
Abstract
Co-occurrence of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and IgA nephropathy (IgAN) is extremely uncommon. To date, only a few case reports have described such patients. Here, we describe the clinical presentation, pathologic features, treatment response, and outcome data of five patients with the rare form of co-existing AAV and IgAN and compared the characteristics of these patients to AAV patients with pauci-immune glomerulonephritis (n = 10) and IgAN patients (n = 10) that were selected as controls by stratified random sampling. In addition, we summarize all the previously reported cases of AAV and IgAN. In total, including the current study, 16 AAV/IgAN overlap cases were reported. Our five patients with the coexistence of AAV and IgAN were younger than the ten AAV patients with pauci-immune glomerulonephritis (22.6 ± 8.2 years versus 48.9 ± 15.7 years, respectively, P = 0.004). Histologically, they had a significantly lower percentage of glomeruli with fibrous crescents compared with AAV patients (0.0% versus 4.0%, P = 0.038). Compared with ten IgAN patients, our five AAV/IgAN patients had higher levels of ESR (P = 0.032) and CRP (P = 0.031). After accepting treatment with a combination of steroid and immunosuppressants, all patients showed a positive response to therapy, except for one patient in our cohort and another previously reported patient. We described the clinical presentation, pathologic features, treatment response, and outcome data of five patients with overlapping AAV and IgAN. They had mild glomerular pathological lesions and a positive response to aggressive immunosuppressive therapy. They were quite similar to pauci-immune AAV patients in clinical features, except for younger age. They had a lower percentage of glomeruli with fibrous crescents compared with AAV patients. In contrast to IgAN patients, they had higher levels of ESR and CRP. The mechanism of the coexistence of IgAN and AAV needs further study.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is typically characterized by the development of autoantibodies to the neutrophil protein leukocyte proteinase 3 (PR3-ANCA) or myeloperoxidase (MPO-ANCA), and is defined as an autoimmune disease [1]. For AAV patients, the kidney and lungs are the commonly affected organs. The hallmark histopathology of renal biopsy is segmental necrosis of the glomerular capillary loops with few or no glomerular immune complex deposits [1].
Immunoglobulin A (IgA) nephropathy (IgAN) is diagnosed by a distinctive pathologic hallmark, the presence of IgA-dominant or co-dominant immune deposits in the glomerular mesangium [2]. IgAN is the most prevalent pattern of primary chronic glomerulonephritis in the world, which typically affects young people [3].
A growing number of IgAN patients with serum ANCA positivity have been reported [4,5,6,7,8,9]. AAV patients with glomerular IgA deposits were also reported in some studies. Yanhong et al. reported that 26 of the 168 AAV patients had glomerular IgA deposition [10]. The coexistence of AAV and IgA nephropathy is extremely rare, and very few case reports described 11 AAV/IgAN overlap patients [11,12,13,14,15,16,17,18].
In this study, we described the clinical presentation, pathologic features, treatment response, and outcome data of five patients with this rare form of combined AAV and IgAN. To understand the features of these rare cases in-depth, we compared them with pauci-immune AAV patients (n = 10) and IgAN patients (n = 10) with the same proportion of crescentic glomeruli. To the best of our knowledge, this is the first report to describe five AAV/IgAN overlap patients and summarize all previously reported cases.
Methods
Patients
The study was approved by the Medical Ethics Committee of the Xiangya Hospital of Central South University for Human Studies (approval number 201711836). We retrospectively screened the clinical and histological data of 151 patients who were diagnosed with AAV and received a renal biopsy in Xiangya Hospital from January 2008 to June 2019. Five of these were diagnosed with IgAN by immunohistochemistry or immunofluorescence according to its diagnostic hallmark, the presence of IgA-dominant or co-dominant immune deposits in the glomerular mesangium. Of the 151 AAV patients, 88 renal biopsies from 88 patients showed pauci-immune glomerulonephritis, with little or no deposition of immunoglobulin or complement detectable by immunofluorescence (IF) or electron microscopy (EM). In order to analyze the features of the five patients in detail, we selected two control groups by stratified random sampling for comparative analysis. Ten patients from the 88 pauci-immune AAV patients were chosen as one group. Another control group was ten IgAN patients who had renal biopsy in Xiangya Hospital from January 2021 to December 2021. Both two control groups were selected by stratified random sampling and met the following criteria: (1) at least ten glomeruli available for histological evaluation and (2) with matching proportions of crescentic glomeruli.
Renal pathology
Renal biopsy specimens of all patients were analyzed by two renal pathologists independently. According to glomerular lesions, renal injury was divided into four patterns, including sclerotic (≥ 50% globally sclerosed glomeruli), crescentic (≥ 50% cellular crescents), focal (≥ 50% normal glomeruli), and mixed (none of the above three types) [19]. The interstitial infiltrates, tubulointerstitial injury score, and MEST-C score were used to evaluate the injury degree of glomeruli.
Statistical analysis
All data were analyzed by SPSS version 26.0. Statistical descriptions for normally distributed data were presented as mean and standard deviation (mean ± SD). Non-normal distribution data were expressed as median with interquartile range (IQR). All quantitative data of the patients in the two groups were compared by independent samples t test for normally distributed data and Mann–Whitney U test for non-normally distributed data. Qualitative data were analyzed by chi-square test or Fisher’s exact test. A P value < 0.05 was considered to be statistically significant.
Literature review
Literature published about AAV/IgAN overlap cases were located using PubMed and Web of Science databases. The following MESH terms were used for searching: IgAN (glomerulonephritides IgA, IgA glomerulonephritis, immunoglobulin A nephropathy, and Berges disease) and ANCA-associated vasculitis (anti-neutrophil cytoplasmic antibody-associated vasculitis, anti-neutrophil cytoplasmic autoantibody, granulomatosis with polyangiitis, Wegener granulomatosis, ANCAc, proteinase 3 ANCA, PR3-ANCA, microscopic polyangiitis, ANCAp, myeloperoxidase ANCA, and MPO-ANCA).
We summarized all case series and single-patient case reports which had the exact diagnosis of AAV and IgAN in the current review. Works of literature were excluded if they were not written in English.
Results
Clinical presentation and laboratory data of 5 AAV/IgAN overlap cases
The laboratory data were summarized in Table 1. All patients were positive for ANCA. Antibodies against the glomerular basement membrane and lupus anticoagulant were both negative in all patients, while patients 1 and 3 had positive antinuclear antibodies (ANA). Complement levels were normal in patients 2 and 5. Patients 3 and 4 had slightly increased sC4 levels. And the level of IgG in patient 1 increased a little. Patients 1 and 4 had markedly elevated serum creatinine levels.
Two patients (nos. 4 and 5) had hypertension. The level of hemoglobin in three patients was low (patients 1, 4, and 5: 68 g/L, 51 g/L, and 104 g/L respectively). All patients had microscopic hematuria. The values of quantification of proteinuria in five patients were all within the non-nephrotic range. The urinary protein level among three patients (nos. 2, 3, and 4) was greater than 1 g/d. Patients 1 and 5 did not have these specific values assessed.
Histologic features
Renal biopsy findings are summarized in Table 2. The number of glomeruli in five biopsy specimens ranged from 10 to 15. Glomeruli with crescent formation were observed in patients 1, 3, and 5. The range of the percentages of the sclerotic glomeruli in five patients was 0–71%. Only two patients (nos. 1 and 4) had 71% sclerotic glomeruli.
No fibrinoid necrosis, Bowman’s capsule rupture, granulomatous lesions, and thrombosis were noted in any of the cases. Glomerular immunofluorescence revealed the intensity of IgA immunostaining in renal biopsies from five patients was more than 2 + . The intensity of IgM staining ranged from 2 + to 4 + . All biopsy specimens indicated that obvious lesions were present in small arteries. The data about interstitial infiltrates, tubulointerstitial injury score, and MEST-C score were analogous in Table 2, showing that all patients had mild renal injury.
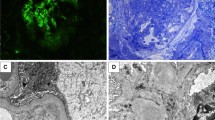
Light microscopy revealed lesions in the mesangium in renal biopsies from all patients. Figure 1 shows histology of representative glomeruli from patient 2. Hematoxylin and eosin (HE × 400) staining revealed diffuse mesangial hypercellularity (Fig. 1A). IF showed IgA staining was diffuse in the glomerular mesangium (Fig. 1B).

Histology of representative glomeruli from patient 2. (A) Hematoxylin and Eosin (HE × 200) stain showing diffuse mesangial hypercellularity. (B) Immunofluorescence microscopy of the renal biopsy specimen demonstrated diffuse mesangial staining for IgA (original magnification × 200)
The detailed proof of diagnoses for both AAV and IgAN in each patient
As evident, the renal pathological characteristics of the five patients described above were consistent with the pathological diagnostic hallmarks of IgA nephropathy: the predominance of IgA deposits in the glomerular mesangium by IF and the visible abnormalities in the mesangial area which can range from mesangial hypercellularity to severe proliferative glomerulonephritis with cellular crescents on light microscopy [20,21,22] (Table 2). The pathological diagnosis of IgAN was valid for every patient.
The diagnosis of MPA/GPA for each patient was made according to the hallmarks of kidney involvement, pulmonary involvement, or other manifestations of systemic vasculitis or renal pathological lesions. The details were listed in Table 3 and described as follows:
Patient 1 had clinical manifestations including microscopic hematuria and proteinuria. And he presented with extrarenal manifestations affecting the lung (diffuse infiltration in both lungs) and eyes (proptosis in the right eye). ANCA testing showed that perinuclear ANCA was positive and the titer of anti-MPO was 98 u/ml. Renal biopsy revealed the percentages of sclerotic glomeruli was 71%, and pathological lesions were observed in the small arteries suggesting vessel wall thickening. Part of the glomerular capillaries showed sclerotic lesions. Inflammatory cell infiltration and fibrous tissue hyperplasia were obvious in the glomerular interstitium. In light of these features, the diagnosis of AAV could be reasonable.
Patient 2 presented with proteinuria (2.66 g/24 h) and mild depressed edema in both lower extremities. His computer tomography revealed small nodules in the posterior basal segment of the right lower lobe. The titer of anti-PR3 was 45 u/ml. In kidney biopsies, 6 of the 15 glomeruli were sclerotic glomeruli but no crescent formation in the glomeruli. However, endothelial hypercellularity was obvious in the glomerular capillary. Additionally, there was inflammatory cell infiltration, fibrous tissue hyperplasia, and granulomatous lesions in the glomerular interstitium; and granulomatous vasculitis was obvious in intrarenal small arteries. In light of these signs, we diagnosed this patient with ANCA-associated vasculitis.
Osphyalgia, hematuria, and proteinuria were the clinical presentations leading to the admission of patient 3. Before these symptoms occurred, he had recurrent bloody nasal discharge and infection in the upper respiratory tract. Renal biopsy showed the percentage of crescentic glomeruli was 20%. Endothelial hypercellularity was obvious in the glomerular capillary. The renal interstitium showed infiltration of plasma cells, monocytes, and lymphocytes, and fibrous tissue hyperplasia. The small arteries had apparent lesions including vessel wall thickening and fibrosis, endothelial hypercellularity, luminal stenosis, and perivascular inflammatory cell infiltration. The laboratory evaluation revealed he was positive for PR3-ANCA and c-ANCA. Taken together, these clinical manifestations and kidney pathological features, patient 3 was considered to have AAV.
The diagnosis of AAV for patient 4 was made according to the result of renal biopsy revealing crescentic glomerulonephritis and her positive result for P-ANCA in 2008. However, she did not receive standard treatment because of other comorbidities such as severe pneumonia, iron deficiency anemia, and intracranial hemorrhage. Over the course of 8 years with an uncertain history of treatment, she had continuous increased creatinine. Eight years later, she presented with recurrent fatigue, cough, blood in the sputum, and mild depressed edema in both lower extremities. She was admitted to our hospital in 2016 and received a second renal biopsy. The result showed she had 10 (71%) sclerotic glomeruli but no crescentic glomeruli. So, the glomerular lesions in patient 4 appeared to be chronic lesions indicative of sclerosis. However, vessel wall thickening and mucoid degeneration could be seen in her renal small arteries. Furthermore, interstitial lesions showed inflammatory cell infiltration and fibrous connective tissue hyperplasia. In view of these findings, the changes were indicative of acute lesions. The imaging data of patient 4 revealed diffuse infiltration in the bilateral lung and alveolar hemorrhage. Combined with the clinical presentation, pathological features, and positive result of p-ANCA, the diagnosis of AAV could be valid.
Patient 5 presented for the first time with a 2-month history of hematuria and proteinuria. She was p-ANCA-positive and anti-MPO-positive. Her chest CT image found no abnormality. She underwent renal biopsy, and the results showed that 8% of glomeruli were sclerotic glomeruli and 23% of glomeruli were crescentic glomeruli. The renal interstitium showed lesions with inflammatory cell infiltration and fibrous tissue hyperplasia. The walls of the small arteries were thickened and the lumen was narrowed. The pathological changes indicated renal injury caused by ANCA-associated vasculitis and a diagnosis of microscopic polyangiitis was made.
According to the 2021 KDIGO CLINICAL PRACTICE GUIDELINE [23] and 2022 ACR/EULAR classification criteria, [24,25,26] three patients could be diagnosed as having microscopic polyangiitis (MPA) and two having granulomatosis with polyangiitis (GPA) (Table 3). The diagnoses of AAV and IgAN were defined concomitantly in four patients (nos. 1, 2, 3, and 5) during their first admission. Patient 4 was diagnosed as having AAV during her first admission, and IgAN was diagnosed by the outcome of the second renal biopsy when she was readmitted to our hospital. The time interval between the two diagnoses was 8 years.
Treatment and follow-up
The treatment and follow-up were summarized in Table 4. Induction and maintenance therapies were undertaken according to the KDIGO CLINICAL PRACTICE GUIDELINE [23] and adjusted according to individual conditions.
Patient 1 received 300 mg methylprednisolone (MP) and discontinued it because of infection. Then he obtained oral prednisone (12.5 mg per day) and mycophenolate mofetil (0.5 g per day) after receiving anti-infection therapy for 5 days. After receiving such treatment, his condition was controlled, and various renal function evaluation indicators decreased. However, 1 month later, an examination showed the level of proteinuria and serum creatinine increased considerably again. When he was readmitted in our hospital, he received the standard treatment for AAV. MP (300 mg/d) for 3 days was administered to him and he quickly went into remission with a prescription of prednisone together with cyclophosphamide.
Patient 2 received immunosuppressive therapy with glucocorticoids (methylprednisolone, 80 mg/day for 3 days; followed by oral prednisone 75 mg/day for 4 days, then 40 mg/day for 7 days, then 30 mg/day tapered to 10 mg/day by 1 month) and intravenous cyclophosphamide (1 g). He then went into remission and had a normal level of serum creatinine at the final date of follow-up.
It is of note that patient 3 had a 5-month history of pulmonary tuberculosis. The dot-like, nodular, and cord-like increased density shadows could be seen in both lungs on his CT image. And during this episode, his tuberculosis was active again. Therefore, he received a low dose of prednisone (20 mg per day) together with antituberculosis drugs. He had a positive response to treatment and remained in stable remission for 48 months. At the final date of follow-up, the level of serum creatinine was 85 umol/L; proteinuria and microscopic hematuria had disappeared.
Patient 4 was diagnosed with AAV in 2008 but she did not receive any standard treatment since her diagnosis. When she was readmitted to our hospital in 2016, she was diagnosed with AAV and IgAN. Then we made a treatment plan comprising of prednisone (50 mg per day for 2 weeks, then tapered to 25 mg/day, then 30 mg/day, then tapered to 5 mg/day by month 3) combined with hydroxychloroquine then cyclophosphamide administered at weeks 16, 19, 21, and 24. However, she discontinued her therapy because of the aggravation of the condition in the eighth week. Her renal function decreased dramatically which led her to begin peritoneal dialysis treatment and remained dialysis-dependent. At the final date of follow-up, the value of serum creatinine was 763.6 umol/L.
Patient 5 obtained prednisone (55 mg/day for 5 days; followed by 20 mg/day for 1 month then tapered to 15 mg/day after 1 month, then 10 mg/day at 3 months, and finally 5 mg/day from 5 to 12 months), mycophenolate mofetil (1.5 g/day for 13 months), and hydroxychloroquine (0.2 g/day for 4 months). The follow-up time of patient 5 was 12 months and her serum creatinine level was normal.
Comparison between patients with coexistence of AAV and IgAN and AAV patients
The baseline clinical data among patients with the coexistence of AAV and IgAN and AAV patients at the time of biopsy are shown in Table 5. The patients with coexistence of AAV and IgAN were younger than the pauci-immune AAV patients (22.6 ± 8.2 years versus 48.9 ± 15.7 years, respectively, P = 0.004). Patients in the AAV/IgAN group also had a trend toward a lower white blood cell count compared with pauci-immune AAV patients (P = 0.058). There were no significant statistical differences in hemoglobin, platelet, serum creatinine, estimated glomerular filtration rate (eGFR), urine protein, complement (C3, C4), or BVAS between the groups. The follow-up time of all patients in this study was at least 1 year ranging from 12 to 78 months. After immunosuppressive therapy, the same percentage of patients (80%) in the two groups achieved remission.
The result of the comparison between the renal pathologic data of the two groups was listed in Table 6. The percent of glomeruli with fibrous crescents in pauci-immune AAV patients was significantly higher than the AAV/IgAN overlap cases (4.0% versus 0.0%, P = 0.038). There were no statistical differences between the two groups in other renal pathologic parameters.
Comparison between patients with the coexistence of AAV and IgAN and IgAN patients
Table 7 showed the comparison of clinical and histological data between AAV/IgAN overlap patients and IgAN patients. Ten IgAN patients all had a normal level of platelet and C4. The levels of platelet and C4 in IgAN groups were lower than that in AAV/IgAN overlap groups (282.6 ± 43.6 versus 214.8 ± 50.7, P = 0.024; 333.6 ± 124.6 versus 223.1 ± 73.1, P = 0.047, respectively). The levels of ESR and CRP were higher in AAV/IgAN overlap cases (P = 0.032, P = 0.031, respectively) when compared with IgAN patients. There were no significant differences in age, white blood cell count, hemoglobin, serum creatinine, estimated glomerular filtration rate (eGFR), urine protein, and C3 between these two groups.
The comparison between the renal pathologic data of the two groups was listed in Table 7. The Oxford classification of IgA nephropathy was used for the assessment of glomerular disease in all patients. The only difference in MEST-C scores was the E score. The percentage of patients with a score of E1 was significantly higher in IgAN group than that in AAV/IgAN overlap group (100% versus 40%, P = 0.022). There were no significant differences between the two groups in other renal pathologic parameters.
Literature review of AAV-IgAN case reports and case series
So far, we retrieved eight studies with a total of 11 cases involving AAV/IgAN overlap syndrome from PubMed and Web of Science databases [11,12,13,14,15,16,17,18]. The clinical and pathologic parameters of the 11 cases are listed in Table 8. Three of the 11 patients are female; the ages ranged from 13 to 83 (49.18 ± 20.80). These 11 patients did not have the same characteristic as our patients, which was on the younger side of onset age. The number of organs involved for these cases was much more than our patients, as ten of the 11 patients had more than one organ involved. However, both our patients and the reported cases had microscopic hematuria. The response to treatment presented well in all patients. For the reported cases, only one of the 11 patients died of multiple organ failure. And the rest of the patients all responded effectively to treatment and achieved partial or complete remission. Our cases were in line with this result because only one patient progressed to ESRD. The reason why she had a worse renal outcome may be because she did not receive effective treatment since her first diagnosis and had the lowest serum C3 level at the second admission (Table 1).
Discussion
AAV patients with renal involvement commonly presented as “pauci-immune” focal necrotizing (and crescentic) glomerulonephritis with little or no immune deposition or complement in the glomeruli [1]. However, an increasing number of articles have reported immunocomplex deposition in the glomeruli of AAV patients [27, 28]. IgAN patients with ANCA positivity were described in quite a few studies, too [4,5,6,7,8,9]. The connection between AAV and IgAN is still not clear. The related literature mainly reported the following conditions: patients with the co-occurrence of AAV and IgAN, AAV patients with IgA deposition, and IgA nephropathy patients with serum positive ANCA.
Yanhong et al. found 26 of the 168 AAV patients had glomerular IgA deposition [10]. There were 55 IgAN patients with serum ANCA positivity in the studies of Yang et al. and Xie et al. [6, 7]. We screened 151 AAV patients and found five patients could be diagnosed as having IgAN simultaneously because of the presence of IgA-dominant or co-dominant immune deposits and the manifest lesions in the glomerular mesangium. Actually, some patients could be diagnosed as AAV and IgAN among the reported 81 patients mentioned above. In this paper, we further identified 11 other case reports of AAV and IgAN by searching in PubMed and Web of Science databases. To the best of our knowledge, our study is the first study to analyze the patients with a co-occurrence of AAV and IgAN by comparing them with AAV patients who had pauci-immune glomerulonephritis up to now.
The typical age of onset of AAV patients in our hospital is 56.70 ± 15.65 years [29]. The patients with co-occurrence of AAV and IgAN in this study were significantly younger. This finding was confirmed by comparison with the control group (22.6 ± 8.2 years versus 48.9 ± 15.7 years, respectively, P = 0.004). However, the 11 previously reported cases (49.18 ± 20.80) did not have a younger age of onset as our patients. Both our patients and the previously reported cases had microscopic hematuria. Renal involvement was seen in all of our patients, but only three patients had multiple organ involvement, in contrast to reported cases in which multiple organ involvement was reported in almost 90.9% (10/11) of the patients. The clinical data, such as serum creatinine, eGFR, CRP, ESR, and the level of complement, did not show any increasing or decreasing tendencies in common (Tables 1 and 8). After accepting treatment with a combination of steroid and immunosuppressants, all patients showed a positive response to therapy (except our patient 4 and reported patient 6).
Compared with ten AAV patients with pauci-immune glomerulonephritis, AAV/IgAN overlap patients had a lower white blood cell count (P = 0.058). Pauci-immune AAV patients had a higher percentage of crescentic glomeruli compared with our patients. But the other clinical and pathological parameters were quite similar between patients with the coexistence of AAV and IgAN and pauci-immune AAV patients. Furthermore, Yang, et al. found that the clinical features were quite similar between ANCA-positive IgAN patients and pauci-immune AAV patients [6]. However, Ma et al. found the patients with IgA deposition had lower eGFR levels, a higher percentage of crescent formation, a tendency toward more proteinuria, and worse renal outcome compared with those pauci-immune AAV patients [10].
Compared with ten IgAN patients, AAV/IgAN overlap patients had higher levels of ESR (P = 0.032) and CRP (P = 0.031). Yang et al. [6] also observed that ANCA-positive IgAN patients had higher levels of ESR and CRP when compared with ANCA-negative IgAN patients. In addition, the comparison between the renal pathologic data of the two groups revealed no significant differences in our study, except for the E score in MEST-C scores.
The pathogenesis of the coexistence of AAV and IgAN remains poorly defined. Ma et al. found that the alternative complement pathway was present in patients with IgAN and those with AAV with or without IgA deposition. However, patients in the AAV and IgA deposition group activated the classical complement pathway which was distinct from patients in the pauci-immune AAV or IgAN groups. Therefore, it is possible that the activation of the classical complement pathway may be linked to the pathogenesis of the co-occurrence of AAV and IgAN [10].
The levels of tumor necrosis factor-α, soluble forms of intercellular adhesion molecule 1, and urinary interleukin-8 were increased in patients with acute or worsening IgAN [30,31,32]. These cytokines could activate polyclonal B cells and augment the formation of ANCA [14]. Therefore, the ANCA seropositivity in ANCA-positive IgAN patients may be promoted by the pro-inflammatory environment caused by IgA deposits. However, the presence of ANCA was rarely seen in IgAN patients who were at the stage of acute onset or exacerbation [6]. Thus, the relationship between IgA deposits and an increase in ANCA titer remains elusive and needs further study.
In our study, the diagnoses of AAV and IgAN were defined concomitantly in four patients (nos. 1, 2, 3, and 5) during their first admission. And since onset, the clinical presentation was quite similar to pauci-immune AAV patients. These indicated that the coexistence of two different pathological processes (IgAN and pauci-immune AAV) may be a coincidence. However, we observed that the age of AAV patients coexisting with IgAN was significantly younger than that of AAV patients. Therefore, it is plausible that there may be a causal relationship between AAV and IgAN.
Our study had several limitations. First, the pathogenesis of the co-occurrence of AAV and IgAN remains unclear. Second, most patients with the co-occurrence of AAV and IgAN enrolled in this study were diagnosed simultaneously by kidney biopsy, pulmonary involvement, or other manifestations of systemic vasculitis. We cannot rule out the possibility that AAV may develop first and then IgAN or vice versa. Third, as a growing number of cases are being reported in the clinic, more research with larger sample sizes is still warranted.
In conclusion, we described the clinical presentation, pathologic features, treatment response, and outcome data of five patients with overlapping of AAV and IgAN, and found that they had a positive response to aggressive immunosuppressive therapy.
Compared with pauci-immune AAV patients, they were younger and had a lower percentage of glomeruli with fibrous crescents. In contrast to IgAN patients, they had higher levels of ESR and CRP. The mechanism of the coexistence of IgAN and AAV needs further study.
References
Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, et al. ANCA-associated vasculitis Nat Rev Dis Primers. 2020;6(1):71. https://doi.org/10.1038/s41572-020-0204-y.
Working Group of the International Ig ANN the Renal Pathology S, Roberts IS, Cook HT, Troyanov S, Alpers CE, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76(5):546–56. https://doi.org/10.1038/ki.2009.168.
Schena FP, Nistor I. Epidemiology of IgA nephropathy: a global perspective. Semin Nephrol. 2018;38(5):435–42. https://doi.org/10.1016/j.semnephrol.2018.05.013.
Allmaras E, Nowack R, Andrassy K, Waldherr R, Fvd Woude, Ritz E. Rapidly progressive IgA nephropathy with anti-myeloperoxidase antibodies benefits from immunosuppression. Clin Nephrol. 1997;48(5–1997):269.
O’Donoghue DJ, Nusbaum P, Noel LH, Halbwachs-Mecarelli L, Lesavre P. Antineutrophil cytoplasmic antibodies in IgA nephropathy and Henoch-Schönlein purpura. Nephrol Dial Transplant. 1992;7(6):534–8.
Yang YZ, Shi SF, Chen YQ, Chen M, Yang YH, Xie XF, et al. Clinical features of IgA nephropathy with serum ANCA positivity: a retrospective case-control study. Clin Kidney J. 2015;8(5):482–8. https://doi.org/10.1093/ckj/sfv078.
Xie L, He J, Liu X, Tang S, Wang W, Li F, et al. Clinical value of systemic symptoms in IgA nephropathy with ANCA positivity. Clin Rheumatol. 2018;37(7):1953–61. https://doi.org/10.1007/s10067-017-3931-z.
Stefan G, Terinte-Balcan G, Stancu S, Zugravu A, Gherghiceanu M, Mircescu G. IgA nephropathy with serum ANCA positivity: case series and literature review. Rheumatol Int. 2021;41(7):1347–55. https://doi.org/10.1007/s00296-021-04888-2.
Bantis C, Stangou M, Schlaugat C, Alexopoulos E, Pantzaki A, Memmos D, et al. Is presence of ANCA in crescentic IgA nephropathy a coincidence or novel clinical entity? A case series. Am J Kidney Dis. 2010;55(2):259–68. https://doi.org/10.1053/j.ajkd.2009.09.031.
Ma Y, Chen L, Xu Y, Han Q, Yu B, Zhao J, et al. The clinicopathologic characteristics and complement activation of antineutrophil cytoplasmic antibody-associated vasculitides with glomerular IgA deposition. Appl Immunohistochem Mol Morphol. 2020;28(10):e87–93. https://doi.org/10.1097/PAI.0000000000000819.
Vrtovsnik F, Queffeulou G, Skhiri H, Nochy D, Walker F, Hayem G, et al. Simultaneous IgA nephropathy and Wegener’s granulomatosis–overlap or coincidence (the role of renal biopsy). Nephrol Dial Transplant. 1999;14(5):1266–7. https://doi.org/10.1093/ndt/14.5.1266.
Richer C, Mouthon L, Cohen PPB, Royer I, Guettier C. IgA glomerulonephritis associated with microscopic polyangiitis or Churg-Strauss syndrome. Clin Nephrol. 1999;52(1):47–50.
Rao N, Bendall A, Lanteri M. ANCA vasculitis and IgA nephropathy linked to silica exposure. Occup Med (Lond). 2020;70(6):445–8. https://doi.org/10.1093/occmed/kqaa122.
Shimizu M, Wada T, Izumiya NSY, Furuichi K, Kobayashi TMK, Goshima S. Clinicopathological features of antineutrophil cytoplasmic antibodiesassociated vasculitis in Japanese patients with IgA nephropathy. Clin Exp Nephrol. 2000;4:251–6.
Joerg L, Kerstin A, Niko B, Dominik A, Martin K, Latus J. A typical Wegeners granulomatosis - but not pauci-immune! Minerva Urol Nefrol. 2012;64:149–52.
Kucuk A, Solak Y, Gaipov A, Bagcaci S, Esen H, Turk S, et al. Co-existing proteinase 3-antineutrophil cytoplasmic antibody-associated vasculitis with immunoglobulin A nephropathy. Korean J Intern Med. 2016;31(1):194–6. https://doi.org/10.3904/kjim.2016.31.1.194.
Galante JR, Daruwalla CP, Roberts ISD, Haynes R, Storey BC, Bottomley MJ. An unusual presentation of propylthiouracil-induced anti-MPO and PR3 positive ANCA vasculitis with associated anti-GBM antibodies, IgA nephropathy and an IgG4 interstitial infiltrate: a case report. BMC Nephrol. 2020;21(1):295. https://doi.org/10.1186/s12882-020-01964-w.
Fukuhara D, Kurayama R, Ito Y, Komagata Y, Arimura Y, Yan K. Granulomatosis with polyangiitis associated with IgA nephropathy. CEN Case Rep. 2013;2(2):204–8. https://doi.org/10.1007/s13730-013-0065-2.
Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21(10):1628–36. https://doi.org/10.1681/ASN.2010050477.
Roberts IS. Pathology of IgA nephropathy. Nat Rev Nephrol. 2014;10(8):445–54. https://doi.org/10.1038/nrneph.2014.92.
Rodrigues JC, Haas M, Reich HN. IgA nephropathy. Clin J Am Soc Nephrol. 2017;12(4):677–86. https://doi.org/10.2215/CJN.07420716.
Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368(25):2402–14. https://doi.org/10.1056/NEJMra1206793.
Kidney disease: improving global outcomes glomerular diseases work G. KDIGO 2021. Clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021;100(4S):S1–276. https://doi.org/10.1016/j.kint.2021.05.021.
Grayson PC, Ponte C, Suppiah R, Robson JC, Craven A, Judge A, et al. 2022 American College of Rheumatology/European alliance of associations for rheumatology classification criteria for eosinophilic granulomatosis with polyangiitis. Ann Rheum Dis. 2022;81(3):309–14. https://doi.org/10.1136/annrheumdis-2021-221794.
Robson JC, Grayson PC, Ponte C, Suppiah R, Craven A, Judge A, et al. 2022 American College of Rheumatology/European alliance of associations for rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis. 2022;81(3):315–20. https://doi.org/10.1136/annrheumdis-2021-221795.
Suppiah R, Robson JC, Grayson PC, Ponte C, Craven A, Khalid S, et al. 2022 American College of Rheumatology/European alliance of associations for rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis. 2022;81(3):321–6. https://doi.org/10.1136/annrheumdis-2021-221796.
Lin W, Shen C, Zhong Y, Ooi JD, Eggenhuizen P, Zhou YO, et al. Glomerular immune deposition in MPO-ANCA associated glomerulonephritis is associated with poor renal survival. Front Immunol. 2021;12:625672. https://doi.org/10.3389/fimmu.2021.625672.
Haas M, Eustace JA. Immune complex deposits in ANCA-associated crescentic glomerulonephritis: a study of 126 cases. Kidney Int. 2004;65(6):2145–52. https://doi.org/10.1111/j.1523-1755.2004.00632.x.
Wu T, Zhong Y, Zhou Y, Chen J, Yang Y, Tang R, et al. Clinical characteristics and prognosis in 269 patients with antineutrophil cytoplasimc antibody associated vasculitis. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2020;45(8):916–22. https://doi.org/10.11817/j.issn.1672-7347.2020.190436.
Wada T, Yokoyama H, Tomosugi N, Hisada Y, Ohta S, Naito T. Detection of urinary interleukin-8 in glomerular diseases. Kidney Int. 1994;46:455–60.
Yokoyama H, Wada T, Furuichi K, Segawa C, Shimizu M, Kobayashi K. Urinary levels of chemokines (MCAF/MCP-1, IL-8) reflect distinct disease activities and phases of human IgA nephropathy. J Leukoc Biol. 1998;63:493–9.
Yokoyama H, Takaeda M, Wada T, Ohta S, Hisada Y, Segawa C. Glomerular ICAM-1 expression related to circulating TNF-alpha in human glomerulonephritis. Nephron. 1997;76:425–33.
Funding
This work was funded by the National Key R&D Program of China (2020YFC2005000 to XX), the Key Research and Development Program of Hunan province (2020WK2008 to YZ), the Science and Technology Innovation Program of Hunan Province (2020RC5002 to JO), the Natural Science Foundation of Hunan Province (2021JJ31130 to YZ and 2020JJ6109 to CS), “Yiluqihang Shenmingyuanyang” Medical Development and Scientific Research Fund Project on Kidney Diseases (SMYY20220301001 to YZ), and Scientific Research Program of Traditional Chinese Medicine in Hunan Province (201761 to XL).
Author information
Authors and Affiliations
Contributions
Y.Z., Q.X., R.T., T.M., W.L., and X.C.X. conceived and designed the research; Y.Z., Q.X., R.T., and X.C.X. wrote the paper; J.D.O., P.J.E., C.J.S., W.N.N., X.L., Q.L.Z., and P.X. revised the paper; Q.X., T.M., W.L., Y.Z., W.N.N., C.J.S., and X.L. collected the clinical and pathologic parameters of patients; Q.X., Y.Z., R.T., T.M., J.B.C, and W.L. analyzed data; all authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Medical Ethics Committee of the Xiangya Hospital of Central South University for Human Studies (approval number 201711836). Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xiong, Q., Lin, W., Shen, C. et al. Coexistence of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and IgA nephropathy. Immunol Res 71, 1–14 (2023). https://doi.org/10.1007/s12026-022-09322-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-022-09322-8