
Abstract
Resistance against glucocorticoids which are used to reduce inflammation and treatment of a number of diseases, including leukemia, is known as the first stage of treatment failure in acute lymphoblastic leukemia. Since these drugs are the essential components of chemotherapy regimens for ALL and play an important role in stop of cell growth and induction of apoptosis, it is important to identify genes and the molecular mechanism that may affect glucocorticoid resistance. In this study, we used the GSE66705 dataset and weighted gene co-expression network analysis (WGCNA) to identify modules that correlated more strongly with prednisolone resistance in type B lymphoblastic leukemia patients. The PPI network was built using the DEGs key modules and the STRING database. Finally, we used the overlapping data to identify hub genes. out of a total of 12 identified modules by WGCNA, the blue module was find to have the most statistically significant correlation with prednisolone resistance and Nine genes including SOD1, CD82, FLT3, GART, HPRT1, ITSN1, TIAM1, MRPS6, MYC were recognized as hub genes Whose expression changes can be associated with prednisolone resistance. Enrichment analysis based on the MsigDB repository showed that the altered expressed genes of the blue module were mainly enriched in IL2_STAT5, KRAS, MTORC1, and IL6-JAK-STAT3 pathways, and their expression changes can be related to cell proliferation and survival. The analysis performed by the WGCNA method introduced new genes. The role of some of these genes was previously reported in the resistance to chemotherapy in other diseases. This can be used as clues to detect treatment-resistant (drug-resistant) cases in the early stages of diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute lymphoblastic leukemia (ALL) is among the most common childhood cancers, accounting for almost two-thirds of childhood acute leukemias [1]. Despite the advances in therapeutic strategies during the last decades and the 5 year event-free survival rate increase from 30 to 85%, a small percentage of patients would still experience treatment failure in the very first stages [2,3,4]. The newly diagnosed patients are usually treated, using multi-drug chemotherapy regimens. Glucocorticoids such as prednisolone and dexamethasone are among the most widely used drugs in treating leukemic malignancies, including ALL, to maintain remission through rapid destruction of leukemia blasts [2, 5]. Glucocorticoids (GCs) are capable of activating apoptosis-inducing genes and inhibiting the transcription of survival genes by binding to glucocorticoid receptors (GRs), resulting in cell death and disruption of the cell cycle in malignant lymphoblastic cells [6,7,8].
Today, using GCs in monotherapy in the first week of treatment is one of the important factors for predicting treatment outcomes. The initial resistance to this class of corticosteroids is known as a prognostic factor of adverse outcome and is associated with the risk of recurrence [9, 10]. Various factors, including polymorphisms and mutations before and during treatment, RNA silencing or suppression by miRNAs and changes in GCs signaling pathways in ALL may be involved in GC resistance [11, 12]. However, the biomolecular mechanisms underlying resistance to GCs in patients with ALL are not completely known. In the present study, we performed a weighted gene co-expression network analysis on the microarray dataset GSE66705 and identified co-expressed modules highly correlated with prednisolone resistance in patients with type B-ALL. Then several gene clusters were obtained by overlapping the obtained data. Finally, the key pathways associated with prednisolone resistance were identified. This study was conducted to identify new genes that may provide new insight to explore the underlying molecular mechanisms of drug resistance in the early stages of B-ALL treatment.
Materials and Methods
Data Preparation and Processing
Gene expression profiles of all prednisolone-treated patients with accession numbers GSE66705 and GSE67684 obtained from the Gene Expression Omnibus (GEO) database [13]. The initial pre-processing of the raw data of both dataset, which are on the same platform gpl570 (Affymetrix hgu133plus2), including background correction and data normalization was performed using the Robust Multi-array Average (RMA) algorithm [14]. Entrez id and median were used, respectively, to remove the probes corresponding to several genes and determine the expression values of the probes assigned to each gene [15, 16]. To construct the WGCNA co-expression network and identify the potential genes involved in prednisolone resistance, only the expression data of samples of patients with lymphoblastic leukemia type B belonging to GSE66705 were used. After data normalization, gene expression changes were compared between the resistant and sensitive samples to prednisolone treatment. |logFC|> 1 and FDR < 0.01 was set to identify differentially expressed genes (DEGs). The gene expression profile of GSE67684 was used to evaluate the effectiveness of prednisolone and confirm the selected genes of GSE66705.
Construction of Weighted Gene Co-Expression Network and Identification of Modules related to Prednisolone Resistance
The WGCNA package of R software includes a comprehensive set of different R functions for analyzing large datasets, constructing a weighted gene co-expression network, and identifying modules related to prednisolone resistance in patients with ALL and the key genes (hub genes) [17].Firstly, Sample clustering was applied based on Pearson's correlation matrix, After that, we used the soft thresholding power β as a parameter to calculate the adjacency matrix and adjust the scale independence of the scale-free network. The lowest value of β, whose its scale-free independence index is above 0.8 was selected. Then, the block-wise Module function was used to manage the network’s construction and to classify co-expressed genes in different modules known as the module eigengene (ME). The minimum number of genes in each module was set to 30 and finally, an integration level cutoff of 0.25 was used as a criterion for integrating modules whose genes are highly correlated [18, 19].
Identification of Key Co-expression Modules Related to Clinical Traits
To identify and investigate the significant relationship between ME and clinical traits, the preliminary information provided about the type of sensitivity of patients to prednisolone was used. Moreover, the heatmap was used to visualize the correlations of each module better, and the modules that were significantly correlated with individual traits (P-value < 0.05) were identified [17]. To evaluate the extent of module preservation and also to select genes that could potentially be related to prednisolone resistance, the module preservation function of the WGCNA package of R software was applied and the Z summary was used to score the level of preservation. In this study modules with Z summary < 2 were considered as poorly preserved modules while modules with Z summary 2 -10 as semi-preserved modules and a Z summary > 10 implied a strongly preserved module. The default number of permutations was defined as P = 100. [20, 21] Since the semi-protected modules have more topological diversity among the case and control groups, modules with 5 < Z summary > 10 were considered as significantly correlated modules. Subsequently, to determine the proximity of a gene to a module, known as module membership, and the relationship between the genes of each module with clinical traits (drug sensitivity), known as Gene Significance, the quantitative association between MEs and gene expression profiles were investigated [20]. Finally, the hub gens of the blue module with the criteria of |MM|≥ 0.80 and |GS|≥ 0.65 were screened.
Module Enrichment Analysis
Enrichment analysis was performed to identify the biological pathways further and explore the function of DEGs of the key modules, which are mostly related to prednisolone resistance, using the Molecular Signature Database (MsigDB).
Construction of the PPI Network of the Clinically Significant Module
The online database (STRING v10.5) was used to assess the interaction between DEGs of the key module at the protein level. The protein–protein interaction (PPI) network was constructed considering the medium confidence score of 0.4 [22]. Cytoscape software (version 3.9) was used for the visualization of the PPI networks. Then, to screen genes with high interaction capability in the PPI network, the degree calculation algorithm in the CytoHubba plugin of Cytoscape software was used [23]. Nodes with connectivity degrees ≥ 10 were used as hub genes for further analysis.
Identifying the Key Genes (Hub Genes)
To identify the key genes that play role in developing resistance to prednisolone, The overlap between the obtained results from the previous steps including identified DEGs of the blue module, hub genes of the PPI network with a minimum connectivity degree of 10, genes of the blue module with |MM|≥ 0.80 and |GS|≥ 0.65, and identified DEG of the GSE67684 platform was investigated. Genes that were common between them were identified as the hub genes.
Statistics and Software
All analysis was performed by R programming language (V 4.1), and an unpaired t-test and One-way ANOVA tests were used to evaluate significant differences between groups. A significance level of less than P < 0.05 was considered for each statistical test. Cytoscape software (V 3.9) was used to visualize the protein networks.
Results
Data Processing
As for both datasets, the initial preprocessing of raw data, including background correction, and data normalization was done using R software. Then, by employing the median of multiple probes, 9299 genes from GSE66705 and 9154 genes from GSE67684, with the highest expression variance (over 25%), were identified. The ratio of gene expression changes between resistant and sensitive samples to prednisolone treatment was compared in the platform GSE66705. Totally, 422 genes with increased expression and 288 genes with decreased expression were distinguished. |logFC|> 1 and FDR < 0.01 was set to identify DEGs (Supplementary Table 1). Due to the existence of the diversity of B-cell subtypes only normalized data of GSE 67,684 were used considering FDR < 0.01(Supplementary Table_2).
Construction of the WGCNA Network
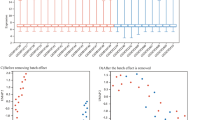
To construct the WGCNA network, only the expression data of treated patients with B-cell ALL of GSE66705 data were used. This dataset includes 45 sensitive samples, 17 samples with intermediate sensitivity, and 24 prednisolone-resistant samples. Average linkage hierarchical clustering was used to remove outliers and subsequently 1 sample was removed from the study due to poor quality. In our study, the soft thresholding power of 12 was chosen to calculate the scale-freeness of the network. To detect and integrate the modules with high correlation, a minimum module size of 30 and a merge Cut Height of 0.25 was set to determine and integrate the modules which have a high correlation (Fig. 1A). Consequently, a total of 12 co-expression modules labeled with different colors were identified (Supplementary Table 3, Fig. 1B). Most genes were grouped in the magenta (2815 genes) and blue (1180 genes) modules except for the gray module. The yellow-green module, with 39 genes, had the lowest number of grouped genes [39].

WGCNA co-expression network analysis and identification of modules related to resistance to prednisolone. A Analysis of fit index of scale independence and average connection for various powers of soft threshold β, B Dendrogram of clustered genes based on β = 12 and merge Cut Height = 0.25, C Identification of modules related to prednisolone resistance based on correlation and p value. Red and blue colors show the highest and lowest correlation, respectively, D Module preservation analysis using Z summary and median rank blue and green lines indicate thresholds of 5 and 10, respectively
Identification of Modules Associated with Prednisolone Resistance
To identify and examine the significant correlation between module eigengenes and clinical traits, we investigated the correlation between each module and the type of patient sensitivity to prednisolone. By comparing all the modules, it was found that the genes of the blue module (cor = 0.76, P = 8.e-08) in particular, were significantly positively correlated with prednisolone resistance followed by black (cor = 0.56, P = 4e-04), purple (cor = 0.42, P = 0.01) and red modules (cor = 0.36, P = 0.04), respectively. On the other hand, the pink (cor = 0.56, P = 4e-04) and magenta modules (cor = − 0.42, P = 0.01) showed a significant negative correlation (Fig. 1C). Due to the lack of significant change in the expression of genes in samples with intermediate sensitivity, their expression data were used only for better identification of modules resistant to prednisolone.
Preservation Assessment and Key Module Selection
The module Preservation function and Z summary were used to check the preservation level of modules and also to select the genes that could potentially be related to disease treatment. The cyan, red, yellow, purple, pink, and black modules were preserved, while the green-yellow module was considered unpreserved (Fig. 1D). Since semi-preserved modules have more topological diversity among the control and disease groups, in this study, GS criteria were used to select the appropriate key genes among blue, black, red, and purple modules.[20] The results of this study showed that the genes of the blue module (cor = 0.62, P = 3.3e-129) have higher importance and a higher module membership than those of other modules. Therefore, only the blue module was considered for further analysis and selection of key genes. Due to the lowest correlation and non-significance, purple and green modules were also excluded from further study. Finally, based on the criteria |MM|≥ 0.80 and |GS|≥ 0.65, 137 genes out of a total of 1180 genes with the possibility of playing role in prednisolone resistance were identified in the blue module ((Supplementary Table_4).
Enriched Key Module Pathways
The blue module pathways enrichment results showed that the genes are mostly upregulated in the following pathways heme metabolism, inflammatory response, IFN-γ, and apoptosis, with a significant level of less than 2e-15. Also, the signaling pathways including IL2_STAT5, KRAS, IL6_JAK_STAT3, and MTORC1 are enriched with a significance of less than 2e-6 (Fig. 2A). Downregulated genes are more enriched in the pathways of hypoxia, adipogenesis, ERL, EMT, and KRAS signaling pathways with a significance level of less than 3e-3 (Fig. 2B). Full details along with the gene names are depicted in (Supplementary Table_5 and 6).

DEGs enrichment analysis in blue module based on MsigDB data and visualization of PPI networks in Cytoscape. A Pathway analysis and ontology of upregulated genes, B Pathway analysis and ontology of downregulated genes, C Co-expression network of GSE66705 differentially expressed genes, The red and blue colors, respectively, indicate the up and downregulated genes in the blue module, and the gray color indicates the rest of the changed genes expressed in the GSE66705 dataset, D Co-expression network of the hub genes is identified in the blue module. The red color indicates that the hub genes and the size of each node is proportional to the displayed degree
Identification of PPI Network Hub Genes
Of a total of 709 genes with altered expression, the only interaction between 568 DEGs was estimated using the STRING database, and a PPI network of 568 nodes and 1959 edges was created by Cytoscape software. A total of 121 nodes with degree ≥ 10 were identified as hubs of PPI network genes (Fig. 2C–D and Supplementary Table_7).
Data Overlap Analysis and Selection of Key Genes
In order to identify the key genes, the overlap between the obtained results of the blue module’s altered expression genes (324 DEGs in total, including 271 upregulated and 53 downregulated genes), the known hub genes of the PPI network (121 genes out of a total of 568 genes), 137 genes selected according to the criteria of |MM|≥ 0.80 and |GS|≥ 0.65 and the statistically significant data extracted out of GSE67684 platform was evaluated (Fig. 3, Supplementary Table8). In total, nine genes consisting of CD82, FLT3, GART, HPRT1, ITSN1, MRPS6, MYC, SOD1, and TIAM1 were identified as the primary hub genes of this study, and MYC gene was identified as the main key gene with the highest degree of connectivity.

Identifying hub genes related to prednisolone resistance in Acute Lymphoblastic Leukemia. Venn diagram of the altered genes of GSE66874, co-expressed genes identified in the PPI network, blue module, and criteria of |MM|≥ 0.80 and |GS|≥ 0.65
Discussion
Although Glucocorticoids including prednisolone are classically served as a key component in the multiagent chemotherapy regimen of ALL, resistance to these agents is frequently observed among patients with ALL [24, 25]. In the present study, to identify the genes and molecular interactions that may contribute to prednisolone resistance development, we used the GSE66705 dataset to analyze the relationship between genes dysregulation and prednisolone resistance in patients with B-ALL. To identify the highly correlated modules and their genes with prednisolone resistance, WGCNA was employed to construct a co-expression network. Among all recognized modules, the blue module was selected as the key module of study. After that, we used the STRING database to identify DEGs out of 709 altered expressed genes of the blue module. 568 DEGs were recognized and a PPI network was constructed. By evaluating the overlapping data between the genes of the blue module with altered expression (324 DEGs in total, including 271 up-regulated genes and 53 down-regulated genes), results obtained from the known PPI network hub genes (121 genes out of a total of 568 genes) and 137 genes according to the criteria |MM|≥ 0.80 and |GS|≥ 0.65, and significantly altered genes of GSE67684 dataset, the overall hub genes of this study were recognized. At last, nine genes, SOD1, CD82, FLT3, GART, HPRT1, ITSN1, TIAM1, MRPS6, and MYC were identified as the primary hub genes of this study and the MYC gene was recognized as the main key gene.
One of the genes selected as a hub gene with increased expression in prednisolone resistance samples in our study is SOD1. The protein encoded by the Superoxide dismutase gene is in charge of destroying reactive oxygen species. The superoxide-free radical undergoes dismutation, and as a result, O2 and H2O2 are produced [26]. It has been shown that SOD1 is upregulated in ovarian cancer cells that are resistant to cisplatin [27]. SOD1 activity was suppressed using a non-cytotoxic dose of TETA, which resulted in a remarkable improved sensitivity to cisplatin in ovarian cancer cells [28]. Several studies have indicated that patients suffering from ALL have increased levels of oxidative stress and decreased levels of antioxidant biomarkers, such as SOD1 [29,30,31]. Nevertheless, a study by Chen et al. showed that overexpression of the SOD1 gene reduced the effect of PMA on myeloid leukemia cell death. This study also suggests that PMA induces cell death via PKD2 activation, and possibly by involving the SOD1-dependent pathways [32]. The next gene which was selected as a hub gene is CD82, a member of the tetraspanin family [33]. This gene is classically known as a metastasis suppressor that P53 can activate. This gene undergoes downregulation in some human cancers such as prostate cancer and breast cancer [34, 35]. However, one study has demonstrated that in pediatric patients with ALL, CD82 gene expression is aberrantly high [36].which is also consistent with our data. In addition, a previous study showed CD82 plays a role in chemoresistance in patients with ALL. Both chemotherapy treatments with daunorubicin and CD82 upregulation together, further increase the overexpression of PKCα. And it is the downstream molecule β1 integrin which in turn promotes AML cells survival [37]. This study also revealed that PKCα and β1 integrins have an integral role in evolving CD82-mediated chemoresistance. Since the inhibition of each of these two molecules reverses chemosensitivity. Our results suggest that increased expression of CD82 in GC-resistant samples compared to sensitive samples may be associated with prednisolone resistance in patients with ALL. The other hub gene, FLT3, is involved in the proliferation, differentiation and survival of hematopoietic stem cells [38]. Previous studies have reported that FLT3 signaling pathways have interacted with PI3K/Akt/mTOR. Akt-mediated phosphorylation which is involved in the later signaling pathway inhibits glucocorticoid receptor activity [24, 39, 40]. On the other hand, HPRT1 has been shown to be overexpressed in various cancers such as breast cancer, endometrial cancer, and head and neck squamous cell carcinoma [41, 42]. One study has shown that HPRT1 upregulation positively correlates to chemoresistance in patients with oral squamous cell carcinoma. In this study HPRT1 and MMP genes were enriched in the MMP1/PI3K/AKT signaling pathway [43]. Since previously PI3K/AKT pathway was previously proved to play a role in chemoresistance, and Akt-mediated phosphorylation which is involved in the later pathway inhibits the GRs activity, the study theorized that FLT3 and HPRT1 could induce glucocorticoid resistance by activating this signaling pathway [43]. The polypeptide encoded by GART gene plays an necessary role in the de novo biosynthesis pathway of the purines. Cell survival and proliferation is enhanced through pathways in which purines are involved [44]. The results of one previous study on mechanisms by which dihydrofolate reductase antifolate binds with the human GART gene, and inhibit purine biosynthesis demonstrated that GART can be potentially used for targeted chemotherapy. It has been reported that one GART inhibitor plays a cytostasis role in cancer cells which are P53 positive [45]. On the other hand, some studies has reported that GART inhibitors are cytotoxic for the tumor cells wether the P53 pathway is intact or not [46].
The mitochondrial ribosomal protein 6 (MRPS6) gene was found to be overexpressed in prednisolone resistance compared to sensitive samples. MRPS6 is shown to be upregulated (differential expression) in breast cancer cells and confers a poor prognosis for patients with breast cancer [47, 48]. However, its role in chemoresistance is still unknown. There are controversial data concerning the Intersectin-1 (ITSN1) gene. On one hand, ITSN1 has been identified as a key anti-apoptotic protein, knocking down the ITSN1 the mitochondrial apoptotic pathways are promoted [49]. ITSN1 exerts its anti-apoptotic effects through the MEK/Erk1/2 signaling pathway. On the other hand, one study showed that overexpression of ITSN1 by modulating the expression levels of Ki67 and cleaved caspase-3 was associated with escalation of cell apoptosis [50]. The next gene which was selected as a hub gene with increased expression in prednisolone resistance samples in our study is Tiam1 which encodes a protein called RAC-1. The RAC-1 signaling pathway is important in carcinogenesis [51, 52]. In one study performed by Izumi et al. indicated that TIAM1 overexpression could cause chemoresistance in colorectal cancer. The study demonstrated two main mechanisms; first the cancer-associated fibroblasts upregulate TIAM1, which in turn enhances resistance against therapeutic agents; the second mechanism is driving chemotherapy resistance by promoting the stemness of cancer cells [53]. Another study showed that the drug NVP-BEZ235 (a chemotherapy agent which confers apoptosis to HER2-positive breast cancer cells) induces upregulation of TIAM1 by activating the FGFR/STAT3 signaling pathway. This cascade of molecular events in turn leads to resistance against BE235 [54].
Pathway analysis of dysregulated genes of the blue module showed that IL2_STAT5 and KRAS signaling pathways were Top enriched in prednisolone resistance. This shows that the above signaling pathways play an important role in resistance to GCs. Previous research on T-type lymphocytes shows that IL2 is able to interfere with glucocorticoid signaling and prevent GRs from entering the nucleus and causing apoptosis [55]. In GC-resistant cells, IL2 can phosphorylate STAT5 through the JAK/STAT pathway, and by forming a heterodimer between GRs and STAT5 proteins, it prevents the transfer of GCR to the nucleus [56, 57]. Activation of STAT5 alone is not enough to induce steroid resistance, but in combination with other cellular defects or through alternative signaling pathways, STAT5 contributes to steroid resistance, This indicates that the expression changes or the mutations created in related genes in this pathway may play an effective role in resistance to prednisolone [58]. One known genes in the IL2_STAT5 pathway is MYC, which was determined as the main hub gene. The MYC family are proto-oncogenes whose phosphorylated proteins encoded by these genes are involved in cell cycle progression, apoptosis, and cell transformation [59]. MYC genes are known to be associated with Burkitt Lymphoma and High-Grade B-Cell Lymphoma Double-Hit/Triple-Hit. The role of MYC upregulation has also been revealed in T-cell ALL. Several studies have demonstrated that MYC overexpression plays a role in resistance to glucocorticoids in B-ALL and T-ALL which is also consistent with our data. Several studies have validated the positive impact of MYC upregulation in GC resistance evolution [6, 25, 60]. A study indicated that patients with c-myc overexpression are more likely to develop a persistent form of B-ALL. Hence it is proposed that the promoted cell growth and declined cellular death caused by increased c-myc regulation may lead to initial treatments [61]. In addition, PI3K/Akt/ mTORC1 is recognized to give rise to MYC overexpression. Silveiral et al. indicated MYC as a downstream gene of PI3K; in fact, the inhibition of PI3K induced a down-regulation in its targeted genes, including MYC. The study reported that in vitro and in vivo experiments showed a PIK3 inhibitor increased the sensitivity of T-ALL cells to glucocorticoids which implies that PI3K3 activity level is positively correlated with resistance to chemotherapy in T-ALL [62]. Therefore, it can be deduced that MYC confers a GC resistance through PI3K/Akt/ mTORC1.
One of the limitations of this study was the selection of key genes based on bioinformatics methods and several advanced statistical methods were applied to the blue module, which may have caused the unintentional deletion of some genes that are involved in prednisolone resistance, on the other hand, no laboratory study on humans or animal has been done and the findings of this study needs final confirmation through experimental tests.
Conclusion
In summary, the available literature demonstrates that some of the hub genes identified by this study have role in chemoresistance through different pathways. CD82, a metastasis suppressor is shown to induce chemoresistance by activating PKCα and β1 integrin in the presence of chemo agent. TIAM1 confers chemoresistance by increasing cancer cell stemness. Additionally, FLT3, HPRT1, and MYC contribute to chemotherapy resistance through the PI3K/Akt signaling pathway. This signifies the contribution of the PI3K3/Akt signaling pathway in the evolution of multi-drug resistance in chemotherapy via regulating various intracellular processes.[63] Although the role of some the hub genes including GART, ITSN1and MRPS6 in developing chemoresistance has not yet been established, it is worth to further studying the role of these genes’ dysregulation in creating drug resistance.
Data Availability
Supporting and raw data are available upon a reasonable request to the corresponding author.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- GC:
-
Glucocorticoids
- GR:
-
Glucocorticoid resistance
- GEO:
-
Gene expression omnibus
- WGCNA:
-
Weighted gene co-expression network analysis
- PPI network:
-
Protein interaction protein network
- DEG:
-
Differentially expressed gene
- logFC:
-
Log2-fold change
- FDR:
-
False discover rate
- GEO:
-
Gene expression omnibus
- SOD1:
-
Superoxide dismutase type 1
- FLT3:
-
Fms-related receptor tyrosine kinase 3
- GART:
-
Glycinamide ribonucleotide transformylase
- HPRT1:
-
Hypoxanthine phosphoribosyl transferase 1
- ITSN1:
-
Intersectin-1
- TIAM1:
-
T-Cell lymphoma invasion and metastasis 1
- MRPS6:
-
Mitochondrial ribosomal protein S6
References
Esparza, S. D., & Sakamoto, K. M. (2005). Topics in pediatric leukemia–acute lymphoblastic leukemia. Medscape General Medicine., 7(1), 23.
Güneş, A. M., Ören, H., Baytan, B., Bengoa, ŞY., Evim, M. S., Gözmen, S., et al. (2014). The long-term results of childhood acute lymphoblastic leukemia at two centers from Turkey: 15 years of experience with the ALL-BFM 95 protocol. Annals of Hematology., 93(10), 1677–1684.
Cooper, S. L., & Brown, P. A. (2015). Treatment of pediatric acute lymphoblastic leukemia. Pediatric Clinics., 62(1), 61–73.
Locatelli, F., Schrappe, M., Bernardo, M. E., & Rutella, S. (2012). How I treat relapsed childhood acute lymphoblastic leukemia Blood. The Journal of the American Society of Hematology., 120(14), 2807–2816.
Pui, C.-H., & Ribeiro, R. C. (2003). International collaboration on childhood leukemia. International Journal of Hematology., 78(5), 383–389.
Renner, K., Ausserlechner, M., & Kofler, R. (2003). A conceptual view on glucocorticoid-induced apoptosis, cell cycle arrest and glucocorticoid resistance in lymphoblastic leukemia. Current Molecular Medicine., 3(8), 707–717.
Sionov, R. V., Spokoini, R., Kfir-Erenfeld, S., Cohen, O., & Yefenof, E. (2008). Mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoid-induced apoptosis. Advances in Cancer Research., 101, 127–248.
Smith, L. K., & Cidlowski, J. A. (2010). Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Progress in Brain Research., 182, 1–30.
Gao, J., & Liu, W. (2018). Prognostic value of the response to prednisone for children with acute lymphoblastic leukemia: A meta-analysis. European Review for Medical and Pharmacological Sciences, 22(22), 7858–7866.
Gaipa, G., Basso, G., Biondi, A., & Campana, D. (2013). Detection of minimal residual disease in pediatric acute lymphoblastic leukemia. Cytometry Part B: Clinical Cytometry., 84(6), 359–369.
Liang, Y.-N., Tang, Y.-L., Ke, Z.-Y., Chen, Y.-Q., Luo, X.-Q., Zhang, H., et al. (2017). MiR-124 contributes to glucocorticoid resistance in acute lymphoblastic leukemia by promoting proliferation, inhibiting apoptosis and targeting the glucocorticoid receptor. The Journal of Steroid Biochemistry and Molecular Biology., 172, 62–68.
Scheijen, B. (2019). Molecular mechanisms contributing to glucocorticoid resistance in lymphoid malignancies. Cancer Drug Resistance., 2(3), 647.
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., et al. (2012). NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Research., 41(D1), D991–D995.
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U., et al. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics, 4(2), 249–264.
Alibés, A., Yankilevich, P., & Díaz-Uriarte, R. (2007). IDconverter and IDClight: Conversion and annotation of gene and protein IDs. BMC Bioinformatics, 8(1), 1–9.
Udhaya Kumar, S., Thirumal Kumar, D., Bithia, R., Sankar, S., Magesh, R., Sidenna, M., et al. (2020). Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: A systematic and bioinformatics approach. Frontiers in Genetics., 11, 734.
Langfelder, P., & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9(1), 1–13.
Langfelder P, Horvath S. Tutorials for the WGCNA Package. UCLA Los Ageles. 2014.
Liu, W., Li, L., & Li, W. (2014). Gene co-expression analysis identifies common modules related to prognosis and drug resistance in cancer cell lines. International Journal of Cancer., 135(12), 2795–2803.
Langfelder, P., Luo, R., Oldham, M. C., & Horvath, S. (2011). Is my network module preserved and reproducible? PLoS Computational Biology., 7(1), e1001057.
Motieghader, H., Kouhsar, M., Najafi, A., Sadeghi, B., & Masoudi-Nejad, A. (2017). mRNA–miRNA bipartite network reconstruction to predict prognostic module biomarkers in colorectal cancer stage differentiation. Molecular BioSystems., 13(10), 2168–2180.
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Research., 43(D1), D447–D452.
Chin, C.-H., Chen, S.-H., Wu, H.-H., Ho, C.-W., Ko, M.-T., & Lin, C.-Y. (2014). cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Systems Biology., 8(4), 1–7.
Olivas-Aguirre, M., Torres-López, L., Pottosin, I., & Dobrovinskaya, O. (2021). Overcoming glucocorticoid resistance in acute lymphoblastic leukemia: Repurposed drugs can improve the protocol. Frontiers in Oncology., 11, 617937.
Chen, Y., Jiang, P., Wen, J., Wu, Z., Li, J., Chen, Y., et al. (2020). Integrated bioinformatics analysis of the crucial candidate genes and pathways associated with glucocorticoid resistance in acute lymphoblastic leukemia. Cancer Medicine., 9(8), 2918–2929.
Li, S., Fu, L., Tian, T., Deng, L., Li, H., Xia, W., et al. (2018). Disrupting SOD1 activity inhibits cell growth and enhances lipid accumulation in nasopharyngeal carcinoma. Cell Communication and Signaling., 16(1), 1–13.
Fitzpatrick, D. P., You, J. S., Bemis, K. G., Wery, J. P., Ludwig, J. R., & Wang, M. (2007). Searching for potential biomarkers of cisplatin resistance in human ovarian cancer using a label-free LC/MS-based protein quantification method. Proteomics-Clinical Applications, 1(3), 246–63.
Brown, D. P., Chin-Sinex, H., Nie, B., Mendonca, M. S., & Wang, M. (2009). Targeting superoxide dismutase 1 to overcome cisplatin resistance in human ovarian cancer. Cancer Chemotherapy and Pharmacology., 63(4), 723–730.
Battisti, V., Maders, L. D., Bagatini, M. D., Santos, K. F., Spanevello, R. M., Maldonado, P. A., et al. (2008). Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clinical Biochemistry., 41(7–8), 511–518.
Rasool, M., Farooq, S., Malik, A., Shaukat, A., Manan, A., Asif, M., et al. (2015). Assessment of circulating biochemical markers and antioxidative status in acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) patients. Saudi Journal of Biological Sciences., 22(1), 106–111.
Mahmoud, L. B., Mdhaffar, M., Ghozzi, H., Ammar, M., Hakim, A., Atheymen, R., et al. (2017). Oxidative stress in Tunisian patients with acute lymphoblastic leukemia and its involvement in leukaemic relapse. Journal of Pediatric Hematology/Oncology., 39(3), e124.
Chen, Y.-L., & Kan, W.-M. (2015). Down-regulation of superoxide dismutase 1 by PMA is involved in cell fate determination and mediated via protein kinase D2 in myeloid leukemia cells. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. https://doi.org/10.1016/j.bbamcr.2015.07.025
Nishioka, C., Ikezoe, T., Yang, J., & Yokoyama, A. (2015). Tetraspanin family member, CD82, regulates expression of EZH2 via inactivation of p38 MAPK signaling in leukemia cells. PLoS ONE, 10(5), e0125017.
Dong, J.-T., Suzuki, H., Pin, S. S., Bova, G. S., Schalken, J. A., Isaacs, W. B., et al. (1996). Down-regulation of the KAI1 metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Research., 56(19), 4387–4390.
Yang, X., Wei, L. L., Tang, C., Slack, R., Mueller, S., & Lippman, M. E. (2001). Overexpression of KAI1 suppresses in vitro invasiveness and in vivo metastasis in breast cancer cells. Cancer research., 61(13), 5284–5288.
Ji, H., Chen, L., Dai, Y., Sun, X., Li, X., Wang, Q., et al. (2017). Aberrant expression of CD133 and CD82 in patients with pediatric acute lymphoblastic leukemia and the clinical significance. Oncology Letters., 14(5), 5811–5818.
Floren, M., Restrepo Cruz, S., Termini, C. M., Marjon, K. D., Lidke, K. A., & Gillette, J. M. (2020). Tetraspanin CD82 drives acute myeloid leukemia chemoresistance by modulating protein kinase C alpha and β1 integrin activation. Oncogene, 39(19), 3910–3925.
Gilliland, D. G., & Griffin, J. D. (2002). The roles of FLT3 in hematopoiesis and leukemia Blood. The Journal of the American Society of Hematology., 100(5), 1532–1542.
Piovan, E., Yu, J., Tosello, V., Herranz, D., Ambesi-Impiombato, A., Da Silva, A. C., et al. (2013). Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell, 24(6), 766–776.
Chougule, R. A., Shah, K., Moharram, S. A., Vallon-Christersson, J., & Kazi, J. U. (2019). Glucocorticoid-resistant B cell acute lymphoblastic leukemia displays receptor tyrosine kinase activation. NPJ Genomic Medicine., 4(1), 1–7.
Townsend, M. H., Robison, R. A., & O’Neill, K. L. (2018). A review of HPRT and its emerging role in cancer. Medical Oncology., 35(6), 1–6.
Ahmadi, M., Eftekhari Kenzerki, M., Akrami, S. M., Pashangzadeh, S., Hajiesmaeili, F., Rahnavard, S., et al. (2021). Overexpression of HPRT1 is associated with poor prognosis in head and neck squamous cell carcinoma. FEBS Open Bio, 11(9), 2525–2540.
Wu, T., Jiao, Z., Li, Y., Su, X., Yao, F., Peng, J., et al. (2022). HPRT1 promotes chemoresistance in oral squamous cell carcinoma via activating MMP1/PI3K/Akt signaling pathway. Cancers, 14(4), 855.
Yin, J., Ren, W., Huang, X., Deng, J., Li, T., & Yin, Y. (2018). Potential mechanisms connecting purine metabolism and cancer therapy. Frontiers in Immunology., 9, 1697.
Zhang, C. C., Boritzki, T. J., & Jackson, R. C. (1997). An inhibitor of glycinamide ribonucleotide formyltransferase is selectively cytotoxic to cells that lack a functional G1 checkpoint. Cancer Chemotherapy and Pharmacology., 41, 223–228.
Bronder, J. L., & Moran, R. G. (2002). Antifolates targeting purine synthesis allow entry of tumor cells into S phase regardless of p53 function. Cancer Research., 62(18), 5236–5241.
Oviya, R. P., Gopal, G., Shirley, S. S., Sridevi, V., Jayavelu, S., & Rajkumar, T. (2021). Mitochondrial ribosomal small subunit proteins (MRPS) MRPS6 and MRPS23 show dysregulation in breast cancer affecting tumorigenic cellular processes. Gene, 790, 145697.
Lin, X., Guo, L., Lin, X., Wang, Y., & Zhang, G. (2022). Expression and prognosis analysis of mitochondrial ribosomal protein family in breast cancer. Scientific Reports., 12(1), 1–13.
Predescu, S. A., Predescu, D. N., Knezevic, I., Klein, I. K., & Malik, A. B. (2007). Intersectin-1s regulates the mitochondrial apoptotic pathway in endothelial cells. Journal of Biological Chemistry., 282(23), 17166–17178.
Xie, C., Xiong, W., Li, J., Wang, X., Xu, C., & Yang, L. (2019). Intersectin 1 (ITSN1) identified by comprehensive bioinformatic analysis and experimental validation as a key candidate biological target in breast cancer. OncoTargets and Therapy., 12, 7079.
Minard, M. E., Herynk, M. H., Collard, J. G., & Gallick, G. E. (2005). The guanine nucleotide exchange factor Tiam1 increases colon carcinoma growth at metastatic sites in an orthotopic nude mouse model. Oncogene, 24(15), 2568–2573.
Liang, J., Oyang, L., Rao, S., Han, Y., Luo, X., Yi, P., et al. (2021). Rac1, a potential target for tumor therapy. Frontiers in Oncology., 11, 674426.
Izumi, D., Toden, S., Ureta, E., Ishimoto, T., Baba, H., & Goel, A. (2019). TIAM1 promotes chemoresistance and tumor invasiveness in colorectal cancer. Cell Death & Disease., 10(4), 1–12.
Qiao, J., Li, M., Li, N., Xu, L., Huang, G., & Yu, J. (2020). TIAM1 upregulation confers NVP-BEZ235 resistance to breast cancer cells through FGFR/STAT3 pathway. Biochemical Genetics., 58(6), 953–965.
Kanagalingam, T., Solomon, L., Vijeyakumaran, M., Palikhe, N. S., Vliagoftis, H., & Cameron, L. (2019). IL-2 modulates Th2 cell responses to glucocorticosteroid: A cause of persistent type 2 inflammation? Immunity, Inflammation and Disease., 7(3), 112–124.
Biola, A., Lefebvre, P., Perrin-Wolff, M., Sturm, M., Bertoglio, J., & Pallardy, M. (2001). Interleukin-2 inhibits glucocorticoid receptor transcriptional activity through a mechanism involving STAT5 (signal transducer and activator of transcription 5) but not AP-1. Molecular Endocrinology., 15(7), 1062–1076.
Goleva, E., Kisich, K. O., & Leung, D. Y. (2002). A role for STAT5 in the pathogenesis of IL-2-induced glucocorticoid resistance. The Journal of Immunology., 169(10), 5934–5940.
Van der Zwet JC, Buijs-Gladdines JG, Hagelaar R, Smits WK, Vroegindeweij E, Graus LT, et al. STAT5 does not drive steroid resistance in T-cell acute lymphoblastic leukemia despite the activation of BCL2 and BCLXL following glucocorticoid treatment. Haematologica. 2020.
Mohamed, A. N. (2018). MYC (MYC proto-oncogene, bHLH transcription factor). Atlas Genet Cytogenet Oncol Haematol., 22(6), 227–232.
Han, S.-S., Han, S., & Kamberos, N. L. (2014). Piperlongumine inhibits the proliferation and survival of B-cell acute lymphoblastic leukemia cell lines irrespective of glucocorticoid resistance. Biochemical and Biophysical Research Communications., 452(3), 669–675.
Allen, A., Gill, K., Hoehn, D., Sulis, M., Bhagat, G., & Alobeid, B. (2014). C-myc protein expression in B-cell acute lymphoblastic leukemia, prognostic significance? Leukemia Research., 38(9), 1061–1066.
Silveira, A. B., Laranjeira, A. B. A., Rodrigues, G. O. L., Leal, P. C., Cardoso, B. A., Barata, J. T., et al. (2015). PI3K inhibition synergizes with glucocorticoids but antagonizes with methotrexate in T-cell acute lymphoblastic leukemia. Oncotarget, 6(15), 13105.
Liu, R., Chen, Y., Liu, G., Li, C., Song, Y., Cao, Z., et al. (2020). PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death & Disease, 11, 797.
Acknowledgements
The authors would like to thank Dr. Behdokht Khani and Hussein Safin for helpful comments on the manuscript.
Funding
This work was supported by the Tehran University of Medical sciences.
Author information
Authors and Affiliations
Contributions
Design and data mining was done by SN and YA, supervision and revision by ZA and YA, and writing of the draft of the manuscript by SN, SF. All authors have read and approved the final version.
Corresponding authors
Ethics declarations
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical Approval
This study was conducted in silico based on the analysis of data obtained from online databases and no laboratory studies were conducted on humans or animals.
Consent for Publication
Not Applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nekoeian, S., Ferdowsian, S., Asgari, Y. et al. Identification of Hub Genes Associated with Resistance to Prednisolone in Acute Lymphoblastic Leukemia Based on Weighted Gene Co-expression Network Analysis. Mol Biotechnol 65, 1913–1922 (2023). https://doi.org/10.1007/s12033-023-00707-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-023-00707-0