the CNS is directly modified [156, 157]. RSNO is mostly created by means of GSNO, wherein NO· from GSNO moves to the protein thiol groups [158, 159]. In this way, GSNO increases bioavailability of NO· and participates in the regulation of NO· depots in the CNS and the regulation of nitrosative stress intensity [160, 161]. GSNO achieves its effect in the process of degradation to NO· and GSSG in the reaction which is spontaneous or mediated by low molecular weight thiols [161]. GSH concentration decline under conditions of NO· overproduction is, among other things, conditioned by the overproduction of RSNO [104, 118]. On one hand, NO·-mediated damages could be prevented in this way [162–164], but since NO· can be further released from RSNO, which can multiply its concentration, its adverse effects could also be potentiated [161, 165]. There are results which indicate that RSNO can also manifest independent immunogenic features, which contribute to the intensification of the inflammatory processes in the CNS [166]. The results of some studies indicate the presence of antibodies on RSNO, or the products of its interaction with other biomolecules, in the plasma or CSF of MS patients [167].
The special importance of thiol redox system in the CNS is in the process of cell signalization in the CNS and in the process of CNS growth and development and control of cell death [143]. As a part of signal pathways in the CNS, produced RSNO is important for the balance of the activities of many enzymes, like S-nitrosoglutathione reductase, thioredoxin reductase, Cu/Zn SOD, carbonyl reductase, and protein disulfide isomerase [129, 157]. RSNO can also mediate S-nitrosylation of the p65 subunit of NF-κB, which inhibits translocation of this transcriptional factor into the nucleus, and thus potentiate the antiinflammatory effect [168]. On the other hand, it is known that proinflammatory cytokines, TNF-α, IFNγ, and IL-1b, lead to demyelination of the p65 subunit, thus intensifying the inflammation process [169]. It seems that the interaction between GSH and NO· is another important place in neuroinflammation pathogenesis where nitrosative and oxidative stress converge. There are results which show that RSNO produced in such way performs covalent modification of all isoenzymatic forms of NOS, thus reducing their activity and disabling the production of new quantities of NO· [170]. In this way, controlled production of NO· is achieved, and that is why the RSNO amount could be the direct indicator of NOS activity [161, 171].
GSH concentration reduction in neurons of MS patients leads to actin polymerization and rearrangement of these filaments, which is confirmed by the histological finding of the analyzed demyelinating plaque. ATP quantity is reduced in this process, which further potentiates prooxidative processes and damages created by their activity [142]. There are results which indicate that the level of GSH concentration declines, which is the consequence of its consumption during the buffering of redox oxidative damages and RSNO formation, which is confirmed by the results of the experimental [172] and clinical research [128]. In the research of Prasad et al. [173], GSH treatment has not shown the protective effect in relation to the intensity of the process of neuroinflammation and its clinical correlates. On the contrary, GSH concentration decline and RSNO concentration increase had an important effect on the reduction of the intensity of inflammatory processes in the CNS, also affecting the reduction of its clinical manifestation [173]. The assumed mechanisms of these effects seem to be the consequence of the overrated role that NO·, released from RSNO, can participate in the processes of posttranslational modification of different signal molecules important for the process of neuroinflammation [174, 175].
The decline of GSH concentration in erythrocytes during the acute attack of MS was shown in some of the earlier studies [176]. There are results which indicate that the antioxidative supplementation based on the large amount of thiol potentiates the increase of antioxidative capacity of erythrocytes [177]. The decline of GSH concentration is interpreted by the accelerated consumption with the aim of compensating the prooxidative condition which exists in these cells [178]. This is supported by results which indicate a negative correlation between the decline of intracellular concentration of GSH, the intensity of oxidative and nitrosative stress, and the length of this state [179].
Under in vitro conditions, it has been shown that the inhibition of inflammation caused by lipopolysaccharides increases the intracellular content of thiol [180]. The assumed mechanism of such effect could be S-nitrosylation of free GSH which modulates the activity of Th17 cells, whose effects are independent of Th1 and Th2 cell response [181]. In this way, a protective effect is achieved on the appearance, the course, and the clinical manifestation of neuroinflammation.
While the content of nonprotein thiol is important for the cell antioxidative capacity, numerous protein SH groups are important components of the antioxidative capacity of the plasma and CSF [163]. These SH groups, as well as SH groups of the cell membrane, are the suitable substrates for the activity of ROS of various alkylation agents, which leads to the reduction of their concentration and the loss of the role they have in the organism [182]. There are results which show the decline of the total SH groups in plasma and CSF in both CIS and RRMS groups [128]. Results of other studies also indicate the decline of total SH group concentration in MS patients during the relapse of the disease, with the increase of SH group concentration after corticosteroid therapy and the establishment of clinical remission [48]. Calabrese et al. [183] point to the big pathogenetic importance of the reduction of SH group concentration in the acute inflammation of the nervous tissue. In the studies of Scapagnini et al. [184], it has been shown that the use of different antioxidants prevents the loss of the total quantity of SH groups. It is assumed that this effect is achieved by modulating the intracellular signalization by the stimulation of HSP32 and heme oxygenase-1, which are the key factors in the earliest phases of neuroinflammation. The reduction of the intensity of neuroinflammation and oxidative and nitrosative stress after the use of interferon therapy leads to the increase of SH group concentration, with the simultaneous improvement of clinical findings [133, 185]. However, there are studies with a larger number of included patients which do not find such a correlation of the changes in SH group concentration and the clinical findings [83].
It has been shown that restoring GSH levels via supplementations based upon thiols could be less effective if GPx activity is impaired [186]. There is evidence that GPx activity approached significant decreased values in MS compared to values obtained in control subjects [22, 187]. There are results which demonstrate that some drugs, being evaluated for therapy in MS, suppress inflammatory activation caused by lipopolysaccharides in vitro, enhancing total thiol levels. That was mediated by distinct mechanisms which were partly dependent on GSH-related enzymes, but other than GPx [180]. There is also a report that states that mitoxantron therapy induces the marked decrease in GPx activity in MS patients, after its initial elevation, which is followed by significant amelioration of clinical severity [188]. There are reports which suggest that both enzymatic and nonenzymatic antioxidants could regulate the function of immunologic cells enrolled in neuroinflammation and its promotion [189]. GPx can act as an inhibitor on 5-lipoxygenase in monocytes and macrophages within inflammatory-mediated processes, in this way decreasing the intensity of neuroinflammation, since 5-lipoxygenase is highly induced in the named inflammatory cells [190].
Pathogenetic Aspects of the Change in Catalase and Superoxide Dismutase Activity in Neuroinflammation
Catalase (CAT) is the main part of the brain antioxidative response [55, 57]. It is an intracellular antioxidant enzyme mainly located in cellular peroxisomes and cytosol which has been found in all cell types of the CNS, including also oligodendroglia which are particularly vulnerable to the effects of oxidative injuries [191]. Some authors demonstrated that various CAT treatment protocols might be useful in suppressing MS progression under experimental [86, 192] and clinical [193] conditions. Lower CAT activity in MS patients might be a consequence of the insufficiency of CNS antioxidative responses due to disease duration [48]. Thus, the CAT activity can be influenced by the limitation of GSH bioavailability, since it is well known that CAT activity is especially important in case of GSH depletion, which is demonstrated in MS [25]. Calabrese et al. [148] demonstrated an approximately threefold increase of CAT activity in the CSF of MS patients compared to control values. Considering CAT activity inducement in disease protection, there is also a demonstration of amelioration of experimental demyelization induced by combining scavenging of superoxide and hydrogen peroxide by viral-mediated gene transfer of the human CAT gene [55]. The findings by van Horssen et al. [21] demonstrate that antioxidative enzymes, including CAT and SOD, are markedly upregulated in active demyelinating MS lesions.
The changes of the activity of SOD in neuroinflammation are expected due to the fact that SOD represents the first line of antioxidative defense of the organism [26, 29, 53]. In the CNS, Cu/ZnSOD (SOD1) is highly expressed in astrocytes, MnSOD (SOD2) is expressed in neurons, while the extracellular Cu/ZnSOD (SOD3) is characterized by a decreased expression compared to the previous two SOD isomorphs [26]. The results of the studies which have tested SOD1 activity changes in neuroinflammation indicate the increase of the gene expression for SOD1 in the active MS plaques [194]. Other studies point to the decrease of the activity of this enzyme with the increase of SOD2 activity in EAE [55]. The studies of Horssen et al. [21] have shown the increase of the total activity of SOD in acute MS attacks, which is supported by the results of the recent studies on SOD activity in plasma [194]. Taking into account the existence of oxidative and nitrosative stress in the CNS [194], the reduction of SOD activity in neuroinflammation could be a part of the reduction of the total antioxidative defense, which is suggested by the results of similar studies [25, 27, 111]. The reduction of SOD activity opens up the possibility for further evolution of oxidative damages inside the CNS by the previously described mechanisms [61]. This is the explanation for the highly important negative correlation between the activity of this enzyme and the severity of the clinical picture and the radiological finding of patients with acute attacks of neuroinflammation [103].
Some earlier studies point to the increase of SOD activity in CSF in acute MS attacks [195], contrary to the results which suggest the decline of SOD activity in these patients [104]. It is possible that ROS, directly or by means of inflammatory mediators, achieve the effect on the gene expression of this enzyme [28], wherein the suppressor effect of ROS on SOD expression is potentiated over time [27]. The increase of SOD activity in erythrocytes during inflammatory processes has also been shown [123, 196]. SOD increase occurs as a consequence of adaptive compensatory reactions on prooxidative processes [21] and indicates the preservation of antioxidative potential, while a smaller increase of the activity or its absence could be a consequence of the inactivation of this enzyme by the prolonged oxidative stress which is a characteristic of MS patients even in the absence of the acute disease attacks [83]. There are studies [123] which find a correlation between SOD activity in erythrocytes and the severity of clinical picture. On the other hand, there are results which deny this type of correlation [197], although these results also suggest that the decline of SOD activity is in a negative correlation with the disease duration. Results of some studies also suggest a strong direct correlation between the decline of both total and cell antioxidative potential and the time during which the organism is exposed to prooxidative effects [122]. This effect is potentiated with the increase of patients’ age [24]. It has been shown that antioxidative potential in a dose- and time-dependent form increases the antioxidative capacity of erythrocytes, thus increasing SOD activity, which prevents the occurrence of various degenerative diseases [198]. This is supported by the results of an experimental study which has shown that SOD activity increases and the clinical presentation of neuroinflammation is mitigated after the antioxidative treatment [99]. It is interesting that erythrocytes, as circulating cells, are always exposed to the adverse effects of ROS, even in the absence of clinical relapses of the disease [22, 82], which makes the change of antioxidative capacity of these cells one of the important links in neuroinflammation pathogenesis [118, 119].
Modulation of Oxidative Stress in Neuroinflammation—Therapeutic Perspectives
The role of oxidative stress in the pathogenesis of neuroinflammation and demyelination is undoubtedly big. The effect of various antioxidative approaches like supplementation of oxidative enzymes in oligodendrocyte culture has been confirmed, thus achieving the protective effect, compared to the damages caused by ROS [148]. The supplementation of cystine and cysteine has shown a favorable effect on this cell line, increasing the intracellular level of GSH [153]. As shown in other experimental studies, by similar mechanisms, NAC achieves protective features in relation to the clinical manifestation of EAE [99]. The earliest supplementation by antioxidants has shown the direct but delayed favorable effect in relation to the clinical presentation of EAE, even after a short application at the beginning of the disease and a further suspension of the use of these enzymes [198]. The existence of protective features of the use of antioxidative enzymes before EAE induction was also indicated because the occurrence of the disease after a premedication with these enzymes was delayed and slow; in one percent of the cases, it did not even occur [86]. The largest number of studies indicates that the use of antioxidants affects the change of neuroinflammation course under experimental conditions, which is reflected in the reduction of the severity of clinical presentation of the disease and the total reversibility of the changes that occurred, faster achieving of remission level, and the delayed and slow course of EAE [118].
This review gives an advanced insight into the roles and the importance of oxidative stress during neuroinflammation and offers the possibility for applying antioxidative treatments in acute attacks of neuroinflammation. In this way, neuroinflammation might be controlled in early phases characterized by reversibility, at the same time delaying later phases which are accompanied with irreversible neurological disabilities.
References
Ames AI (2000) CNS energy metabolism as related to function. Brain Res Rev 34:42–68
Galea I, Bechmann I, Perry VH (2007) What is immune privilege (not)? Trends Immunol 28:12–18
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7:41–53
Bailey SL, Carpentier PA, McMahon EJ, Begolka WS, Miller SD (2006) Innate and adaptive immune responses of the central nervous system. Crit Rev Immunol 26:149–188
Owens T, Bechmann I, Engelhardt B (2008) Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67:1113–1121
Chan WY, Kohsaka S, Rezaie P (2007) The origin and cell lineage of microglia: new concepts. Brain Res Rev 53:344–354
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76(2):77–98
Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26(3):349–354
Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B (2005) Microglial cell population dynamics in the injured adult central nervous system. Brain Res Rev 48:196–206
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14:1189–1197
Fabriek BO, Van Haastert ES, Galea I, Polfliet MM, Dopp ED, Van Den Heuvel MM, Van Den Berg TK, De Groot CJ, Van DV, Dijkstra CD (2005) CD163 positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia 51:297–305
Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9(6):429–439
Kivisakk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, Khoury SJ (2009) Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol 65:457–469
Fetler L, Amigorena S, Neuroscience (2005) Brain under surveillance: the microglia patrol. Science 309:392–393
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(5):1175–1189
Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH (1999) Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci 11:3648–3658
Ponomarev ED, Shriver LP, Maresz K, Dittel BN (2005) Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res 81:374–389
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000) Multiple sclerosis. N Engl J Med 343:938–952
Hafler DA (2004) Multiple sclerosis. J Clin Invest 113:788–94
Rizvi SI, Maurya PK (2007) Alterations in antioxidant enzymes during aging in humans. Mol Biotechnol 37:58–61
van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, de Vries HE (2008) Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med 45(12):1729–1737
Tasset I, Agüera E, Sánchez-López F, Feijóo M, Giraldo AI, Cruz AH, Gascón F, Túnez I (2012) Peripheral oxidative stress in relapsing-remitting multiple sclerosis. Clin Biochem 45(6):440–444
Ullevig S, Kim HS, Asmis R (2013) S-glutathionylation in monocyte and macrophage (dys)function. Int J Mol Sci 14(8):15212–15232
Erden Inal M, Kanbak G, Sunal E (2001) Antioxidant enzyme activities and malondialdehyde levels related to aging. Clin Chim Acta 305:75–80
Schreibelt G, van Horssen J, van Rossum S, Dijkstra CD, Drukarch B, de Vries HE (2007) Therapeutic potential and biological role of endogenous antioxidant enzymes in multiple sclerosis pathology. Brain Res Rev 56(2):322–330
Miller AF (2004) Superoxide dismutases: active sites that save, but a protein that kills. Curr Opin Chem Biol 8(2):162–168
Namaki S, Mohsenzadegan M, Mirshafiey A (2009) Superoxide dismutase: a light horizon in treatment of multiple sclerosis. J Chin Clin Med 4(10):585–591
Afonso V, Champy R, Mitrovic D, Collin P, Lomri A (2007) Reactive oxygen species and superoxide dismutases: role in joint disease. Joint Bone Spine 74:324–329
Johnson F, Giulivi C (2005) Superoxide dismutases and their impact upon human health. Mol Aspects Med 26(4–5):340–52
Ramming T, Appenzeller-Herzog C (2013) Destroy and exploit: catalyzed removal of hydroperoxides from the endoplasmic reticulum. Int J Cell Biol 2013:180906
Woolley JF, Stanicka J, Cotter TG (2013) Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem Sci 38(11):556–565
Piloni NE, Fermandez V, Videla LA, Puntarulo S (2013) Acute iron overload and oxidative stress in brain. Toxicology 314(1):174–182
Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D (2013) The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol 64(4):409–421
Ozyazgan S, Andican G, Erman H, Tuzcu A, Uzun H, Onal B, Ozyazgan Y (2013) Relation of protein oxidation parameters and disease activity in patients with Behçet’s disease. Clin Lab 59(7–8):819–825
Feligioni M, Nisticò R (2013) SUMO: a (oxidative) stressed protein. Neuromolecular Med 15(4):707–719
Sitar ME, Aydin S, Cakatay U (2013) Human serum albumin and its relation with oxidative stress. Clin Lab 59(9–10):945–952
Sadowska-Bartosz I, Adamczyk-Sowa M, Galiniak S, Mucha S, Pierzchala K, Bartosz G (2013) Oxidative modification of serum proteins in multiple sclerosis. Neurochem Int 63(5):507–516
Go YM, Jones DP (2013) Thiol/disulfide redox states in signaling and sensing. Crit Rev Biochem Mol Biol 48(2):173–181
Boersma BJ, D’Alessandro T, Benton MR, Kirk M, Wilson LS, Prasain J, Botting NP, Barnes S, Darley-Usmar VM, Patel RP (2003) Neutrophil myeloperoxidase chlorinates and nitrates soy isoflavones and enhances their antioxidant properties. Free Radic Biol Med 35(11):1417–1430
Perez Gutierrez RM, Flores Cotera LB, Gonzalez AM (2012) Evaluation of the antioxidant and anti-glycation effects of the hexane extract from piper auritum leaves in vitro and beneficial activity on oxidative stress and advanced glycation end-product-mediated renal injury in streptozotocin-treated diabetic rats. Molecules 17(10):11897–11919
Ferretti G, Bacchetti T (2011) Peroxidation of lipoproteins in multiple sclerosis. J Neurol Sci 311(1–2):92–97
Nam TG (2011) Lipid peroxidation and its toxicological implications. Toxicol Res 27(1):1–6
Yeagle PL (2013) Non-covalent binding of membrane lipids to membrane proteins. Biochim Biophys Acta. doi:10.1016/j.bbamem.2013.11.009
Ren R, Hashimoto T, Mizuno M, Takigawa H, Yoshida M, Azuma T, Kanazawa K (2013) A lipid peroxidation product 9-oxononanoic acid induces phospholipase A2 activity and thromboxane A2 production in human blood. J Clin Biochem Nutr 52(3):228–233
Koch M, Mostert J, Arutjunyan AV, Stepanov M, Teelken A, Heersema D, De Keyser J (2007) Plasma lipid peroxidation and progression of disability in multiple sclerosis. Eur J Neurol 14(5):529–533
Quintana FJ, Yeste A, Weiner HL, Covacu R (2012) Lipids and lipid-reactive antibodies as biomarkers for multiple sclerosis. J Neuroimmunol 248(1):53–57
Pandey KB, Rizvi SI (2009) Protective effect of resveratrol on formation of membrane protein carbonyls and lipid peroxidation in erythrocytes subjected to oxidative stress. Appl Physiol Nutr Metab 34:1093–1097
Mitosek-Szewczyk K, Gordon-Krajcer W, Walendzik P, Stelmasiak Z (2010) Free radical peroxidation products in cerebrospinal fluid and serum of patients with multiple sclerosis after glucocorticoid therapy. Folia Neuropathol 48(2):116–122
Pandey KB, Rizvi SI (2010) Protective effect of resveratrol on markers of oxidative stress in human erythrocytes subjected to in vitro oxidative insult. Phytother Res 24(S1):S11–S14
Sims-Robinson C, Hur J, Hayes JM, Dauch JR, Keller PJ, Brooks SV, Feldman EL (2013) The role of oxidative stress in nervous system aging. PLoS One 8(7):e68011
Sarsour EH, Louise Kalen A, Goswami P (2013) Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antioxid Redox Sign. doi:10.1089/ars.2013.5303
Sheng Y, Chattopadhyay M, Whitelegge J, Valentine JS (2012) SOD1 aggregation and ALS: role of metallation states and disulfide status. Curr Top Med Chem 12(22):2560–2572
Mirshafiey A, Mohsenzadegan M (2009) Antioxidant therapy in multiple sclerosis. Immunopharmacol Immunotoxicol 31(1):13–29
Carillon J, Rouanet JM, Cristol JP, Brion R (2013) Superoxide dismutase administration, a potential therapy against oxidative stress related diseases: several routes of supplementation and proposal of an original mechanism of action. Pharm Res 30(11):2718–2728
Qi X, Hauswirth WW, Guy J (2007) Dual gene therapy with extracellular superoxide dismutase and catalase attenuates experimental optic neuritis. Mol Vis 13:1–11
Heather LM, Teismann P (2009) Glutathione—a review on its role and significance in Parkinson’s disease. FASEB J 23(10):3263–3272
Baud O, Greene AE, Li J, Wang H, Volpe JJ, Rosenberg PA (2004) Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci 24:1531–1540
Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q (2004) Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol 36:2376–2391
Halldorsdottir SM, Kristinsson HG, Sveinsdottir H, Thorkelsson G, Hamaguchi PY (2013) The effect of natural antioxidants on haemoglobin-mediated lipid oxidation during enzymatic hydrolysis of cod protein. Food Chem 141(2):914–919
Penga F, Yanga Y, Liua J, Jianga Y, Zhua C, Denga X, Hua X, Chena X, Zhongb X (2012) Low antioxidant status of serum uric acid, bilirubin and albumin in patients with neuromyelitis optica. Eur J Neurol 19:277–283
Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451(7182):1076–1081
Pitt D, Werner P, Raine CS (2000) Glutamate excitotoxicity in a model of multiple sclerosis. Nature Med 6:67–70
Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4:181–189
Ilhan A, Akyol O, Gurel A, Armutcu F, Iraz M, Oztas E (2004) Protective effects of caffeic acid phenethyl ester against experimental allergic encephalomyelitis-induced oxidative stress in rats. Free Radic Biol Med 37:386–394
Witte ME, Bo L, Rodenburg RJ, Belien JA, Musters R, Hazes T, Wintjes LT, Smeitink JA, Geurts JJ, De Vries H, Van DV, van Horssen J (2009) Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol 219:193–204
Pahan K, Mondal S (2012) Crosstalk between nitric oxide and T helper cells. J Clin Cell Immunol 3:e109
Satoh T, Lipton SA (2007) Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci 1:37–45
Yokoyama H, Yano R, Aoki E, Kato H, Araki T (2008) Comparative pharmacological study of free radical scavenger, nitric oxide synthase inhibitor, nitric oxide synthase activator and cyclooxygenase inhibitor against MPTP neurotoxicity in mice. Metab Brain Dis 23:335–349
Charil A, Filippi M (2007) Inflammatory demyelination and neurodegeneration in early multiple sclerosis. J Neurol Sci 259:7–15
Macco R, Pelizzoni I, Consonni A, Vitali I, Giacalone G, Martinelli Boneschi F, Codazzi F, Grohovaz F, Zacchetti D (2013) Astrocytes acquire resistance to iron-dependent oxidative stress upon proinflammatory activation. J Neuroinflamm 10(1):130
Ortiz GG, Pacheco-Moisés FP, Bitzer-Quintero OK, Ramírez-Anguiano AC, Flores-Alvarado LJ, Ramírez-Ramírez V, Macias-Islas MA, Torres-Sánchez ED (2013) Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol 2013:708659
Ljubisavljevic S, Stojanovic I (2014) Neuroinflammation and demyelination from the point of nitrosative stress as a new target for neuroprotection. Rev Neurosci. doi:10.1515/revneuro-2014-0060
Garden GA, Moller T. Microglia biology in health and disease. J. Neuroimmune. Pharmacol. 2006; 1: 127–137
Kornek B, Storch M, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive and remyelinated lesions. Amer J Pathol 157:267–276
Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, Nataf S (2010) Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol 68:465–476
Knoferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tönges L, Stadelmann C, Brück W, Bähr M, Lingor P (2010) Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A 107:6064–6069
Shah D, Kiran R, Wanchu A, Bhatnagar A (2010) Oxidative stress in systemic lupus erythematosus: relationship to Th1 cytokine and disease activity. Immunol Lett 129:7–12
Fukuda M, Kanou F, Shimada N, Sawabe M, Saito Y, Murayama S, Hashimoto M, Maruyama N, Ishigami A (2009) Elevated levels of 4-hydroxynonenal-histidine Michael adduct in the hippocampi of patients with Alzheimer’s disease. Biomed Res 30:227–233
Miller E, Mrowicka M, Saluk-Juszczak J, Ireneusz M (2011) The level of isoprostanes as a non-invasive marker for in vivo lipid peroxidation in secondary progressive multiple sclerosis. Neurochem Res 36(6):1012–1016
Hendrickx DA, Koning N, Schuurman KG, van Strien ME, van Eden CG, Hamann J, Huitinga I (2013) Selective upregulation of scavenger receptors in and around demyelinating areas in multiple sclerosis. J Neuropathol Exp Neurol 72(2):106–118
Zhu X, Su B, Wang X, Smith MA, Perry G (2007) Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 64:2202–2210
Xiang W, Weisbach V, Sticht H, Seebahn A, Bussmann J, Zimmermann R, Becker CM (2013) Oxidative stress-induced posttranslational modifications of human hemoglobin in erythrocytes. Arch Biochem Biophys 529:34–44
Oliveira SR, Kallaur AP, Simao ANC, Morimoto HK, Lopes J, Panis C, Petenucci DL, da Silva E, Cecchini R, Kaimen-Maciel DR, Reiche EM (2012) Oxidative stress in multiple sclerosis patients in clinical remission: association with the expanded disability status scale. J Neurol Sci 321(1–2):49–53
Przedborski S, Ischiropoulos H (2005) Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal 7:685–693
Kim SU, Park YH, Min JS, Sun HN, Han YH, Hua JM, Lee TH, Lee SR, Chang KT, Kang SW, Kim JM, Yu DY, Lee SH, Lee DS (2013) Peroxiredoxin I is a ROS/p38 MAPK-dependent inducible antioxidant that regulates NF-κB-mediated iNOS induction and microglial activation. J Neuroimmunol 259(1–2):26–36
Singh I, Paintlia AS, Khan M, Stanislaus R, Paintlia MK, Haq E, Singh AK, Contreras MA (2004) Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res 1022(1–2):1–11
Zeis T, Graumann U, Reynolds R, Schaeren-Wiemers N (2008) Normal-appearing white matter in multiple sclerosis is in a subtle balance between inflammation and neuroprotection. Brain 131:288–303
Sullivan GM, Mierzwa AJ, Kijpaisalratana N, Tang H, Wang Y, Song SK, Selwyn R, Armstrong RC (2013) Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J Neuropathol Exp Neurol 72(12):1106–1125
Fancy SP, Kotter MR, Harrington EP, Huang JK, Zhao C, Rowitch DH, Franklin RJ (2010) Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp Neurol 225:18–23
Chaitanya GV, Omura S, Sato F, Martinez NE, Minagar A, Ramanathan M, Guttman BW, Zivadinov R, Tsunoda I, Alexander JS (2013) Inflammation induces neuro-lymphatic protein expression in multiple sclerosis brain neurovasculature. J Neuroinflamm 10(1):125
Agrawal SM, Williamson J, Sharma R, Kebir H, Patel K, Prat A, Yong VW (2013) Extracellular matrix metalloproteinase inducer shows active perivascular cuffs in multiple sclerosis. Brain 136(6):1760–1777
Uccelli A, Pedemonte E, Narciso E, Mancardi G (2003) Biological markers of the inflammatory phase of multiple sclerosis. Neurol Sci 24(5):S271–274
Willard SS, Koochekpour S (2013) Glutamate signaling in benign and malignant disorders: current status, future perspectives, and therapeutic implications. Int J Biol Sci 9(7):728–742
Mandolesi G, Musella A, Gentile A, Grasselli G, Haji N, Sepman H, Fresegna D, Bullitta S, De Vito F, Musumeci G, Di Sanza C, Strata P, Centonze D (2013) Interleukin-1β alters glutamate transmission at Purkinje cell synapses in a mouse model of multiple sclerosis. J Neurosci 33(29):12105–12121
Arend C, Brandmann M, Dringen R (2013) The antiretroviral protease inhibitor ritonavir accelerates glutathione export from cultured primary astrocytes. Neurochem Res 38(4):732–741
Fernandez-Fernandez S, Almeida A, Bolaños JP (2012) Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem J 443(1):3–11
Bizzozero OA, Ziegler JL, De Jesus G, Bolognani F (2006) Acute depletion of reduced glutathione causes extensive carbonylation of rat brain proteins. J Neurosci Res 83(4):656–667
Rumzan R, Wang JJ, Zeng C, Chen X, Li Y, Luo T, Lv F, Wang ZP, Hou H, Huang F (2013) Iron deposition in the precentral grey matter in patients with multiple sclerosis: a quantitative study using susceptibility-weighted imaging. Eur J Radiol 82(2):95–99
Abo-Krysha N, Rashed L (2008) The role of iron dysregulation in the pathogenesis of multiple sclerosis: an Egyptian study. Mult Scler J 14(5):602–608
Besler HT, Comoglu S (2003) Lipoprotein oxidation, plasma total antioxidant capacity and homocysteine level in patients with multiple sclerosis. Nutr Neurosci 6:189–196
Salemi G, Gueli MC, Vitale F, Battaglieri F, Guglielmini E, Ragonese P, Trentacosti A, Massenti MF, Savettieri G, Bono A (2010) Blood lipids, homocysteine, stress factors, and vitamins in clinically stable multiple sclerosis patients. Lipids Health Dis 9:19–21
Wallberg M, Bergquist J, Achour A, Breij E, Harris RA (2007) Malondialdehyde modification of myelin oligodendrocyte glycoprotein leads to increased immunogenicity and encephalitogenicity. Eur J Immunol 37:1986–1995
Ljubisavljevic S, Stojanovic I, Pavlovic D, Milojkovic M, Sokolovic D, Stevanovic I, Petrovic A (2013) Suppression of the lipid peroxidation process in the CNS reduces neurological expression of experimentally induced autoimmune encephalomyelitis. Folia Neuropathol 51(1):51–57
Ljubisavljevic S, Stojanovic I, Vojinovic S, Stojanov D, Stojanovic S, Kocic G, Savic D, Cvetkovic T, Pavlovic D (2013) Cerebrospinal fluid and plasma oxidative stress biomarkers in different clinical phenotypes of neuroinflammatory acute attacks. Conceptual accession: from fundamental to clinic. Cell Mol Neurobiol 33(6):767–777
Bongarzone ER, Pasquini JM, Soto EF (1995) Oxidative damage to proteins and lipids of CNS myelin produced by in vitro generated reactive oxygen species. J Neurosci Res 41:213–221
Wuttge DM, Bruzelius M, Stemme S (1999) T-cell recognition of lipid peroxidation products breaks tolerance to self proteins. Immunology 98:273–279
Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HPN, Issa PC, Cano M, Brandstätter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Christoph J (2011) Binder complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478:76–81
Kalyvas A, David S (2004) Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron 41:323–335
Marusic S, Leach MW, Pelker JW, Azoitei ML, Uozumi N, Cui J, Shen MW, DeClercq CM, Miyashiro JS, Carito BA, Thakker P, Simmons DL, Leonard JP, Shimizu T, Clark JD (2005) Cytosolic phospholipase A2α-deficient mice are resistant to experimental autoimmune encephalomyelitis. J Exp Med 202:841–851
Jana A, Pahan K (2007) Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: implications for multiple sclerosis. J Neuroimmune Pharmacol 2:184–193
Tavazzi B, Batocchi AP, Amorini AM, Nociti V, D’Urso S, Longo S, Gullotta S, Picardi M, Lazzarin G (2011) Serum metabolic profile in multiple sclerosis patients. Multiple Sclerosis International. doi:10.1155/2011/167156
Keles MS, Taysi S, Sen N, Aksoy H, Akçay F (2001) Effect of corticosteroid therapy on serum and CSF malondialdehyde and antioxidant proteins in multiple sclerosis. Can J Neurol Sci 28(2):141–143
Sbardella E, Greco A, Stromillo ML, Prosperini L, Puopolo M, Cefaro LA, Pantano P, De Stefano N, Minghetti L, Pozzilli C (2012) Isoprostanes in clinically isolated syndrome and early multiple sclerosis as biomarkers of tissue damage and predictors of clinical course. Mult Scler J. doi:10.1177/1352458512457721
Joseph JA, Shukitt-Hale B, Casadesus G, Fisher D (2005) Oxidative stress and inflammation in brain aging: nutritional considerations. Neurochem Res 30(6–7):927–935
Ferretti G, Bacchetti T, Principi F, Di Ludovico F, Viti B, Angeleri VA, Danni M, Provinciali L (2005) Increased levels of lipid hydroperoxides in plasma of patients with multiple sclerosis: a relationship with paraoxonase activity. Mult Scler J 11:677–682
Tsuda K (2012) Associations of oxidative stress and inflammation and their role in the regulation of membrane fluidity of red blood cells in hypertensive and normotensive men: an electron spin resonance investigation. Adv Biosci Biotechnol 3:1020–1027
Hon GM, Hassan MS, van Rensburg SJ, Abel S, Marais DW, van Jaarsveld P, Smuts CM, Henning F, Erasmus RT, Matsha T (2009) Erythrocyte membrane fatty acids in patients with multiple sclerosis. Mult Scler J 15(6):759–762
Koch M, Ramsaransing GS, Arutjunyan AV, Stepanov M, Teelken A, Heersema DJ, De Keyser J (2006) Oxidative stress in serum and peripheral blood leukocytes in patients with different disease courses of multiple sclerosis. J Neurol 253:483–487
de Freitas MV, de Oliveira MR, dos Santos DF, de Cássia Mascarenhas Netto R, Fenelon SB, Penha-Silva N (2010) Influence of the use of statin on the stability of erythrocyte membranes in multiple sclerosis. J Membr Biol 233(1–3):127–134
Pasichna EP, Morozova RP, Donchenko HV, Vinychuk SM, Kopchak OO (2007) Lipid peroxidation and antioxidant defence enzyme activity in multiple sclerosis. Ukr Biokhim Zh 79(5):165–174
Miler E, Walczak A, Majsterek I, Kędziora J (2013) Melatonin reduces oxidative stress in the erythrocytes of multiple sclerosis patients with secondary progressive clinical course. J Neuroimmunol. doi:10.1016/j.jneuroim.2013.02.012
Vani R, Shiva CS, Devi SA (2002) Oxidative stress in erythrocytes: a study on the effect of antioxidant mixtures during intermittent exposures to high altitude. Int J Biometeorol 54(5):553–562
Acar A, Ugur Cevik M, Evliyaoglu O, Uzar E, Tamam Y, Arıkanoglu A, Yucel Y, Varol S, Onder H, Taşdemir N (2012) Evaluation of serum oxidant/antioxidant balance in multiple sclerosis. Acta Neurol Belg 112(3):275–280
Witko-Sarsat V, Friedlander M, Capeillere-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers P, Descamps-Latscha B (1996) Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 49:1304–1313
Ghafourifar P, Mousavizadeh K, Parihar MS, Nazarewicz RR, Parihar A, Zenebe WJ (2008) Mitochondria in multiple sclerosis. Front Biosci 13:3116–3126
Fernandez O, Vermersch P (2011) From the fundamentals of multiple sclerosis to clinical management. J Neurol Sci 311:S1–2
Pentón-Rol G, Cervantes-Llanos M, Martínez-Sánchez G, Cabrera-Gómez JA, Valenzuela-Silva CM, Ramírez-Nuñez O, Casanova-Orta M, Robinson-Agramonte MA, Lopategui-Cabezas I, López-Saura PA (2009) TNF-α and IL-10 downregulation and marked oxidative stress in neuromyelitis optica. J Inflamm 6:18
Ljubisavljevic S, Stojanovic I, Vojinovic S, Stojanov D, Stojanovic S, Cvetkovic T, Savic D, Pavlovic D (2013) The patients with clinically isolated syndrome and relapsing remitting multiple sclerosis show different levels of advanced oxidation protein products and total thiol content in plasma and CSF. Neurochem Int 62(7):988–997
Park B, Lee S, Kim E, Cho K, Riddell SR, Cho S, Ahn K (2006) Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell 127:369–382
Heinecke JW, Li W, Daehnke HD, Goldstein JA (1993) Dityrosine, a specific marker of oxidation, in synthesized by the myeloperoxidase hydrogen peroxide system of human neutrophils and macrophages. J Biol Chem 268:4069–4077
Davies U (1987) Protein damage and degradation by oxygen radicals. I. General aspects. J Biol Chem 262:9895–9901
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717
Lucas M, Rodríguez MC, Gata JM, Zayas MD, Solano F, Izquierdo G (2003) Regulation by interferon beta-1a of reactive oxygen metabolites production by lymphocytes and monocytes and serum sulfhydryls in relapsing multiple sclerosis patients. Neurochem Int 42:67–71
Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis—the plaque and its pathogenesis. N Engl J Med 354:942–55
Bramow S, Frischer JM, Lassmann H, Koch-Henriksen N, Lucchinetti CF, Sorensen PS, Laursen H (2010) Demyelination versus remyelination in progressive multiple sclerosis. Brain 133:2983–2998
Bisaga GN, Odinak MM, Boiko AN, Melnik YB, Popova NF (2012) Treatment of exacerbations of multiple sclerosis without the use of corticosteroids: the role of metabolic and antioxidant therapy. Neurosci Behav Physiol 42(2):123–127
Bjartmar C, Trapp BD (2001) Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol 14:271–278
De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, Matthews PM, Arnold DL (2001) Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol 58:65–70
Pandey KB, Rizvi SI (2012) Markers of oxidative stress in erythrocytes and plasma during aging in humans. Oxid Med Cell Longev 3(1):2–12
Fiorini A, Koudriavtseva T, Bucaj E, Coccia R, Foppoli C, Giorgi A, Schininà ME, Di Domenico F, De Marco F, Perluigi M (2013) Involvement of oxidative stress in occurrence of relapses in multiple sclerosis: the spectrum of oxidatively modified serum proteins detected by proteomics and redox proteomics analysis. PLoS One 8(6):e65184
Shelton MD, Mieyal JJ (2008) Regulation by reversible S-glutathionylation: molecular targets implicated in inflammatory diseases. Mol Cell 25:332–346
Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E (2010) Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem 285:39646–39654
Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30:42–59
Castegna A, Palmieri L, Spera I, Porcelli V, Palmieri F, Fabis-Pedrini MJ, Kean RB, Barkhouse DA, Curtis MT, Hooper DC (2011) Oxidative stress and reduced glutamine synthetase activity in the absence of inflammation in the cortex of mice with experimental allergic encephalomyelitis. Neuroscience 185:97–105
Tao F, Lu SD, Zhang LM, Huang YL, Sun FY (2001) Role of excitatory amino acid transporter 1 in neonatal rat neuronal damage induced by hypoxia-ischemia. Neuroscience 102(3):503–513
Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A (2010) Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc¯ as a neuroprotective drug target. CNS Neurol Disord Drug Targets 9(3):373–382
Werner P, Pitt D, Raine CS (2001) Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 50(2):169–180
Calabrese V, Scapagnini G, Ravagna A, Bella R, Foresti R, Bates TE, Giuffrida Stella AM, Pennisi G (2002) Nitric oxide synthase is present in the cerebrospinal fluid of patients with active multiple sclerosis and is associated with increases in cerebrospinal fluid protein nitrotyrosine and S-nitrosothiols and with changes in glutathione levels. J Neurosci Res 70:580–587
Moss DW, Bates TE (2001) Activation of murine microglial cell lines by lipopolysaccharide and interferon-γ causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci 13:529–538
Kronke M, Adam-Klages S (2002) Role of caspases in TNF-mediated regulation of cPLA2. FEBS Lett 531:18–22
Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H (2010) Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 23(2):75–93
Steinert JR, Chernova T, Forsythe ID (2010) Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 16:435–452
Ouyang M, Shen X (2006) Critical role of ASK1 in the 6- hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Neurochem 97:234–244
Whiteman M, Chua YL, Zhang D, Duan W, Liou YC, Armstrong JS (2006) Nitric oxide protects against mitochondrial permeabilization induced by glutathione depletion: role of S-nitrosylation? Biochem Biophys Res Commun 339:255–262
Paige JS, Xu G, Stancevic B, Jaffrey SR (2008) Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem Biol 15:1307–1316
Romero JM, Bizzozero OA (2009) Intracellular glutathione mediates the denitrosylation of protein nitrosothiols in the rat spinal cord. J Neurosci Res 87:701–709
Marozkina NV, Gaston B (2011) S-Nitrosylation signaling regulates cellular protein interactions. Biochim Biophys Acta. doi:10.1016/j.bbagen.2011.06.017
Motterlini R, Foresti R, Bassi R, Calabrese V, Clark JE, Green CJ (2000) Endothelial heme oxygenase-1 induction by hypoxia. Modulation by inducible nitric-oxide synthase and S-nitrosothiols. J Biol Chem 275:13613–13620
Muller B, Kleschyov AL, Alencar JL, Vanin A, Stoclet JC (2002) Nitric oxide transport and storage in the cardiovascular system. Ann NY Acad Sci 962:131–139
Kim SF, Huri DA, Snyder SH (2005) Inducible nitric oxide synthase binds, S-nitrosylates and activates cyclooxygenase-2. Science 310:1966–1970
Knott AB, Bossy-Wetzel E (2009) Nitric oxide in health and disease of the nervous system. Antioxid Redox Signal 11:541–53
Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS (2004) Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116:617–628
Foster MW, McMahon TJ, Stamler JS (2003) S-Nitrosylation in health and disease. Trends Mol Med 9:160–168
Witherick J, Wilkins A, Scolding N, Kemp K (2010) Mechanisms of oxidative damage in multiple sclerosis and a cell therapy approach to treatment. Autoimmune Diseases. doi:10.4061/2011/164608
Liu JS, Zhao ML, Brosnan CF, Lee SC (2001) Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol 158:2057–2066
Sparaco M, Gaeta LM, Tozzi G, Bertini E, Pastore A, Simonati A, Santorelli FM, Piemonte F (2006) Protein glutathionylation in human central nervous system: potential role in redox regulation of neuronal defense against free radicals. J Neurosci Res 83:256–263
Boullerne AI, Rodrıguez JJ, Touil T, Brochet B, Schmidt S, Abrous NR, Le Moal M, Pua JR, Jensen MA, Mayo W, Arnason BGW, Petry KG (2002) Anti-S-nitrosocysteine antibodies are a predictive marker for demyelination in experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neurosci 22(1):123–132
Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, Elango C, Singh AK, Singh I (2005) S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab 25(2):177–192
Hendriks JJ, Teunissen CE, De Vries HE, Dijkstra CD (2005) Macrophages and neurodegeneration. Brain Res Rev 48:185–195
Ischiropoulos H, Beckman JS (2003) Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest 111:163–169
Foster MW, Hess DT, Stamler JS (2009) Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med 15:391–404
Ljubisavljevic S, Stojanovic I, Pavlovic R, Stojnev S, Stevanovic I, Sokolovic D, Pavlovic D (2012) The reduced glutathione and S-nitrosothiols levels in acute phase of experimental demyelination—pathophysiological approach and possible clinical relevancy. Neuroscience 219:175–182
Prasad R, Giri S, Nath N, Singh I, Singh AK (2007) GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia 55:65–77
Richter-Addo GB, Legzdins P, Burstyn J (2002) Introduction: nitric oxide chemistry. Chem Rev 102(4):857–860
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345(1–2):91–104
Kidd PM (2001) Multiple sclerosis, an autoimmune inflammatory disease: prospects for its integrative management. Altern Med Rev 6:54066
Grinberg L, Fibach E, Amer J, Atlas D (2005) N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radical Bio Med 38:1136–1145
Deneke SM (2000) Thiol-based antioxidants. Curr Top Cell Regul 36:151–180
Hvaring C, Vujicic S, Aasly JO, Feinstein DL, White LR, Boullerne AI (2013) IgM to S-nitrosylated protein is found intrathecally in relapsing-remitting multiple sclerosis. J Neuroimmunol 256(1–2):77–83
Lin SX, Lisi L, Dello Russo C, Polak PE, Sharp A, Weinberg G, Kalinin S, Feinstein DL (2011) The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 3(2):75–84. doi:10.1042/AN20100033
Nath N, Morinaga O, Singh I (2010) S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J Neuroimmune Pharm 2:240–251
Staron A, Makosa G, Koter-Michalak M (2012) Oxidative stress in erythrocytes from patients with rheumatoid arthritis. Rheumatol Int 32:331–334
Calabrese V, Scapagnini G, Ravagna A, Bella R, Butterfield DA, Calvani M, Pennisi G, Giuffrida Stella AM (2003) Disruption of thiol homeostasis and nitrosative stress in the cerebrospinal fluid of patients with active multiple sclerosis: evidence for a protective role of acetylcarnitine. Neurochem Res 28(9):1321–1328
Scapagnini G, Foresti R, Calabrese V, Giuffrida Stella AM, Green CJ, Motterlini R (2002) Caffeic acid phenethyl ester and curcumin: a novel class of heme oxygenase-l inducers. Mol Pharmacol 61:554–561
Stojanovic I, Ljubisavljevic S, Stevanovic I, Pavlovic R, Cvetkovic T, Djordjevic V, Pavlovic D, Vojinovic S, Basic J (2012) Nitric oxide-mediated signalization and nitrosative stress in neuropathology. J Med Biochem 31(4):295–300
van Meeteren ME, Teunissen CE, Dijkstra CD, van Tol EAF (2005) Antioxidants and polyunsaturated fatty acids in multiple sclerosis. Eur J Clin Nutr 59:1347–1361
Ljubisavljevic S, Stojanovic I, Cvetkovic T, Vojinovic S, Stojanov D, Stojanovic D, Bojanic V, Stokanovic D, Pavlovic D (2014) The glutathione homeostasis disruption of erythrocytes, but not glutathione peroxidase activity changes, is closely accompanied with neurological and radiological scoring of acute CNS inflammation. Neuroimmunomodulation 21:13–20
Adamczyksowa M, Sowa P, Pierzchala K, Polaniak R, Labuzroszak B (2012) Antioxidative enzymes activity and malondialdehyde concentration. During mitoxantrone therapy in multiple sclerosis patients. J Physiol Pharmacol 63(6):683–690
Miller E, Mrowicka M, Zołyński K, Kedziora J (2009) Oxidative stress in multiple sclerosis. Pol Merkur Lekarski 162:499–502
Straif D, Werz DO, Kellner R, Bahr U, Steinhilber D (2000) Glutathione peroxidase-1 but not -4 is involved in the regulation of cellular 5-lipoxygenase activity in monocytic cells. Biochem J 349:455–461
Jana A, Hogan EL, Pahan K (2009) Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci 278(1):5–15
Liu Y, Hao W, Letiembre M, Walter S, Kulanga M, Neumann H, Fassbender K (2006) Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J Neurosci 26(50):12904–12913
van Horssen J, Witte ME, Schreibelt G, de Vries HE (2011) Radical changes in multiple sclerosis pathogenesis. BBA – Mol Basis Dis 1812(2):141–150
Tajouri L, Mellick AS, Ashton KJ, Tannenberg AEG, Nagra RM, Tourtellotte WW, Griffiths LR (2003) Quantitative and qualitative changes in gene expression patterns characterize the activity of plaques in multiple sclerosis. Mol Brain Res 119(2):170–183
Yoshida E, Mokunoa K, Aoki S, Takahashi A, Riku S, Murayama T, Yanagi T, Kato K (1994) Cerebrospinal fluid levels of superoxide dismutases in neurological diseases detected by sensitive enzyme immunoassays. J Neurol Sci 124:25–31
Gallan PM, Carrascosa A, Gussinye M, Dominguez C (2003) Biomarkers of diabetes associated oxidative stress and antioxidant status in young diabetic patients with or without subclinical complication. Free Radical Bio Med 34:1563–1574
Zagórski T, Dudek I, Berkan L, Mazurek M, Kedziora J, Chmielewski H (1991) Superoxide dismutase (SOD-1) activity in erythrocytes of patients with multiple sclerosis. Neurol Neurochir Pol 25(6):725–730
Rowiński R, Kozakiewicz M, Kędziora-Kornatowska K, Hübner-Woźniak E, Kędziora J (2013) Markers of oxidative stress and erythrocyte antioxidant enzyme activity in older men and women with differing physical activity. Exp Gerontol 48:1141–1146
Acknowledgments
This work was supported by the grant from the scientific project number 41018 financed by the Ministry of Education and Science, Republic of Serbia.
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ljubisavljevic, S. Oxidative Stress and Neurobiology of Demyelination. Mol Neurobiol 53, 744–758 (2016). https://doi.org/10.1007/s12035-014-9041-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-9041-x