
Abstract
The intestinal epithelium undergoes rapid cell turnover to maintain the integrity of the mucosal barrier, which is driven by the proliferation and differentiation of intestinal stem cells (ISCs). Due to their properties, ISCs are not only vulnerable targets during intestinal damage, but also act as the resources responsible for repair and regeneration. Moreover, the intestinal tract is the largest immune organ in the body, with the greatest number of immune cells including, but not limited to, macrophages, innate lymphoid cells and T cells. With the advance of intestinal organoid culture systems and single-cell RNA sequencing, the effects of immune cells on ISCs have been initially explored. As a component of the stem cell niche, these activated immune cells and their corresponding cytokines directly modulate apoptosis or survival of ISCs, leading to either destruction or protection of the intestinal epithelium in immune-mediated diseases, such as inflammatory bowel disease and graft-versus-host disease. In this review, we describe the effects of various immune cells on ISCs, as well as the mechanisms underlying these effects. We also highlight the remarkable role of ISCs in intestinal pathogenesis and raise the possibility of developing novel and effective therapeutic strategies for immune-mediated diseases based on ISCs.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human intestinal tract contains the second largest epithelium in the body, with a surface area of >30 m2, separating the host from the external environment [1]. The intestinal epithelium is in direct contact with contents of the lumen, which also contains a large number of toxins and microorganisms [1,2,3]. Persistent assult from luminal contents induces a remarkably high rate of intestinal epithelial cell (IEC) death; therefore, rapid epithelial renewal is required to maintain integrity of the mucosal barrier. This process is highly dependent on the proliferation and differentiation of intestinal stem cells (ISCs) [3], which divide every 24 h and develop into transit-amplifying (TA) daughter cells. These cells continue to differentiate and migrate, gradually rising from the base to the tip of the villi to replace damaged cells [2].
Types of differentiated cells derived from ISCs include enterocytes, Paneth cells, goblet cells, enteroendocrine cells, tuft cells, and M cells [2,3,4]. In addition to generating multiple lineages, ISCs also divide to maintain self-renewal of the ISC pool. These characteristics are essential for intestinal epithelial regeneration and homeostasis, especially in cases of injury and inflammation [1]. Increasing numbers of studies have focused on the impact of ISCs on intestinal inflammatory injury and repair, as well as on tumorigenesis, which has become an area of interest in recent years.
Importantly, the intestinal tract is not only critical for digestion, but is also the largest immune organ of the body [5], comprising the greatest numbers of immune cells, including macrophages, T cells and B cells in the human body [6]. These immune cells mostly reside in the lamina propria just beneath the crypts [7]. Together with other stromal cells, such as fibroblasts and endothelial cells, immune cells constitute the ISC microenvironment and precisely modulate the complicated processes of ISC self-renewal and differentiation [1, 4, 8, 9]. Moreover, intestinal microbes and their metabolites also regulate the crypt homeostasis, which have been reviewed in detail previously [10,11,12,13,14,15,16].
Immune cells regulate ISCs through cytokines and other stimulatory factors, thereby further affecting integrity of the intestinal barrier, which plays an important role in the pathogenesis of intestinal and systemic diseases [17]. However, the mechanism by which immune cells influence ISCs are poorly understood. Recent development of intestinal organoids culture system and single-cell RNA sequencing (scRNA-seq) have enabled some studies into the effects of immune cells and cytokine signals on ISCs in immune-mediated diseases, such as graft-versus-host disease (GVHD), inflammatory bowel disease (IBD), and chronic inflammation-associated tumorigenesis, thus greatly contributing to our understanding of the immune regulation of ISCs.
In this review, we discuss the basic structure and classic signaling pathways of the intestinal epithelium and ISCs, and summarize how immune cells and their cytokine repertoires influence ISCs in response to injury and inflammation. Lastly, we highlight the role of ISCs in intestinal diseases and propose potential therapeutic strategies based on ISCs.
Intestinal Epithelium and Stem Cells
Intestinal Epithelium
The intestinal epithelium constitutes the barrier surface separating the host from the external environment (Fig. 1) [11, 18,19,20]. The epithelium differs in architecture, as well as in cellular composition between the small intestine (SI) and the colon to adapt to their respective functions [19]. In the SI, the epithelium projects into the lumen in the form of finger-like villi, which increase the mucosal surface area to facilitate nutrient absorption. At the base of, and continuous with, the villi are the crypts. ISCs reside at the base of the crypts and give rise to multiple daughter cells to replenish the intestinal epithelium. The inward structure of the crypts protects ISCs from the harsh luminal environment [13, 19]. In contrast, villi are completely absent from the colon, resulting in a flat epithelial surface that minimizes potential damage inflicted by stool transiting through the large bowel [19].

Intestinal epithelial structure and immune cell distribution. Left: Structure of the small intestine. Intestinal epithelial cells (IECs) constitute the barrier surface to separate the host from the external environment. Intestinal stem cells (ISCs) reside at the base of the crypts and give rise to multiple daughter cells to replenish the intestinal epithelium, with the IECs eventually being shed into the lumen at the tops of villi. Transit-amplifying (TA) daughter cells continue to differentiate and migrate, gradually moving from base of the intestine to the villi region. Types of differentiated cells include enterocytes, Paneth cells, goblet cells, enteroendocrine cells, tuft cells, and M cells covering the Payer’s patches [1,2,3]. With the exception of intraepithelial lymphocytes (IELs) distributed among IECs, innate lymphoid cells (ILCs), T cells, macrophages, dendritic cells (DCs), eosinophils, mast cells, and B cells all reside at the lamina propria [6]. AMPs: anti-microbial peptides, sIgA: secretory immunoglobulin A. Right: Structure of the colon. Paneth cells are absent. Instead, deep secretory cells are located between ISCs. The thick mucus layer reduces epithelial contact with luminal microorganisms. The distribution of immune cells is similar to that in the small intestine [1, 6]
The SI epithelium consists of six types of cells, each with a different and specialized function (Fig. 1). The cells can be categorized into two main groups: the absorptive lineage, consisting of enterocytes and M cells, and the secretory lineage, comprising goblet cells, enteroendocrine cells, Paneth cells, and tuft cells [1, 21,22,23]. Whereas the colon epithelium consists of enterocytes, enteroendocrine cells, goblet cells, and tuft cells, Paneth cells and M cells are unique to the SI [6]. Alternatively, deep secretory cells reside among ISCs to serve as functional equivalents to Paneth cells [24].
Intestinal Stem Cells
Rapid turnover of the intestinal epithelium depends on the active proliferation and differentiation of ISCs. Crypt base columnar (CBC) cells were first described by Cheng and Leblond in 1974 as continuously cycling cells at the bottom of the crypt [25]. The Wnt target gene Lgr5 (leucine-rich repeat-containing G protein-coupled receptor 5) is an excellent marker for CBC cells that can be used to assess their position in the cell hierarchy by genetic lineage tracing [26]. Notably, Lgr5+ CBC cells can give rise to all the different epithelial cell types and thus are considered self-renewing, long-lived, multipotent stem cells [2].
In addition to CBC cells, a population of DNA-label-retaining cells residing at “position four” (directly above the uppermost Paneth cells), namely “+4 cells”, are considered quiescent or reserve stem cells [27] (Fig. 1). Several markers have been used to identify these +4 cells, such as Bmi1, Hopx, mTert, Lrig1 [3]. Using lineage tracing, studies have shown that +4 cells are able to regenerate other epithelial cells, including Lgr5+ CBC cells, under conditions of injury [1]. Although somewhat controversial, +4 cells are now considered reserve stem cells with high resistance to radiation, which replenish the pool of continuously cycling Lgr5+ CBC cells when required [1]. Because culture conditions for +4 cells have not yet been defined [2] and active stem cells (the Lgr5+ CBC cells) are primarily responsible for daily epithelial homeostasis, “ISC” here mainly refers to Lgr5+ CBC cells.
Intestinal Stem Cell Niche and Signaling Pathways
The ISC microenvironment, or niche, modulates ISC self-renewal and differentiation to maintain homeostasis of the ISC pool. The ISC niche consists of an extracellular matrix component and a cellular component of Paneth cells, myofibroblasts, fibroblasts, smooth muscle cells, endothelial cells, pericytes, neural cells and immune cells (Fig. 1). These cells not only provide structural support but also send important signals to dynamically regulate ISC behavior [1, 28]. For example, Paneth cells express Wnt3, epidermal growth factor (EGF) and the Notch ligands DLL1 and DLL4, all of which are required for stem cell maintenance and development [1, 2].
The canonical key signals of ISCs include the Wnt, Notch, EGF, and bone morphogenetic protein (BMP) pathways [4]. Canonical Wnt signaling is the most important pathway in ISC fate, and is tightly linked to ISC maintenance and differentiation, as well as to tumorigenesis upon dysregulation [1]. When Wnt signals bind to the Frizzled-LRP5/6 co-receptors, β-catenin is stabilized against degradation and subsequently translocates into the nucleus and binds to transcription factor TCF4, forming a complex that regulates stemness genes [2, 13]. Wnt signal deletion leads to complete loss of ISCs and breakdown of the epithelium [1]. Further, Wnt signaling is augmented by the agonist R-spondin, which binds to LGR4/5 and enhances the efficiency of Wnt receptors Frizzled and LRPs [1].
Notch signaling is essential for maintaining the undifferentiated state of ISCs through lateral inhibition [2, 4]. When Notch ligands DLL1/4, which are expressed on the surface of Paneth cells, bind to receptors Notch1–Notch4 through cell-cell contact, Notch pathway activation induces a series of proteolytic cleavages to generate the Notch intracellular domain (NID). The NID then translocates to the nucleus, interacts with the transcription factor CSL, and activates target gene transcription [2, 4]. Inhibition of Notch activity in the intestine results in loss of Lgr5+ ISCs and the differentiation of proliferating TA cells into secretory cells [13].
EGF is a crucial component of intestinal organoid culture medium and exerts a strong mitogenic effect on ISCs and TA cells upon binding to EGF receptors (EGFRs), which are highly expressed on ISCs. EGF-EGFR interactions lead to phosphorylation of the receptor and activation of the membrane protein Ras, as well as the sequential activation of proteins Raf and MEK. Subsequently, MEK phosphorylates cytosolic ERK1/2, which translocates into the nucleus and activates cell fate regulators [13]. Furthermore, ISCs also express high level of Lrig1, a negative regulator of the EGFR family, which co-regulates ISC proliferation [1, 29]. Blocking EGF signaling in intestinal organoids leads to quiescence of proliferative Lgr5+ ISCs. Nevertheless, the cells maintain their stemness and can resume proliferation once EGF signaling is restored [1, 29].
BMP signaling counteracts the proliferative signals of Wnt/β-catenin in the ISC niche and promotes ISC differentiation via Smad-mediated gene repression. BMP inhibitors, such as Gremlin 1, Gremlin 2, and Noggin, are secreted primarily by myofibroblasts and smooth muscle cells below the crypt, which creates a gradient of increasing BMP activity from the bottom of the crypt toward the tip of the villus. Decreasing Wnt and increasing BMP signals guide cells from an undifferentiated to a differentiated state during their migration along the crypt-villus axis [1, 4].
The signaling pathways described above determine the differentiation of mature cell types synergistically. For example, active Notch signaling promotes absorptive fate, whereas absence of Notch signals determines secretory characteristics [1]. In the absence of Notch and Wnt signaling, the vast majority of secretory progenitors become goblet cells. Otherwise, secretory progenitors become Paneth cells under conditions of high Wnt signaling. Low Wnt levels, a lack of Notch signals and reduced EGF pathway activation lead to the enteroendocrine cell fate. An increasing gradient of BMP signals allow absorptive progenitor cells to differentiate into enterocytes [1].
Immune Cells and their Effects on Intestinal Stem Cells
As stated previously, the intestinal tract is the largest immune organ in the body and contains a great number of immune cells (Fig. 1) [6]. Immune responses are critical for maintaining homeostasis of the intestinal environment and protecting against infection and toxins during dynamic interactions between the host and microorganisms [10, 30, 31]. It is noteworthy that cytokines primarily control immune-related events and exert a wide range of immunoregulatory effects by binding to their corresponding receptors [32]. Thus, cytokine receptors determine the targets of immune action. Importantly, ISCs express multiple cytokine receptors, such as the interferon gamma (IFN-γ), interleukin (IL)-13, IL-17, IL-10, and IL-22 receptors, which ultimately determine the crosstalk between immune cells and ISCs (Table. 1) [33]. However, the specific effects of these interactions and their underlying mechanisms are still unclear. Recently, several significant studies elucidated the role of immune cells in ISC homeostasis, which has greatly expanded our understanding of immunological impacts on ISCs (Fig. 2) [7, 33,34,35,36]. Thus, we will review the effects of immune cells on ISCs and their related mechanisms below.

The effects of immune cells on ISCs. Left: T cells can be recruited to the ISC compartment by MAdCAM-1 and release IFN-γ to directly act on ISCs [42] after allogeneic hematopoietic stem cell transplantation (allo-HSCT). IFN-γ and TNF-α have dual effects on ISCs; short-term exposure promotes regeneration, whereas prolonged exposure inhibits proliferation and promotes apoptosis [38]. They also act on reserve ISCs to activate epithelial regeneration during inflammation [43]. IL-17, IL-13 and IL-2 promote ISC differentiation, whereas IL-10 maintains ISC self-renewal [33]. Group 3 innate lymphoid cells (ILC3s) secrete IL-22 to promote ISC proliferation and epithelial regeneration in immune-mediated processes [35, 50]. IL-22 also acts on enterocytes to produce REG3γ to prevent apoptosis of ISCs and Paneth cells [53]. Moreover, ILC3s preserve ISCs and promote crypt cell proliferation through YAP1 signaling to drive tissue repair after damage [62]. Tuft-cell-derived IL-25 activates ILC2s to secrete IL-13, resulting in ISC self-renewal and differentiation toward tuft and goblet cells to initiate type 2 mucosal immunity against helminths [63, 64]. In addition, macrophages promote ISC proliferation and survival through Wnts, Prostaglandin E2 (PGE2) and neuregulin 1 (NRG1) [66, 68,69,70]. Monocytes move close to ISCs by the epithelial MyD88-signaling pathway and increase ISC proliferation [71]. Mast cells and eosinophils may impact ISCs, but no direct evidence confirms this. Right: Mechanisms underlying IL-22 and IFN-γ action on ISCs. Upper: IL-22 interacts with IL-22 receptor on ISCs and induces STAT3 phosphorylation, thereby increasing epithelial regeneration and repair [35]. In addition, phosphorylated STAT3 promotes ATM gene expression, subsequently initiates the DNA damage response (DDR) cascade, and reduces mutation accumulation [57]. Lower: IFN-γ dimer binds to IFN-γ receptor on ISCs and induces JAK1/2 phosphorylation to activate STAT1 signaling, resulting in Bak/Bax-dependent ISC apoptosis [36]. In addition, short-term exposure to IFN-γ leads to early activation of PI3K-AKT-β-catenin signaling to promote ISC proliferation, whereas extended exposure induces apoptosis via the Wnt inhibitor Dkk1 [38]
Lamina Propria T Cells
Both CD4+ and CD8+ T cells are found in the lamina propria. They are thought to be derived from T cells which have been primed in secondary lymphoid organs. Therefore, most lamina propria T cells show effector or memory phenotypes [6]. Studies have shown that T cells can promote intestinal epithelial proliferation to repair injury and maintain intestinal homeostasis in a short time [37, 38]. Using mouse colitis models and crypt staining, Nava and Lee et al. showed that short-term exposure (5 h) to T cells or IFN-γ and tumor necrosis factor (TNF)-α lead to early and sustained activation of AKT-mediated β-catenin, resulting in promoted activation of ISCs and populations of proliferative progenitor cells within the TA zone. This process is initiated by phosphorylation of PI3K through IFN-γ receptor-associated JAK1 and JAK2 (Fig. 2) [37, 38].
However, prolonged inflammation promotes ISC death. Extended exposure (72 h) to IFN-γ and TNF-α inhibits epithelial cell proliferation and promotes apoptosis, despite continued AKT-β-catenin signaling [38, 75]. This inhibitory effect is achieved by induction of the Wnt inhibitor Dkk1, a direct transcriptional target of β-catenin-TCF4 [38]. In mouse intestinal organoids co-cultured with T cells (72 h), activated T cells induce organoid damage, reduce Lgr5+ ISC mRNA levels and numbers of goblet cells [39], as well as induce Paneth cell death through a caspase-3/7–dependent mechanism [40]. Further, Lgr5+ ISCs undergo cell death earlier than Paneth cells in response to IFN-γ as assessed by simultaneous tracking [76], suggesting that ISC depletion is directly induced by IFN-γ.
Beyond the duration of exposure, T cell numbers or cytokine dose also determines the fate of ISCs. ScRNA-seq of fetal intestinal CD4+ T effector memory (Tem) cells shows a Th1 phenotype in these cells, which are characterized by TNF-α production [44]. Organoid co-cultures reveal a dose-dependent, TNF-α-mediated effect of fetal CD4+ T cells on ISC development. Low T cell numbers or low-dose TNF-α (0.2 ng/mL) supports the development of organoids, whereas large numbers or a high dose (≥ 20 ng/mL) significantly impair ISC proliferation, along with increased expression of apoptosis-associated genes and decreased Wnt signaling [44]. This dose-dependent effect is important for human intestinal development, as TNF-α-producing CD4+ T cells are preferentially presented in fetal intestine to support intestinal development, while premature exposure to the external environment can result in increased CD4+ Tem cells and lead to necrotizing enterocolitis (NEC) [44].
Moreover, activated T cells can aggregate in crypts and directly damage the ISCs. Acute GVHD (aGVHD) is the most common life-threatening complication after allo-HSCT. It occurs when immunocompetent T cells in the donor tissue (the graft) recognize the recipient (the host) as a foreign organism (non-self) and attack vital recipient organs, classically the skin, liver and gastrointestinal tract [50, 77], leading to dysregulation of the inflammatory cytokine cascade [78]. (Fig. 3) Emerging studies indicate that ISCs and their niche Paneth cells are targeted during aGVHD [36, 50, 79, 80]. the ISC compartment is the primary intestinal location invaded by allogeneic T cells (including CD4+ and CD8+ T cells) after allogeneic hematopoietic stem cell transplantation (allo-HSCT) in a major histocompatibility complex (MHC)-mismatched murine bone marrow transplant (BMT) model [36, 42]. This recruitment is directly correlated with adhesion molecule MAdCAM-1 expression, which is known to contribute to T cell recruitment to the gut [42]. Further, T cell-derived IFN-γ directly targets ISCs, activates JAK1-dependent STAT1 within the epithelium, and results in Bak/Bax-dependent ISC apoptosis by acting on the IFN-γ receptor of ISCs (Fig. 2) [36]. Moreover, both CD4+ and CD8+ T cells are able to contribute to injury of the ISC compartment [36, 42]. The accumulation of T cells in crypt and ISC apoptosis are also confirmed in mice injected with death-inducing T cells [41].

The mechanism of ISC injury in GVHD. (1) APCs are activated by host tissues damage and subsequent inflammatory cytokine release [78]. (2) Donor T cells are activated and proliferate, with upregulated integrin α4β7 [78]. Activated T cells migrate to the ISC compartment via MAdCAM-1, which is primarily expressed on vessels near the crypt base. The ISC compartment is an early site of pathologic T cell invasion and remains the most densely infiltrated region even after widespread T cell infiltration throughout the mucosa [42]. (3) Alloreactive T cell-derived IFN-γ directly targets ISCs to induce their apoptosis in a JAK/STAT-dependent manner [36]. In addition, ILC3s are eliminated during GVHD, leading to decreased IL-22 production. Deficiency of recipient-derived IL-22 increases crypt apoptosis [35, 50]. Thus, the damaged ISCs fail to regenerate and maintain the epithelial barrier, resulting in translocation infection and amplified destruction of host tissues [78]
In addition to effector cells, T helper (Th) cells and cytokines are capable of modulating the balance of ISCs between self-renewal and differentiation. Using co-culture of intestinal organoids with T cells, Biton et al. showed that IECs, including ISCs, express receptors for Th cytokines IFN-γ, IL-13, IL-17A, IL-10, and IL-22. Both co-culture with induced Treg cells (iTregs) and stimulation with IL-10 (10 ng/ml, 72 h) lead to ISC expansion within organoids [24, 48], whereas Treg depletion leads to increased apoptosis and necrosis within the ISC niche [45]. Co-culture with Th1, Th2, and Th17 cells or treatment with IL-13 (secreted by Th2 cells, 20 ng/ml, 72 h) or IL-17 (secreted by Th17 cells, 20 ng/ml, 72 h) result in depletion of the ISC pool and expansion of cells with differentiated features as assessed by scRNA-seq [33, 45]. Specifically, IL-17 treatment expands TA cell numbers [33], while IL-13 treatment increases tuft cell and goblet cell numbers [81]. Treatment with IFN-γ (produced by Th1 cells) increases Paneth cell numbers, and IL-22 treatment amplifies enterocyte numbers [33]. Addition of IL-2 to cultured organoids induces their maturation, which includes development of differentiated cells to carry out epithelial physiological functions [49]. Whereas T cell ablation leads to ISC accumulation, possibly due to reduced differentiation capacity, particularly toward the absorptive lineage [33].
However, in colon organoids, Th17 cells promote colon organoid growth, upregulate Lgr5 expression via IL-17 (20 ng/ml), and enhance colon stem cell tumorigenesis in mice [46, 47], the latter of which seems to activate STAT3 signaling through NF-κB [47]. An obvious inconsistency surrounds whether IL-17 leads to depletion of the ISC pool and differentiation or whether it promotes ISC proliferation and tumorigenesis. We believe this inconsistency might be explained by the different tissue responses to external stimuli due to the varied microenvironments of SI and colon. For instance, the degree of radiation resistance in colonic stem cells is distinct from that of SI stem cells [82]. However, difference in exposure time to IL-17 cannot be ruled out. Further work is required to verify the above hypothesis.
The response of +4 cells to T cells is quite different from that of ISCs. +4 cells seem to be more resistant to inflammatory signals than Lgr5+ ISCs. In a murine model of αCD3-antibody-induced acute SI inflammation, +4 cells are induced to proliferate and functionally contribute to epithelial regeneration whereas Lgr5+ ISCs undergo apoptosis [43]. The inflammatory cytokines IFN-γ and TNF-α lead to +4 cell activation in organoid co-culture (48 h), which requires JAK/STAT1 signaling [43]. The different responses may be explained by the expression of both MHC class I (MHCI) and MHC class II (MHCII) molecules on Lgr5+ ISCs [33, 41]. Lgr5+ ISCs are quick-cycling stem cells and, therefore, cannot evade immune surveillance, leading to further clearance by activated T cells [42]. In contrast, +4 cells are quiescent tissue stem cells and may escape immune clearance. These results highlight a reserve and repair role of +4 cells in response to intestinal inflammation.
Moreover, the relationship between T cells and ISCs is not unidirectional. ISCs may regulate the differentiation and activation of immune cells in turn. In fact, ISCs can act as nonconventional antigen-presenting cells (APCs) to activate T cells. Using scRNA-seq, Biton et al. observed Lgr5+ ISCs with enriched expression of MHCII, which can interact and activate Th cells via MHCII presentation of peptides once stimulated with antigen. In MHCII-deleted mice, numbers of CD4+ cells in the crypt lamina propria are strongly reduced, and ablation of MHCII eliminates the ability of ISCs to activate T cells in co-culture. Interestingly, the expression of MHCII genes in ISCs is induced by Th1 cells in the organoid co-culture system. Thus, T cell-Lgr5+ ISC crosstalk is far more complicated than the effects of cytokines on ISCs [33]. The organoid-immune cell co-culture system can not only be used to clarify the effects of immune cells on ISCs in vitro, but also investigate the impacts of ISCs on the differentiation and activation of immune cells, whether under homeostasis or pathological conditions.
In summary, moderate pro-inflammatory cytokines promote the directed differentiation of ISCs and coordinate immune responses. However, excessive cytokines lead to apoptosis of ISCs and destruction of the intestinal barrier, thereby aggravating or initiating immune-mediated intestinal damage, such as GVHD and NEC. These works largely extend our knowledge on the crosstalk between the T cells and stem cells, and may provide promising therapeutic strategies for intestinal diseases.
Innate Lymphoid Cells
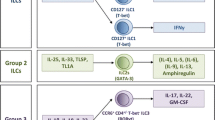
Innate lymphoid cells (ILCs) are a recently identified subset of innate immune cells of lymphoid origin thought to be the “gatekeepers” of mucosa-associated tissues. They are specifically located in the lamina propria of the small and large intestines [83]. Unlike adaptive lymphocytes, such as T and B cells, ILCs lack antigen-specific receptors. They rapidly respond to cytokine stimulation and release additional cytokines to facilitate control of infection [83, 84].
Based on their phenotypic and functional traits, ILCs can be generally divided into cytolytic and noncytolytic ILCs. Cytolytic ILCs refer to the conventional natural killer (cNK) cells. While noncytolytic or “helper” ILCs can be further classified into three subgroups according to their cytokine and transcription factors expression: group 1 (ILC1), group 2 (ILC2), and group 3 (ILC3) [84, 85]. ILC1s produce IFN-γ when stimulated by IL-12, IL-15, or IL-18; ILC2s release IL-5 and IL-13 when stimulated with IL-25, or IL-33; and ILC3s secrete IL-17, IL-22, TNF and granulocyte macrophage colony-stimulating factor (GM-CSF) when activated by IL-1β, IL-18, or IL-23. RAR-related orphan receptor gamma t (RORγt) and aryl hydrocarbon receptor (AHR) are key transcription factors that drive the development, maintenance, and function of ILC3s [83, 84, 86].
ILC3s are crucial for maintaining intestinal epithelial integrity during intestinal injury [87]. As the major cellular source of IL-22 in the SI [88], ILC3s are eliminated during GVHD, thus leading to decreased IL-22 production. Deficiency of recipient-derived IL-22 increases crypt apoptosis. Treatment with IL-22 in vivo after mouse allo-BMT enhances the recovery of ISCs and reduces mortality from GVHD. Using ex vivo organoid cultures, Lindemans et al. showed that ILC3s increase the growth of mouse SI organoids in an IL-22-dependent fashion. Recombinant IL-22 directly targets ISCs and induces ATF-STAT3 phosphorylation, rather than enhances Wnt or Notch signaling (Fig. 2) [35, 89]. The same effect can be seen in a mouse model of ethanol and burn injury [51]. Notably, although IL-22 can be produced by other lymphoid populations, such as Th17 cells and NK cells, only CD3−RORγt+CCR6+ ILC3s demonstrate significant IL-22 expression after BMT [50]. These results show the crucial role of ILC3s in protection against GVHD via IL-22.
Although IL-22 promotes organoid growth and epithelial regeneration, its more important role appears to prevent ISCs from apoptosis caused by GVHD via induction of regenerating islet-derived 3γ (REG3γ) [53]. REG3γ is a key AMP secreted by IECs to protect crypts from luminal microbes and mediated by IL-22-induced STAT3 signaling [35, 54, 90]. IL-22 administration restores REG3γ production and prevents apoptosis of both ileum ISCs and Paneth cells, but this protection is completely abrogated in Reg3g deleted mice [53]. Thus, REG3γ might act as a survival signal for ISCs and Paneth cells, preventing their apoptosis in GVHD. However, in contrast to findings from Zhao et al., we believe that IL-22-induced REG3γ is expressed by enterocytes, rather than Paneth cells, as another previous study shows [91], because no evidence of pSTAT3 is found in Paneth cells in response to IL-22 [35]. Since the regulatory mechanisms of REG3γ are still unclear, further research is necessary to confirm the underlying mechanism and potential role of REG3γ beyond AMP.
Moreover, recent studies have showed that long noncoding RNA (lncRNA) is also involved in the immunomodulatory effects of IL-22. H19, an evolutionarily conserved and maternally expressed imprinted lncRNA, is induced by IL-22 during inflammation via both STAT3 signaling [55] and PKA activation [56], and is localized to Lgr5+ CBCs and Lgr5− epithelial cells of the SI [56]. Inflammation-induced H19 lncRNA increases the expression of a subset of cell growth–promoting genes by inhibiting p53 and multiple cell growth-inhibitory miRNAs, thereby increasing ISC proliferation to promote intestinal epithelial regeneration [56]. Taken together, these findings support a novel model whereby H19 lncRNA is an important intermediate signal linking IL-22 to other regulatory networks that control the proliferation and repair of the intestinal epithelium under inflammatory conditions. In recent years, increasing evidence has emerged that lncRNA acts as a novel class of regulators of intestinal epithelial homeostasis [92,93,94], which may inform a promising research direction involving identifying novel biomarkers and selecting therapeutic targets.
In addition, IL-22 acts as an important regulator of DNA damage response (DDR) machinery in colon ISCs, thus preventing the accumulation of potentially dangerous mutations. Specifically, IL-22 is required to initiate the DDR via STAT3 activation after DNA damage (Fig. 2). ISCs deprived of IL-22 signals escape DDR-controlled apoptosis, contain more mutations and are more likely to give rise to colon cancer [57]. Therefore, this work reveals a novel role of IL-22 in maintaining the genomic integrity of stem cells through initiation of DDR machinery.
As IL-22 is a key factor of ILC3, the proper production of IL-22 is essential for ILC3 function. ILC3s can be activated not only by cytokines, but also by some metabolites through AHR, such as the Lactobacillus metabolite indole-3-aldehyde [52] and metabolites of glucosinolates, a group of phytochemicals contained in cruciferous vegetables [57]. Indole-3-aldehyde stimulates ILC3s to secrete IL-22 through AHR, thereby inducing STAT3 phosphorylation to accelerate ISC regeneration [52]. While mice on a glucosinolate-free diet have low IL-22 levels and impaired DDR in epithelial cells [57]. Hence, AHR is an important “switch” for ILC3 activation. AHR deficiency has detrimental consequences associated with loss of ILC3s and absence of IL-22 production [95], as well as unrestricted ISC proliferation and malignant transformation [96]. Actually, AHR is widely expressed in immune and non-immune cells of the bowel and can be activated by several dietary components, xenobiotics, and microbial metabolites. Upon activation, AHR participates in multiple regulatory mechanisms such as those involving IL-10, IL-22, and IFN-γ, AMPs, and is associated with intestinal inflammation and tumorigenesis [57, 97]. Thus, AHR-based therapies, as well as the metabolites, may provide options for preventing and treating intestinal diseases.
In addition to IL-22, ILC3s are also required to protect ISCs from the deleterious effects of the chemotherapeutic agent methotrexate (MTX) through yes-associated protein (YAP) signals. Maintenance of ISCs after MTX-induced damage is severely impaired in absence of ILC3s [62, 98]. Although IL-22 is an ILC3 signature cytokine involved in communication between ILC3s and ISCs, crypt proliferation and tissue regeneration after MTX-induced damage are IL-22 independent [61, 62]. In contrast, ILC3s induce YAP1 nuclear translocation and magnify Hippo-YAP1 signals in crypt cells, ensuring adequate initiation of tissue repair. YAP transiently reprograms Lgr5+ ISCs by suppressing Wnt signaling and excessive Paneth cell differentiation, while promoting ISC survival [99]. However, whether an intermediate exists between ILC3s and ISCs, such as mesenchymal stromal cells, to activate YAP1 signaling remains unknown. Organoid co-culture system may provide an excellent platform to elucidate the precise ILC3-YAP1 repair mechanism.
However, under normal circumstances, studies using SI ISCs culture [59] and mice treated with recombinant IL-22 [60] show that IL-22 promotes TA cell proliferation but reduces the proliferative capacity and survival of Lgr5+ ISCs [58] by inhibiting Notch and Wnt signaling in vitro and in vivo. These results indicate that the maintenance of ISCs regulated by IL-22 is dependent on the niche environment. In the case of immune-mediated intestinal inflammation, IL-22 acts as a protective factor to preserve ISCs and promote epithelial repair. Whereas under normal circumstances, IL-22 itself can inhibit ISCs, highlighting the dual roles of IL-22.
In addition to ILC3s, ILC2s also impact ISCs through different pathways (Fig. 2). As reported previously, ILC2s are required to protect the host against helminth infection [83]. Tuft cells are the primary source of the parasite-induced cytokine, IL-25, which further activates ILC2s to secrete IL-13 and induces a type 2 immune response [23, 63]. IL-13 acts on epithelial crypt progenitors to promote differentiation toward tuft and goblet cells [63, 64]. Tuft cells, ILC2s and epithelial progenitors therefore compose a response circuit that mediates epithelial remodeling associated with type 2 immunity. However, the underlying mechanism of these cellular activities is still unknown, although it is hypothesized that the circuit is modulated by altering the balance of Notch signaling, as the Notch signaling inhibitor also induces tuft cell hyperplasia in organoids [64].
Besides, ILC2-derived IL-13 promotes the self-renewal of ISCs through the circular RNA circPan3. Immune cell–associated circPan3 is highly expressed in mouse and human Lgr5+ ISCs. Ablation of circPan3 in Lgr5+ ISCs impairs their capacity for self-renewal and epithelial regeneration. Mechanistically, circPan3 increases the expression of IL-13 receptor, which enables the ILC2s-derived IL-13 to initiate STAT6-Foxp1-β-catenin pathway to induce self-renewal of Lgr5+ ISCs [28]. Moreover, although IL-4 and IL-13 are both type 2 cytokines, IL-13, other than IL-4, plays a critical role in ISC homeostasis [28, 63, 64].
Altogether, these results indicate that ILCs are involved in different regulatory networks to control ISC maintenance and differentiation, thereby strengthening the fundamental defense system provided by the integrity of epithelium. Further research is needed to elucidate the precise immunological properties of ILCs and the crosstalk between ILCs and ISCs to offer novel strategies for immunologically mediated diseases of the bowel.
Mononuclear Phagocytes
Mononuclear phagocytes (MPs) consist of macrophages and dendritic cells (DCs). Intestinal macrophages constitute the largest pool of macrophages in the body, and are located mainly in the lamina propria [100]. Due to their high degree of plasticity, macrophages can adopt different phenotypes depending on their microenvironment (i.e., pro-inflammatory phenotype-M1 macrophages and anti-inflammatory phenotype-M2 macrophages) [67]. Meanwhile, intestinal DCs are located diffusely throughout the intestinal lamina propria, within gut-associated lymphoid tissues and intestinal-draining lymph nodes [101].
The role of macrophages in ISC niche has been increasingly recognized. Macrophage ablation following colony-stimulating factor 1 receptor blockade hinders Paneth cell differentiation, impairs crypt cell proliferation and results in a reduction of Lgr5+ ISCs [102, 103]. Mechanistically, recent studies show that macrophages have emerged as a potential source of Wnt ligands [67]. Murine peritoneal macrophages treated with IL-4 and polarized toward an M2 phenotype overexpress Wnt2b, Wnt7b, and Wnt10a in a STAT6-dependent manner [65]. Furthermore, using a macrophage-restricted ablation of Porcupine, Saha et al. demonstrated that macrophage-derived extracellular vesicle-packaged Wnts rescue ISCs and enhance ISC survival after radiation injury [66].
In addition to Wnt, macrophages produce prostaglandin E2 (PGE2) to promote ISC proliferation and crypt fission. PGE2 production is induced by extracellular hyaluronic acid (HA) through cyclooxygenase 2 (COX2) [68]. Subsequently, PGE2 stimulates the proliferation of ISCs and reduces radiation-induced apoptosis via transactivation of EGFR and enhanced activation of AKT [69]. A recent study shows that macrophages express neuregulin 1 (NRG1), a key EGF family ligand, which is upregulated after injury. NRG1 drives ISC proliferation and regeneration of damaged epithelium in part through elevated activation of mitogen-activated protein kinase (MAPK) and AKT [70]. Hence, as a component of the ISC niche, macrophages participate in ISC renewal via several pathways.
Intestinal DCs play a central role in the initiation and differentiation of adaptive immune responses by promoting effector T cell differentiation and directing migration of activated T cells [101]. However, research on the impact of DCs on ISCs is lacking. Intestinal organoids co-cultured with bone marrow-derived dendritic cells (BMDCs) show morphological changes of organoids and goblet cell depletion with Notch signal activation, which is promoted by E-cadherin–mediated BMDC adhesion [104]. Nevertheless, the exact mechanisms are still unclear. A primary human macrophage-enteroid or DC-enteroid co-culture model has been established and may provide a promising platform for investigating in vitro effects and mechanisms, thus beneficial to future study [105, 106].
Monocytes and Neutrophils
Monocytes and neutrophils are usually the first lines of defense against infection and play important roles in wound healing [107, 108]. Using a mouse colonic mucosal explant model and crypt-monocyte co-culture model, Skoczek et al. showed that the epithelial MyD88-signaling pathway drives the movement of Ly6C+7/4+ monocytes closer to ISCs, leading to increased crypt cell proliferation and ISC numbers. Reduced numbers of tissue Ly6C+ monocytes suppress Lgr5+ stem cell expression [71]. However, the specific effect and the underlying mechanism remain unclear.
In most cases, neutrophils are the first cells to reach sites of tissue injury. However, analyzing the impact of neutrophils on ISCs is difficult due to their short lifespan. With the development of a novel photoconverter reporter system, researchers can now monitor the migration and colonization of neutrophils after injury [109], which may provide a solution for studying their effects on ISCs. An established co-culture system of enteroid/colonoid with monocytes and neutrophils may also be useful for studying the specific effects on ISCs [106].
Eosinophils and Mast Cells
Gastrointestinal tract has the largest population of eosinophils and mast cells in the body. Eosinophils are multipotent innate immune cells located in the lamina propria of the gastrointestinal tract, while mast cells are long-living granulated immune cells that derive from bone marrow. They both participate in host immunity to maintain the protective mucosal barrier [110,111,112,113].
Until now, no evidence has shown that eosinophils or mast cells can regulate ISCs directly. However, some studies suggest that mast cells may impact the crypt, for example, intestinal barrier function was significantly decreased in mice lacking mast cells or mast cell chymase Mcpt4, which is associated with decreased intestinal epithelial cell migration along the crypt-villus axis and dysregulated claudin-3 crypt expression [72]. These results suggest that mast cells may play a role in the migration of ISCs or TA cells along the crypt-villus axis. Mast cells are enhanced in chronically inflamed rabbit SI, along with the stimulation of brush border membrane Na-glutamine co-transporting in crypt cells. These effects are reversed by ketotifen, a mast cell stabilizer [73]. However, determining whether mast cells act on ISCs in a direct manner requires further exploration.
Moreover, eosinophils are armed with the ability to synthesize and release a number of immunomodulatory and/or signaling molecules such as IL-3, IL-5, and TGF-β to act on different target cells including mast cells, lymphocytes, neurons, smooth muscle cells, fibroblasts, endothelial cells, goblet cells, and others [114]. Moreover, eosinophils are the most important source of IL-22 binding protein (IL-22BP), an endogenous inhibitor of IL-22, in the human gut. In IBD patients, an increased number of eosinophils express IL-22BP at a high level, thereby blocking the protective effect of IL-22 [74]. Thus, eosinophils may also influence ISCs in a direct or indirect manners, with further research required to investigate the effect of eosinophils and mast cells on ISCs.
Prospects for Therapeutic Targets
Intestinal mucosal damage is the key pathogenetic factor in many intestinal and systemic diseases, therefore, promoting mucosal healing is the primary goal of treatment for these diseases. ISCs have an extraordinary capacity for self-renewal and multi-directional differentiation to refresh the epithelium and maintain the mucosal barrier. Therefore, therapies targeting ISC survival and renewal are of great significance and may reveal a promising future direction of treatment (Table. 2).
Graft-Versus-Host Disease
GVHD is a severe complication of allo-HSCT. Clinical data show that GVHD continues to be a leading cause of morbidity and mortality after allo-HSCT, of which gastrointestinal GVHD is the major cause of mortality [79]. The pathophysiology of GVHD consists of three major steps involving both innate and adaptive immune cells: (1) activation of APCs by host tissue damage and subsequent inflammatory cytokine release; (2) donor alloreactive T cell activation, proliferation, differentiation and migration; and (3) destruction of host tissues (Fig. 3) [78].
As previously mentioned, GVHD causes the most severe damage to ISCs. After BMT, T cells are recruited to the ISC compartment by MAdCAM-1 [42] and directly target ISCs via IFN-γ, leading to ISC depletion and epithelial barrier destruction [36]. Thus, reducing T cell infiltration is quite important for preventing and treating GVHD. Indeed, researchers have demonstrated that MAdCAM-1 blockade specifically reduces crypt base infiltration by donor T cells and protects the ISC compartment [42], as well as alleviates GVHD-associated intestinal injury [115, 116]. In fact, inhibition of α4β7/MAdCAM-1 axis has been used for IBD treatment to reduce T cells infiltration. Vedolizumab, a monoclonal antibody directed against α4β7, has been approved for ulcerative colitis (UC) and Crohn’s disease (CD) [117]. Other α4β7/MAdCAM-1 monoclonal antibodies (e.g., Abrilumab, Etrolizumab, Ontamalimab) also proved safety and efficacy for patients with moderate-to-severe UC in their clinical trials [118,119,120]. These therapies may be directly transplanted to GVHD to reduce intestinal damage.
Furthermore, reducing the response of ISCs to cytokines can block the effects of cytokines. JAK proteins mediate the intracellular signaling of a wide range of cytokines. Ruxolitinib, a selective JAK1/2 inhibitor of IFN-γ signaling, also affords protection against IFN-γ-induced ISC apoptosis [36]. Ruxolitinib reduces the loss of Paneth cells and ISCs by increasing regulatory T cell numbers and altering T cell trafficking, thereby ameliorating GVHD in mice [40, 122]. Hence, JAK inhibitors provide clinically efficacious immunosuppression by decreasing ISC responses to pathologic signals from the immune system. Several clinical trials have shown that ruxolitinib and other JAK1/JAK2 inhibitors are effective in the treatment of steroid-refractory (SR) acute and chronic GVHD and are currently being tested in prospective randomized studies [121, 123,124,125,126, 136]. Although studies of JAK1/JAK2 inhibitors did not initially focus on ISCs, we believe that their effect on ISCs is also an important aspect for the protective effects. The therapeutic impact on ISCs must also be taken into account in the future.
Protection of the ISC niche is another key approach to restoring intestinal homeostasis and relieving GVHD. A recent study showed that IFN-λ (IL-28/IL-29) may be an attractive and rapidly testable approach for GVHD treatment. PEG-rIL-29, an adjunctive therapy for hepatitis C that is currently in phase IIb clinical trials (NCT01001754) [165], enhances ISC growth and reduces GVHD severity after BMT [127]. Therefore, PEG-rIL-29 may be a promising treatment for GVHD to rapidly translate to the clinic. Moreover, in vivo treatment with IL-22 after mouse BMT enhances the recovery of ISCs, increases epithelial regeneration, and reduces mortality from GVHD by directly acting on ISCs [35, 80]. F-652, a rhIL-22-dimer and Fc-fusion protein, promotes mouse epithelial regeneration without evidence of toxicity. Treating allogeneic BMT recipients with F-652 starting 1 week after transplantation significantly reduces systemic signs of GVHD and GVHD-related mortality [35]. In a clinical trial (NCT02406651), F-652 is being administered to patients with grade II-IV aGVHD in the lower intestinal tract [77].
However, IL-22 has both protective and pro-inflammatory properties. In addition to ILC3s, other cells can also secrete IL-22, such as donor T cells [131]. Studies have shown that IL-22 deficiency in donor T cells or administration of IL-22 antibody increases Foxp3+ Treg cells and decreases the severity of aGVHD [128, 130, 133, 134]. IL-22 from Th/Tc22 cells causes dysbiosis in a Reg3γ-dependent manner in SR-aGVHD [166]. Its effect depends on the timing, the target tissue, and the origin of the producing cells (donor/host) [131]. Therefore, although IL-22 has a protective effect on ISCs, its pro-inflammatory nature hinders the therapeutic application in GVHD. Further work is required to comprehensively understand the role of IL-22 in allo-HSCT before clinical application.
In addition to cytokines, other small molecules also acts as a survival signal for ISCs [53]. R-spondin-1, a Wnt agonist, reduced murine GVHD by protecting ISCs from injury and by expanding Paneth cells to secrete antimicrobial α-defensins, thereby inhibiting GVHD-associated changes to the microbiome [77]. REG3α/γ, a key AMP, reduces the apoptosis of ISCs and Paneth cells, maintains the integrity of intestinal epithelium [53], and inhibits the contact between microbes and the mucosal barrier [135], thereby reducing the activation of T cells and alleviating GVHD. Although these approaches are still in preclinical testing, they may provide a more physiologic approach to the prevention and treatment of GVHD through maintaining the integrity of mucosal barrier rather than by intensifying systemic immunosuppression [53, 77].
To sum up, GVHD is characterized by T cell response and loss of ISCs, thus, reducing T cell infiltration, inhibiting the response of ISCs and preserving ISCs are all of great importance for the prevention and treatment of GVHD. Inhibition of α4β7/MAdCAM-1 has achieved significant benefits in the treatment of IBD and can be applied to GVHD. Although the protective effect of IL-22 on ISCs has been widely recognized, its pro-inflammatory nature hinders the therapeutic application in GVHD. Small molecules, such as R-spondin-1 and REG3α/γ, are still in preclinical testing, but they may be more effective than systemic immunosuppressive strategies.
Inflammatory Bowel Disease
IBD is characterized by chronic immune-mediated intestinal inflammation and mucosal barrier damage comprising UC and CD [167]. A large proportion of inflammatory cell infiltration and prolonged exposure to cytokines actively hinders ISC proliferation and epithelial regeneration [38]. Therefore, inhibition of immune cell trafficking has emerged as a major therapeutic principle in IBD. Vedolizumab is an effective and approved treatment for CD and UC [117]. Ontamalimab, a monoclonal antibody that binds and inhibits the action of MAdCAM-1, has already demonstrated safety and efficacy for moderate-to-severe UC patients in phase II clinical trials [120]. Moreover, JAK/STAT pathway is also targeted for the treatment of IBD [138]. the JAK inhibitor ruxolitinib protects ISCs through suppressing IFN-γ signaling [40]. Tofacitinib, a pan-JAK inhibitor, has been recently approved for the treatment of moderate-to-severe UC [168], and several second-generation selective JAK inhibitors are under development to improve the benefit-risk ratio [137, 139]. However, none of these treatment strategies have considered the impact on ISCs up to now. Considering the key role of ISCs in the repair of mucosal damage, we believe the response of ISCs must be taken into consideration.
In addition, rebuild or reset the patient immune system through hematopoietic stem cell (HSC) transplantation may be an althernative treatment for IBD. Replacing the pathogenic immune cells with fresh, un-sensitized immune cells may rebuild the immune response, and reduce their damage to ISCs as well as mucosal barrier. Bone marrow transplantation or HSC transplantation represents the most widely used cell-based regenerative therapy. The combination of high-dose immunosuppression and autologous HSC transplantation induces disease remission and may benefit some patients with refractory CD in a phase I study [169]. However, due to its high incidence of serious adverse events such as viral infection and neutropenic sepsis, it may not be recommended as a good and safe alternative therapy for refractory CD [151, 170].
Moreover, reconstruction of the mucosal barrier by ISC transplantation has been feasible, since Sato et al. successfully established a long-term culture method for both mice and human ISCs [171]. The donor organoid cells can engraft and cover the ulcer lesions of the DSS-colitis mice, and maintain for a prolonged period of 6 months after transplantation [146,147,148]. The successful transplantation and their beneficial effects on clinical outcome of colitis are encouraging, thus, we suppose that ISC transplantation may provide a new therapeutic approach in IBD patients to promote healing of inflamed mucosa. The ISCs can be collected from healthy intestinal mucosa in IBD patients or healthy donors via the endoscopic biopsy, then expanded in vitro by the standard organoid culture method. Subsequently, the expanded organoids are delivered to the desired sites by endoscopy after pathogenic and tumorigenicity assessment [7, 149, 150]. However, several problems have to be resolved before any further clinical application. For example, a standard culture method for human ISCs with fully defined factors has not been settled, and it remains unclear whether the ISCs derived from the patient mucosa have the same in vitro expansion capacity compared to those derived from a healthy donor [151].
Importantly, although ISC transplantation may promote the epithelial healing process, a monotherapy based on ISC transplantation remains impractical, as cells presumably will have difficulties engrafting during ongoing inflammation [150]. Hence, considering the crosstalk between immune cells and ISCs, we suppose that immunomodulatory mediators (e.g., IL-22, REG3α, PGE2, M2 macrophages, ILC3s) may be administrated together or signaling pathways (e.g., Wnt, STAT, EGF, JAK) can be targeted to promote the survival and proliferation of the transplanted cells.
Nevertheless, mesenchymal stem cell (MSC) transplantation is the most promising cell therapy available, which combines anti-inflammatory and regenerative properties. MSCs refer to cells that can differentiate into various mesoderm lineage cell types and also retain their ability to self-renew. They can be found in almost any tissue, but the main tissue sources used for MSC transplantation are adipose tissues, umbilical cord and bone marrow [117, 151]. Upon stimulation by pro-inflammatory cytokines, MSCs secrete a variety of immunosuppressive molecules, thus decreasing the overall inflammation. In addition, MSCs promote wound healing and tissue regeneration by secretion of TGF-β and fibroblast growth factor, and MSCs can also differentiate into fibroblasts or endothelial cells to form granulation tissue. Moreover, MSCs do not express MHC II or stimulate T cells, thus enabling escape from immune surveillance and low rejection after transplantation [117, 152]. Therefore, MSCs can be used for the treatment of active complex perianal fistula in CD patients.
Several clinical trials have focused on the efficacy and safety of MSC transplantation. A phase 3 randomized, double-blind controlled trial (NCT01541579) using allogeneic adipose-derived MSCs (Cx601) for complex perianal fistulas in CD shows that a significantly greater proportion of patients treated with Cx601 versus placebo achieve combined remission at week 24 (53 of 107 [50%] vs 36 of 105 [34%]; p = 0.024), with less treatment-related adverse events (18 (17%) of 103 vs 30 (29%) of 103) [153], and it remains safe and effective in closing external openings after 52 weeks [154]. Another prospective, randomized, clinical trial (NCT02445547) using umbilical cord MSCs also demonstrated the efficacy in the treatment of CD with mild side effects after 12 months [155]. Moreover, allogeneic bone marrow-derived MSCs is also safe and effective for CD-associated perianal fistulas after 24 weeks and 4 years (NCT01144962) [143]. In total, 28 animal works (n = 567) and 18 human trials (n = 360) have proved that MSC transplantation reduced the CD activity index [144]. Therefore, MSC is currently the most promising cell therapy for CD.
In Summary, the most recent strategy of IBD treatment aims not only to control mucosal inflammation, but also to acquire ‘mucosal healing’. Inhibiting immune cell trafficking may act as a major therapeutic principle in IBD, while JAK inhibitors can be approved for moderate-to-severe UC. Cell-based therapies, including HSC, ISC and MSC transplantation, show huge potential for certain active complex CD, among which MSC therapy may be introduced to clinical practice first as a supplementary therapy for CD.
Colorectal Cancer
Disruption of the delicate balance between proliferation and differentiation of ISCs governed by key signaling pathways in the crypt can lead to hyperproliferation and ultimately tumorigenesis [145]. Particularly under long-term chronic inflammation, inappropriate self-renewal and survival signals from the niche may result in excessive proliferation and mutation accumulation to yield colorectal cancer (CRC) [172]. Therefore, considering the high responsiveness of ISCs to immune cell activation, these immunomodulatory points may serve as effective targets for CRC prevention and treatment. Wnt/β-catenin is the most important signal for ISC self-renewal and is strongly associated with the development of CRC [173, 174]. Several inflammatory pathways, including NF-κB, PI3K and AKT, drive β-catenin nuclear accumulation without any mutations [156, 158]. Exposure to cytokines such as IFN-γ, TNF-α and IL-22 or soluble mediators such as PGE2 during inflammation can activate the NF-κB, PI3K-AKT, and STAT3 pathways, thereby enhancing β-catenin signaling and contributing to tumorigenesis [37, 69, 157, 159]. Thus, targeting Wnt pathway may have great therapeutic potential in treatment of colitis-associated cancer (CAC). However, the Wnt-pathway therapy has been fraught with the challenges of targeting complex intracellular signalling hubs and the toxicity associated with inhibiting the “ubiquitous” Wnt pathway [175].
Moreover, Th17 cells are thought to contribute to colon tumorigenesis by promoting colon ISC growth via IL-17. For example, IL-17 neutralizing antibody abrogates Citrobacter rodentium-induced colon stem cell proliferation and tumorigenesis [47]. The microRNA-34a (miR-34a), a known tumor suppressor that targets genes associated with cell proliferation and apoptosis, can inhibit Th17 cell differentiation, expansion, and recruitment and induce ISC proliferation [47]. Hence, miR-34a is likely to act as a safeguard for the inflammatory stem cell niche by modulating immune responses to inflammation and subsequently mitigating CAC. In addition to general inhibitors, the effects of miR-34a provide evidence that microRNAs may represent new therapeutic strategies against various tumors. In fact, MRX34, the first tumor-targeted microRNA drug, which is based on miR-34a mimics, has been tested in a phase I clinical trial (NCT01829971) of 155 participants with one of seven cancer types, including several solid tumors and hematopoietic malignancies [162]. Although some adverse immune responses occurred, the extensive potential of MRX34 in cancer therapy cannot be neglected [162, 163]. As miR-34a moves into clinical trials, this is the first application of microRNA in cancer treatment [160, 161].
Furthermore, the most important hallmark of tumorigenesis is mutation, which can be induced by genotoxic factors [176]. Mutations that occur in ISCs are more likely to result in malignant transformation and cancer development [173], whereas these are prevented by DDR in most cases. AHR expressed by ILC3 and γδ T cells serves as a “sensor” of genotoxic factors for on-demand production of IL-22, thereby effectively initiating DDR following DNA damage in ISCs [57]. In addition, a recent study shows that AHR activation protects the stem cell niche and prevents tumorigenesis by inhibiting Wnt-β-catenin signaling and restricting ISC proliferation [96]. Therefore, AHR is an important node for preventing colorectal tumorigenesis. Of note, metabolites such as the glucosinolates, indole-3-aldehyde and indole-3-carbinol are AHR ligands that activate the AHR pathway, thus can be used as chemopreventive agents for IBD-associated CRC in human patients [52, 57, 164]. The activation of AHR by treatment of indole-3-carbinol prevents the development of CAC in mice, highlighting the protective role of AHR [164]. More preclinical trials are in urgent need to confirm the safety and efficacy.
In summary, targeting Wnt pathway has demonstrated great therapeutic potential, but huge challenges due to the intracellular signalling hubs and toxicity remain to be settled. MiR-34a may represent new therapeutic strategies against various tumors, which has been tested in a phase I clinical trial. AHR ligands can be used as chemopreventive agents for IBD-associated CRC in human patients after safety and efficacy are proved.
Conclusions and Perspectives
In this review, we discussed the effects and mechanisms of immune cells and their cytokine repertoires on ISCs. Current studies mainly focus on T cells and ILCs. T cells usually eliminate ISCs, leading to severe intestinal diseases such as GVHD, NEC, and IBD, whereas ILCs preserve ISCs and reduce intestinal immune damage through multiple signaling networks. However, the interaction between ISCs and immune cells is much more complicated than what is understood to date and involves more than distinguishing simply between pro-inflammatory and anti-inflammatory effects. Future research must be more specific regarding the context, concentration, and duration of cytokine action to gain a more comprehensive understanding of the diverse roles of cytokines in health and disease, and render them more suitable for clinical, real-world conditions. Such studies represent a new research frontier and will truly benefit clinical practice related to intestinal disease.
Therapies targeting the survival and renewal of ISCs are of great significance for multiple intestinal diseases, such as GVHD, IBD, and CRC. Reducing T cell infiltration, inhibiting the response of ISCs and preserving ISCs can be considered for the treatment of GVHD and IBD. Cell-based therapies show huge potential for some active complex CD patients and can serve as a supplementary therapy. MiR-34a represents new therapeutic strategies against CRC, while AHR ligands can be used as chemopreventive agents in human patients. Overall, the essential role of ISCs in epithelial repair and regeneration must be fully recognized, and the therapeutic impacts on ISCs must also be considered seriously in the future.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Gehart, H., & Clevers, H. (2019). Tales from the crypt: New insights into intestinal stem cells. Nature Reviews. Gastroenterology & Hepatology, 16, 19–34.
Sato, T., & Clevers, H. (2013). Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science, 340, 1190–1194.
Barker, N. (2014). Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nature Reviews. Molecular Cell Biology, 15, 19–33.
Santos, A. J. M., Lo, Y. H., Mah, A. T., et al. (2018). The intestinal stem cell niche: Homeostasis and adaptations. Trends in Cell Biology, 28, 1062–1078.
Arrazuria, R., Pérez, V., Molina, E., et al. (2018). Diet induced changes in the microbiota and cell composition of rabbit gut associated lymphoid tissue (GALT). Scientific Reports, 8, 14103.
Mowat, A. M., & Agace, W. W. (2014). Regional specialization within the intestinal immune system. Nature Reviews. Immunology, 14, 667–685.
Hou, Q., Huang, J., Ayansola, H., et al. (2020). Intestinal stem cells and immune cell relationships: Potential therapeutic targets for inflammatory bowel diseases. Frontiers in Immunology, 11, 623691.
Yeung, T. M., Chia, L. A., Kosinski, C. M., et al. (2011). Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cellular and Molecular Life Sciences, 68, 2513–2523.
Hou, Y., Wei, W., Guan, X., et al. (2021). A diet-microbial metabolism feedforward loop modulates intestinal stem cell renewal in the stressed gut. Nature Communications, 12, 271.
Brown, E. M., Sadarangani, M., & Finlay, B. B. (2013). The role of the immune system in governing host-microbe interactions in the intestine. Nature Immunology, 14, 660–667.
Martens, E. C., Neumann, M., & Desai, M. S. (2018). Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nature Reviews. Microbiology, 16, 457–470.
Peck, B. C. E., Shanahan, M. T., Singh, A. P., et al. (2017). Gut microbial influences on the mammalian intestinal stem cell niche. Stem Cells International, 2017, 5604727.
Xing, P. Y., Pettersson, S., & Kundu, P. (2020). Microbial metabolites and intestinal stem cells tune intestinal homeostasis. Proteomics, 20, e1800419.
Hou, Q., Ye, L., Huang, L., et al. (2017). The research Progress on intestinal stem cells and its relationship with intestinal microbiota. Frontiers in Immunology, 8, 599.
Moossavi, S., Zhang, H., Sun, J., et al. (2013). Host-microbiota interaction and intestinal stem cells in chronic inflammation and colorectal cancer. Expert Review of Clinical Immunology, 9, 409–422.
Bonfini, A., Liu, X., & Buchon, N. (2016). From pathogens to microbiota: How Drosophila intestinal stem cells react to gut microbes. Developmental and Comparative Immunology, 64, 22–38.
Naik, S., Larsen, S. B., Cowley, C. J., et al. (2018). Two to tango: Dialog between immunity and stem cells in health and disease. Cell, 175, 908–920.
Turner, J. R. (2009). Intestinal mucosal barrier function in health and disease. Nature Reviews. Immunology, 9, 799–809.
Allaire, J. M., Crowley, S. M., Law, H. T., et al. (2018). The intestinal epithelium: Central coordinator of mucosal immunity. Trends in Immunology, 39, 677–696.
Rees, W. D., Sly, L. M., & Steiner, T. S. (2020). How do immune and mesenchymal cells influence the intestinal epithelial cell compartment in inflammatory bowel disease? Let's crosstalk about it! Journal of Leukocyte Biology, 108, 309–321.
Knoop, K. A., & Newberry, R. D. (2018). Goblet cells: Multifaceted players in immunity at mucosal surfaces. Mucosal Immunology, 11, 1551–1557.
Clevers, H. C., & Bevins, C. L. (2013). Paneth cells: Maestros of the small intestinal crypts. Annual Review of Physiology, 75, 289–311.
Howitt, M. R., Lavoie, S., Michaud, M., et al. (2016). Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science, 351, 1329–1333.
Sasaki, N., Sachs, N., Wiebrands, K., et al. (2016). Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proceedings of the National Academy of Sciences of the United States of America, 113, E5399–E5407.
Cheng, H., & Leblond, C. P. (1974). Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian theory of the origin of the four epithelial cell types. The American Journal of Anatomy, 141, 537–561.
Barker, N., van Es, J. H., Kuipers, J., et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449, 1003–1007.
Potten, C. S., Hume, W. J., Reid, P., et al. (1978). The segregation of DNA in epithelial stem cells. Cell, 15, 899–906.
Zhu, P., Zhu, X., Wu, J., et al. (2019). IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nature Immunology, 20, 183–194.
Takahashi, T., & Shiraishi, A. (2020). Stem cell signaling pathways in the small intestine. International Journal of Molecular Sciences, 21, 2032.
Kamada, N., & Núñez, G. (2014). Regulation of the immune system by the resident intestinal Bacteria. Gastroenterology, 146, 1477–1488.
Xiong, N., & Hu, S. (2015). Regulation of intestinal IgA responses. Cellular and Molecular Life Sciences, 72, 2645–2655.
Spangler, J. B., Moraga, I., Mendoza, J. L., et al. (2015). Insights into cytokine-receptor interactions from cytokine engineering. Annual Review of Immunology, 33, 139–167.
Biton, M., Haber, A. L., Rogel, N., et al. (2018). T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell, 175, 1307–1320 e22.
Xue, X., & Falcon, D. M. (2019). The role of immune cells and cytokines in intestinal wound healing. International Journal of Molecular Sciences, 20, 6097.
Lindemans, C. A., Calafiore, M., Mertelsmann, A. M., et al. (2015). Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature, 528, 560–564.
Takashima, S., Martin, M. L., Jansen, S. A., et al. (2019). T cell-derived interferon-gamma programs stem cell death in immune-mediated intestinal damage. Science Immunology, 4, eaay8556.
Lee, G., Goretsky, T., Managlia, E., et al. (2010). Phosphoinositide 3-kinase signaling mediates beta-catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology, 139, 869–881, 881 e1–9.
Nava, P., Koch, S., Laukoetter, M. G., et al. (2010). Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity, 32, 392–402.
Evans, C. M., Phillips, A. D., Walker-Smith, J. A., et al. (1992). Activation of lamina propria T cells induces crypt epithelial proliferation and goblet cell depletion in cultured human fetal colon. Gut, 33, 230–235.
Eriguchi, Y., Nakamura, K., Yokoi, Y., et al. (2018). Essential role of IFN-gamma in T cell-associated intestinal inflammation. JCI Insight, 3.
Agudo, J., Park, E. S., Rose, S. A., et al. (2018). Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity, 48, 271–285 e5.
Fu, Y. Y., Egorova, A., Sobieski, C., et al. (2019). T cell recruitment to the intestinal stem cell compartment drives immune-mediated intestinal damage after allogeneic transplantation. Immunity, 51, 90–103 e3.
Richmond, C. A., Rickner, H., Shah, M. S., et al. (2018). JAK/STAT-1 signaling is required for reserve intestinal stem cell activation during intestinal regeneration following acute inflammation. Stem Cell Reports, 10, 17–26.
Schreurs, R., Baumdick, M. E., Sagebiel, A. F., et al. (2019). Human fetal TNF-alpha-cytokine-producing CD4(+) effector memory T cells promote intestinal development and mediate inflammation early in life. Immunity, 50, 462–476 e8.
Nino, D. F., Sodhi, C. P., Egan, C. E., et al. (2017). Retinoic acid improves incidence and severity of necrotizing enterocolitis by lymphocyte balance restitution and repopulation of LGR5+ intestinal stem cells. Shock, 47, 22–32.
Whetstone, R. D., & Gold, B. (2015). T-cells enhance stem cell mutagenesis in the mouse colon. Mutation Research, 774, 1–5.
Wang, L., Wang, E., Wang, Y., et al. (2018). miR-34a is a microRNA safeguard for Citrobacter-induced inflammatory colon oncogenesis. Elife, 7, e39479.
Wei, L., Leibowitz, B. J., Epperly, M., et al. (2018). The GS-nitroxide JP4-039 improves intestinal barrier and stem cell recovery in irradiated mice. Scientific Reports, 8, 2072.
Jung, K. B., Lee, H., Son, Y. S., et al. (2018). Interleukin-2 induces the in vitro maturation of human pluripotent stem cell-derived intestinal organoids. Nature Communications, 9, 3039.
Hanash, A. M., Dudakov, J. A., Hua, G., et al. (2012). Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity, 37, 339–350.
Hammer, A. M., Morris, N. L., Cannon, A. R., et al. (2017). Interleukin-22 prevents microbial Dysbiosis and promotes intestinal barrier regeneration following acute injury. Shock, 48, 657–665.
Hou, Q., Ye, L., Liu, H., et al. (2018). Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death and Differentiation, 25, 1657–1670.
Zhao, D., Kim, Y. H., Jeong, S., et al. (2018). Survival signal REG3alpha prevents crypt apoptosis to control acute gastrointestinal graft-versus-host disease. The Journal of Clinical Investigation, 128, 4970–4979.
Zheng, Y., Valdez, P. A., Danilenko, D. M., et al. (2008). Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature Medicine, 14, 282–289.
Chen, W., Zai, W., Fan, J., et al. (2020). Interleukin-22 drives a metabolic adaptive reprogramming to maintain mitochondrial fitness and treat liver injury. Theranostics, 10, 5879–5894.
Geng, H., Bu, H. F., Liu, F., et al. (2018). In inflamed intestinal tissues and epithelial cells, interleukin 22 signaling increases expression of H19 long noncoding RNA, which promotes mucosal Regeneration. Gastroenterology, 155, 144–155.
Gronke, K., Hernandez, P. P., Zimmermann, J., et al. (2019). Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature, 566, 249–253.
Zwarycz, B., Gracz, A. D., Rivera, K. R., et al. (2019). IL22 inhibits epithelial stem cell expansion in an Ileal organoid model. Cellular and Molecular Gastroenterology and Hepatology, 7, 1–17.
Zhang, X., Liu, S., Wang, Y., et al. (2019). Interleukin22 regulates the homeostasis of the intestinal epithelium during inflammation. International Journal of Molecular Medicine, 43, 1657–1668.
Zha, J. M., Li, H. S., Lin, Q., et al. (2019). Interleukin 22 expands transit-amplifying cells while depleting Lgr5(+) stem cells via inhibition of Wnt and notch signaling. Cellular and Molecular Gastroenterology and Hepatology, 7, 255–274.
Aparicio-Domingo, P., Romera-Hernandez, M., Karrich, J. J., et al. (2015). Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. The Journal of Experimental Medicine, 212, 1783–1791.
Romera-Hernandez, M., Aparicio-Domingo, P., Papazian, N., et al. (2020). Yap1-driven intestinal repair is controlled by group 3 innate lymphoid cells. Cell Reports, 30, 37–45 e3.
Gerbe, F., Sidot, E., Smyth, D. J., et al. (2016). Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature, 529, 226–230.
von Moltke, J., Ji, M., Liang, H. E., et al. (2016). Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature, 529, 221–225.
Cosin-Roger, J., Ortiz-Masia, D., Calatayud, S., et al. (2016). The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunology, 9, 986–998.
Saha, S., Aranda, E., Hayakawa, Y., et al. (2016). Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nature Communications, 7, 13096.
Cosin-Roger, J., Ortiz-Masia, M. D., & Barrachina, M. D. (2019). Macrophages as an emerging source of Wnt ligands: Relevance in mucosal integrity. Frontiers in Immunology, 10, 2297.
Riehl, T. E., Alvarado, D., Ee, X., et al. (2020). Hyaluronic acid promotes Lgr5(+) stem cell proliferation and crypt fission through TLR4 and PGE2 transactivation of EGFR. American Journal of Physiology. Gastrointestinal and Liver Physiology, 319, G63–G73.
Tessner, T. G., Muhale, F., Riehl, T. E., et al. (2004). Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. The Journal of Clinical Investigation, 114, 1676–1685.
Jarde, T., Chan, W. H., Rossello, F. J., et al. (2020). Mesenchymal niche-derived Neuregulin-1 drives intestinal stem cell proliferation and regeneration of damaged epithelium. Cell Stem Cell, 27, 646–662 e7.
Skoczek, D. A., Walczysko, P., Horn, N., et al. (2014). Luminal microbes promote monocyte-stem cell interactions across a healthy colonic epithelium. Journal of Immunology, 193, 439–451.
Groschwitz, K. R., Ahrens, R., Osterfeld, H., et al. (2009). Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America, 106, 22381–22386.
Singh, S., Arthur, S., Talukder, J., et al. (2015). Mast cell regulation of Na-glutamine co-transporters B0AT1 in villus and SN2 in crypt cells during chronic intestinal inflammation. BMC Gastroenterology, 15, 47.
Martin, J., Bériou, G., Heslan, M., et al. (2016). IL-22BP is produced by eosinophils in human gut and blocks IL-22 protective actions during colitis. Mucosal Immunology, 9, 539–549.
Sun, L., Rollins, D., Qi, Y., et al. (2020). TNFalpha regulates intestinal organoids from mice with both defined and conventional microbiota. International Journal of Biological Macromolecules, 164, 548–556.
Yokoi, Y., Adachi, T., Sugimoto, R., et al. (2021). Simultaneous real-time analysis of Paneth cell and intestinal stem cell response to interferon-γ by a novel stem cell niche tracking method. Biochemical and Biophysical Research Communications, 545, 14–19.
Zeiser, R., & Blazar, B. R. (2017). Acute graft-versus-host disease - biologic process, prevention, and therapy. The New England Journal of Medicine, 377, 2167–2179.
Ferrara, J. L. M., Levine, J. E., Reddy, P., et al. (2009). Graft-versus-host disease. The Lancet, 373, 1550–1561.
Teshima, T., Reddy, P., & Zeiser, R. (2016). Acute graft-versus-host disease: Novel biological insights. Biology of Blood and Marrow Transplantation, 22, 11–16.
Karrich, J. J., & Cupedo, T. (2016). Group 3 innate lymphoid cells in tissue damage and graft-versus-host disease pathogenesis. Current Opinion in Hematology, 23, 410–415.
Ishikawa, N., Wakelin, D., & Mahida, Y. R. (1997). Role of T helper 2 cells in intestinal goblet cell hyperplasia in mice infected with Trichinella spiralis. Gastroenterology, 113, 542–549.
Hua, G., Wang, C., Pan, Y., et al. (2017). Distinct levels of Radioresistance in Lgr5(+) colonic epithelial stem cells versus Lgr5(+) small intestinal stem cells. Cancer Research, 77, 2124–2133.
Bostick, J. W., & Zhou, L. (2016). Innate lymphoid cells in intestinal immunity and inflammation. Cellular and Molecular Life Sciences, 73, 237–252.
Wu, J., Lv, X., Zhu, S., et al. (2019). Critical roles of balanced innate lymphoid cell subsets in intestinal homeostasis, chronic inflammation, and Cancer. Journal of Immunology Research, 2019, 1325181.
Castellanos, J. G., & Longman, R. S. (2019). The balance of power: Innate lymphoid cells in tissue inflammation and repair. The Journal of Clinical Investigation, 129, 2640–2650.
Diefenbach, A., Gnafakis, S., & Shomrat, O. (2020). Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity, 52, 452–463.
Sonnenberg, G. F., & Artis, D. (2015). Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nature Medicine, 21, 698–708.
Mizoguchi, A., Yano, A., Himuro, H., et al. (2018). Clinical importance of IL-22 cascade in IBD. Journal of Gastroenterology, 53, 465–474.
Glal, D., Sudhakar, J. N., Lu, H. H., et al. (2018). ATF3 sustains IL-22-induced STAT3 phosphorylation to maintain mucosal immunity through inhibiting phosphatases. Frontiers in Immunology, 9, 2522.
Choi, S. M., McAleer, J. P., Zheng, M., et al. (2013). Innate Stat3-mediated induction of the antimicrobial protein Reg3γ is required for host defense against MRSA pneumonia. The Journal of Experimental Medicine, 210, 551–561.
Peterson, L. W., & Artis, D. (2014). Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nature Reviews. Immunology, 14, 141–153.
Zhu, P., Wu, J., Wang, Y., et al. (2018). LncGata6 maintains stemness of intestinal stem cells and promotes intestinal tumorigenesis. Nature Cell Biology, 20, 1134–1144.
Xiao, L., Gorospe, M., & Wang, J. Y. (2019). Long noncoding RNAs in intestinal epithelium homeostasis. American Journal of Physiology. Cell Physiology, 317, C93–c100.
Wang, J. Y., Xiao, L., & Wang, J. Y. (2017). Posttranscriptional regulation of intestinal epithelial integrity by noncoding RNAs. Wiley Interdisciplinary Reviews: RNA, 8, 10.1002/wrna.1399.
Lee, J. S., Cella, M., McDonald, K. G., et al. (2011). AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of notch. Nature Immunology, 13, 144–151.
Metidji, A., Omenetti, S., Crotta, S., et al. (2018). The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity, 49, 353–362 e5.
Pernomian, L., Duarte-Silva, M., & de Barros Cardoso, C. R. (2020). The aryl hydrocarbon receptor (AHR) as a potential target for the control of intestinal inflammation: Insights from an immune and Bacteria sensor receptor. Clinical Reviews in Allergy and Immunology, 59, 382–390.
Eberl, G. (2015). ILC3s protect intestinal stem cells from chemotherapy. The Journal of Experimental Medicine, 212, 1756.
Gregorieff, A., Liu, Y., Inanlou, M. R., et al. (2015). Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature, 526, 715–718.
Na, Y. R., Stakenborg, M., Seok, S. H., et al. (2019). Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nature Reviews. Gastroenterology & Hepatology, 16, 531–543.
Bekiaris, V., Persson, E. K., & Agace, W. W. (2014). Intestinal dendritic cells in the regulation of mucosal immunity. Immunological Reviews, 260, 86–101.
Akcora, D., Huynh, D., Lightowler, S., et al. (2013). The CSF-1 receptor fashions the intestinal stem cell niche. Stem Cell Research, 10, 203–212.
Sehgal, A., Donaldson, D. S., Pridans, C., et al. (2018). The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nature Communications, 9, 1272.
Ihara, S., Hirata, Y., Hikiba, Y., et al. (2018). Adhesive interactions between mononuclear phagocytes and intestinal epithelium perturb Normal epithelial differentiation and serve as a therapeutic target in inflammatory bowel disease. Journal of Crohn's & Colitis, 12, 1219–1231.
Noel, G., Baetz, N. W., Staab, J. F., et al. (2017). A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Scientific Reports, 7, 45270.
Staab, J. F., Lemme-Dumit, J. M., Latanich, R., et al. (2020). Co-culture system of human Enteroids/Colonoids with innate immune cells. Current Protocols in Immunology, 131, e113.
Habtezion, A., Nguyen, L. P., Hadeiba, H., et al. (2016). Leukocyte trafficking to the small intestine and Colon. Gastroenterology, 150, 340–354.
Perez-Lopez, A., Behnsen, J., Nuccio, S. P., et al. (2016). Mucosal immunity to pathogenic intestinal bacteria. Nature Reviews. Immunology, 16, 135–148.
Hülsdünker, J., Ottmüller, K. J., Neeff, H. P., et al. (2018). Neutrophils provide cellular communication between ileum and mesenteric lymph nodes at graft-versus-host disease onset. Blood, 131, 1858–1869.
Albert-Bayo, M., Paracuellos, I., Gonzalez-Castro, A. M., et al. (2019).Intestinal mucosal mast cells: Key modulators of barrier function and homeostasis. Cells, 8, 135.
Powell, N., Walker, M. M., & Talley, N. J. (2010). Gastrointestinal eosinophils in health, disease and functional disorders. Nature Reviews. Gastroenterology & Hepatology, 7, 146–156.
Berek, C. (2016). Eosinophils: Important players in humoral immunity. Clinical and Experimental Immunology, 183, 57–64.
Wouters, M. M., Vicario, M., & Santos, J. (2016). The role of mast cells in functional GI disorders. Gut, 65, 155–168.
Loktionov, A. (2019). Eosinophils in the gastrointestinal tract and their role in the pathogenesis of major colorectal disorders. World Journal of Gastroenterology, 25, 3503–3526.
Murai, M., Yoneyama, H., Ezaki, T., et al. (2003). Peyer's patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nature Immunology, 4, 154–160.
Ueha, S., Murai, M., Yoneyama, H., et al. (2007). Intervention of MAdCAM-1 or fractalkine alleviates graft-versus-host reaction associated intestinal injury while preserving graft-versus-tumor effects. Journal of Leukocyte Biology, 81, 176–185.
Misselwitz, B., Juillerat, P., Sulz, M. C., et al. (2020). Emerging treatment options in inflammatory bowel disease: Janus kinases, stem cells, and more. Digestion, 101(Suppl 1), 69–82.
Sandborn, W. J., Cyrille, M., Hansen, M. B., et al. (2019). Efficacy and safety of Abrilumab in a randomized, placebo-controlled trial for moderate-to-severe ulcerative colitis. Gastroenterology, 156, 946–957.e18.
Peyrin-Biroulet, L., Hart, A., Bossuyt, P., et al. (2021). Etrolizumab as induction and maintenance therapy for ulcerative colitis in patients previously treated with tumour necrosis factor inhibitors (HICKORY): a phase 3, randomised, controlled trial. The Lancet Gastroenterology & Hepatology, 7, 128–140.
Picardo, S., & Panaccione, R. (2020). Anti-MADCAM therapy for ulcerative colitis. Expert Opinion on Biological Therapy, 20, 437–442.
Maffini, E., Giaccone, L., Festuccia, M., et al. (2016). Ruxolitinib in steroid refractory graft-vs.-host disease: a case report. Journal of Hematology & Oncology, 9, 67.
Spoerl, S., Mathew, N. R., Bscheider, M., et al. (2014). Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood, 123, 3832–3842.
Ali, H., Salhotra, A., Modi, B., et al. (2020). Ruxolitinib for the treatment of graft-versus-host disease. Expert Review of Clinical Immunology, 16, 347–359.
Khandelwal, P., Teusink-Cross, A., Davies, S. M., et al. (2017). Ruxolitinib as salvage therapy in steroid-refractory acute graft-versus-host disease in pediatric hematopoietic stem cell transplant patients. Biology of Blood and Marrow Transplantation, 23, 1122–1127.
Schroeder, M. A., Khoury, H. J., Jagasia, M., et al. (2020). A phase 1 trial of itacitinib, a selective JAK1 inhibitor, in patients with acute graft-versus-host disease. Blood Advances, 4, 1656–1669.
von Bubnoff, N., Ihorst, G., Grishina, O., et al. (2018). Ruxolitinib in GvHD (RIG) study: A multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versus-host disease (aGvHD) (NCT02396628). BMC Cancer, 18, 1132.
Henden, A. S., Koyama, M., Robb, R. J., et al. (2021). IFNlambda therapy prevents severe gastrointestinal graft-versus-host disease. Blood, 138(8), 722–737.
Couturier, M., Lamarthee, B., Arbez, J., et al. (2013). IL-22 deficiency in donor T cells attenuates murine acute graft-versus-host disease mortality while sparing the graft-versus-leukemia effect. Leukemia, 27, 1527–1537.
Dudakov, J. A., Mertelsmann, A. M., O’Connor, M. H., et al. (2017). Loss of thymic innate lymphoid cells leads to impaired thymopoiesis in experimental graft-versus-host disease. Blood, 130, 933–942.
Lamarthee, B., Malard, F., Gamonet, C., et al. (2016). Donor interleukin-22 and host type I interferon signaling pathway participate in intestinal graft-versus-host disease via STAT1 activation and CXCL10. Mucosal Immunology, 9, 309–321.
Lamarthee, B., Malard, F., Saas, P., et al. (2016). Interleukin-22 in Graft-Versus-Host Disease after Allogeneic Stem Cell Transplantation. Frontiers in Immunology, 7, 148.
Pan, B., Wang, D., Li, L., et al. (2019). IL-22 Accelerates thymus regeneration via Stat3/Mcl-1 and decreases chronic graft-versus-host disease in mice after allotransplants. Biol Blood Marrow Transplant, 25, 1911–1919.
Wu, J., Gu, J., Zhou, S., et al. (2018). Anti-IL-22 antibody attenuates acute graft-versus-host disease via increasing Foxp3(+) T cell through modulation of CD11b(+) cell function. Journal of Immunology Research, 2018, 1605341.
Zhao, K., Zhao, D., Huang, D., et al. (2014). Interleukin-22 aggravates murine acute graft-versus-host disease by expanding effector T cell and reducing regulatory T cell. Journal of Interferon & Cytokine Research, 34, 707–715.
Loonen, L. M., Stolte, E. H., Jaklofsky, M. T., et al. (2014). REG3gamma-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal Immunology, 7, 939–947.
Zeiser, R., von Bubnoff, N., Butler, J., et al. (2020). Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. The New England Journal of Medicine, 382, 1800–1810.
Danese, S., Argollo, M., Le Berre, C., et al. (2019). JAK selectivity for inflammatory bowel disease treatment: Does it clinically matter? Gut, 68, 1893–1899.
Salas, A., Hernandez-Rocha, C., Duijvestein, M., et al. (2020). JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nature Reviews. Gastroenterology & Hepatology, 17, 323–337.
Sandborn, W. J., Ghosh, S., Panes, J., et al. (2020). Efficacy of Upadacitinib in a randomized trial of patients with active ulcerative colitis. Gastroenterology, 158, 2139–2149.e14.
Pelczar, P., Witkowski, M., Perez, L. G., et al. (2016). A pathogenic role for T cell-derived IL-22BPin inflammatory bowel disease. Science, 354, 358–362.
Kirchberger, S., Royston, D. J., Boulard, O., et al. (2013). Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. Journal of Experimental Medicine, 210, 917–31.
Patnaude, L., Mayo, M., Mario, R., et al. (2021). Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sciences, 271, 119195.
Barnhoorn, M. C., Wasser, M., Roelofs, H., et al. (2020). Long-term evaluation of allogeneic bone marrow-derived mesenchymal stromal cell therapy for Crohn's disease perianal fistulas. Journal of Crohn's & Colitis, 14, 64–70.
Wang, R., Yao, Q., Chen, W., et al. (2021). Stem cell therapy for Crohn's disease: Systematic review and meta-analysis of preclinical and clinical studies. Stem Cell Research & Therapy, 12, 463.
Spit, M., Koo, B. K., & Maurice, M. M. (2018). Tales from the crypt: Intestinal niche signals in tissue renewal, plasticity and cancer. Open Biology, 8, 180120.
Yui, S., Nakamura, T., Sato, T., et al. (2012). Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nature Medicine, 18, 618–623.
Fordham, R. P., Yui, S., Hannan, N. R., et al. (2013). Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell, 13, 734–744.
Sugimoto, S., Ohta, Y., Fujii, M., et al. (2018). Reconstruction of the human Colon epithelium in vivo. Cell Stem Cell, 22, 171–176.e5.
Nakamura, T., & Watanabe, M. (2017). Intestinal stem cell transplantation. Journal of Gastroenterology, 52, 151–157.
Holmberg, F. E., Seidelin, J. B., Yin, X., et al. (2017). Culturing human intestinal stem cells for regenerative applications in the treatment of inflammatory bowel disease. EMBO Molecular Medicine, 9, 558–570.
Okamoto, R., & Watanabe, M. (2016). Investigating cell therapy for inflammatory bowel disease. Expert Opinion on Biological Therapy, 16, 1015–1023.
Carvello, M., Lightner, A., Yamamoto, T., et al. (2019). Mesenchymal stem cells for perianal Crohn's disease. Cells, 8, 764.
Panés, J., García-Olmo, D., Van Assche, G., et al. (2016). Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn's disease: A phase 3 randomised, double-blind controlled trial. Lancet, 388, 1281–1290.
Panés, J., García-Olmo, D., Van Assche, G., et al. (2018). Long-term efficacy and safety of stem cell therapy (Cx601) for complex perianal fistulas in patients with Crohn's disease. Gastroenterology, 154, 1334–1342.e4.
Zhang, J., Lv, S., Liu, X., et al. (2018). Umbilical cord mesenchymal stem cell treatment for Crohn's disease: A randomized controlled clinical trial. Gut Liver, 12, 73–78.
Grivennikov, S. I. (2013). Inflammation and colorectal cancer: Colitis-associated neoplasia. Seminars in Immunopathology, 35, 229–244.
Kaler, P., Godasi, B. N., Augenlicht, L., et al. (2009). The NF-kappaB/AKT-dependent induction of Wnt signaling in Colon Cancer cells by macrophages and IL-1beta. Cancer Microenvironment, 2, 69–80.
Svrcek, M., Borralho Nunes, P., Villanacci, V., et al. (2018). Clinicopathological and molecular specificities of inflammatory bowel disease-related colorectal neoplastic lesions: The role of inflammation. Journal of Crohn's & Colitis, 12, 1486–1498.
Wei, H. X., Wang, B., & Li, B. (2020). IL-10 and IL-22 in mucosal immunity: Driving protection and pathology. Frontiers in Immunology, 11, 1315.
Farooqi, A. A., Fayyaz, S., Shatynska-Mytsyk, I., et al. (2016). Is miR-34a a well-equipped swordsman to conquer Temple of molecular oncology? Chemical Biology & Drug Design, 87, 321–334.
Slabakova, E., Culig, Z., Remsik, J., et al. (2017). Alternative mechanisms of miR-34a regulation in cancer. Cell Death & Disease, 8, e3100.
Zhang, L., Liao, Y., & Tang, L. (2019). MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. Journal of Experimental & Clinical Cancer Research, 38, 53.
Hong, D. S., Kang, Y. K., Borad, M., et al. (2020). Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. British Journal of Cancer, 122, 1630–1637.
Díaz-Díaz, C. J., Ronnekleiv-Kelly, S. M., Nukaya, M., et al. (2016). The aryl hydrocarbon receptor is a repressor of inflammation-associated colorectal tumorigenesis in mouse. Annals of Surgery, 264, 429–436.
Muir, A. J., Arora, S., Everson, G., et al. (2014). A randomized phase 2b study of peginterferon lambda-1a for the treatment of chronic HCV infection. Journal of Hepatology, 61, 1238–1246.
Song, Q., Wang, X., Wu, X., et al. (2021). IL-22-dependent dysbiosis and mononuclear phagocyte depletion contribute to steroid-resistant gut graft-versus-host disease in mice. Nature Communications, 12, 805.
Neurath, M. F. (2019). Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nature Immunology, 20, 970–979.
Danese, S., Fiorino, G., & Peyrin-Biroulet, L. (2020). Positioning therapies in ulcerative colitis. Clinical Gastroenterology and Hepatology, 18, 1280–1290.e1.
Oyama, Y., Craig, R. M., Traynor, A. E., et al. (2005). Autologous hematopoietic stem cell transplantation in patients with refractory Crohn's disease. Gastroenterology, 128, 552–563.
DiNicola, C. A., Zand, A., & Hommes, D. W. (2017). Autologous hematopoietic stem cells for refractory Crohn's disease. Expert Opinion on Biological Therapy, 17, 555–564.
Sato, T., Vries, R. G., Snippert, H. J., et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 459, 262–265.
Nadeem, M. S., Kumar, V., Al-Abbasi, F. A., et al. (2020). Risk of colorectal cancer in inflammatory bowel diseases. Seminars in Cancer Biology, 64, 51–60.
Barker, N., Ridgway, R. A., van Es, J. H., et al. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature, 457, 608–611.
Farooqi, A. A., de la Roche, M., Djamgoz, M. B. A., et al. (2019). Overview of the oncogenic signaling pathways in colorectal cancer: Mechanistic insights. Seminars in Cancer Biology, 58, 65–79.
Sphyris, N., Hodder, M. C., & Sansom, O. J. (2021). Subversion of niche-Signalling pathways in colorectal Cancer: What makes and breaks the intestinal stem cell. Cancers, 13, 1000.
Pilié, P. G., Tang, C., Mills, G. B., et al. (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nature Reviews. Clinical Oncology, 16, 81–104.
Funding
This work was supported by the National Natural Science Foundation of China (81630016) and Fudan’s Undergraduate Research Opportunities Program (18016).
Author information
Authors and Affiliations
Contributions
H.Z., B.Z. and J.L. designed and approved the initial draft. J.Z., D.Y. and F.L. revised the manuscript and provided suggestions. B.W. and B.J. collected and reviewed the documents. L.M. and J.Y. contributed equally in writing the paper. The final manuscript was approved by all authors.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflicts of Interest/Competing Interests
The authors declare no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, L., Yu, J., Zhang, H. et al. Effects of Immune Cells on Intestinal Stem Cells: Prospects for Therapeutic Targets. Stem Cell Rev and Rep 18, 2296–2314 (2022). https://doi.org/10.1007/s12015-022-10347-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-022-10347-7