
Abstract
Intestinal stem cells (ISC) are characterized by their ability to continuously self-renew and differentiate into various functionally distinct intestinal epithelial cell types. Impaired stem cell proliferation and differentiation can cause severe dysfunction of the gastrointestinal tract and lead to the development of several clinical disorders. Animal mouse models provide a valuable platform to study ISC function, disease mechanisms, and the intestinal epithelium’s regenerative capacity upon tissue damage. However, advanced in vitro systems that are more relevant to human physiology are needed to understand better the diverse disease-triggering factors and the heterogeneity in clinical manifestations. Intestinal biopsies from patients might serve as potent starting material for such “gut-in-a-dish” approaches. While many promising tools for intestinal tissue processing, in vitro expansion, and downstream analysis have been developed in recent years, a comprehensive guide with recommendations to successfully launch or improve intestinal stem cell culture is missing. In this review, we present a selection of currently established methods, highlight recent publications and discuss the potential and limitations of those methodological approaches to facilitate and support the future design of novel and more personalized therapeutic options.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The intestinal epithelial is composed of a single-cell layer that is primarily involved in breaking down and absorbing nutrients but also acts as a physical barrier against potentially pathogenic microbes. Although the intestinal immune system has developed tolerance mechanisms towards harmless commensal or dietary antigens, many diseases such as inflammatory bowel disease (IBD) or celiac disease result from a disruption of this tightly balanced gut immune homeostasis. The exact pathogenesis of these chronic diseases is still not completely understood, and many patients suffer from poor quality of life due to lacking curative therapies. While animal studies provide insight on pathophysiological mechanisms and serve as models for testing potential therapeutic compounds, follow-up clinical trials often fail due to low animal-to-human translational success rates. Therefore, pre-clinical models with improved predictive power and which also meet the future demands for more personalized medicine are needed. The development of in vitro models that are capable of mimicking the complex human gut environment is challenging. Recent advances in 2D and 3D cell culture technology have allowed studying stem cell biology, organ regeneration, and disease pathogenesis on living patient-derived material that retained its original donor’s genetic and biological properties. The culture conditions for 2D epithelial monolayer and 3D organoid systems are constantly optimized to generate more predictive and physiologically relevant in vitro models. Better knowledge about the function of important stem cell niche factors has contributed to the development of improved growth medium formulation and more chemically defined extracellular matrix composition. In parallel, considerable efforts were allocated to advance downstream analysis, including 3D live-imaging, high-throughput workflows for automated screens, and genome editing with the CRISPR-Cas9 system. By advancing these technologies, animal experimentation could, in the future, be partially replaced by human-derived, self-sustaining intestinal models, which might also increase the success rates in the mandatory phase of clinical trials.
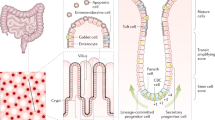
Human intestinal biopsies serve as a valuable source for isolating and expanding patient-derived intestinal epithelial cells. Due to the low amount of starting material, the successful generation of human intestinal monolayers or organoids from biopsies requires an efficient dissociation of the epithelial tissue and a growth environment that provides the important niche factors for expanding the culture. Over the years, many methodological papers have been published with approaches to optimize growth conditions and experimental procedures, starting from the digestion of the biopsy and ending with the handling of highly sophisticated multi-omics data. However, finding suitable methodological input in this fast-growing field of research has become increasingly challenging. Additionally, many publications only cover specific steps of the entire process in detail. Therefore, this review aims to provide a framework with a comprehensive selection of currently established methods and tools, including an evaluation of their advantages and disadvantages. This review primarily focuses on promising techniques to study intestinal stem cell biology, but we also summarize the basic recommendations for successfully culturing human intestinal cells from biopsies (Fig. 1). By linking the different methodological steps and describing available options for downstream analysis, we hope to guide natural scientists who consider building their own pipeline to study human intestinal pathophysiology and thereby contribute to the promotion of these emerging research platforms (Fig. 2).

Graphical summary of methods to study intestinal stem cells that are described in the review

Interconnections between methods described in the review
Tissue Processing
Transport and Storage of Collected Biopsy
Upon collection of biopsies from a patient, it is recommended to place them immediately in an ice-cold collection buffer. The collection buffer can be prepared in advance and stored at 4 °C for at least one month. PBS or basal culture medium is often used, supplemented with an antibody cocktail (see Table 1).
Once the collected tissue is placed in the collection buffer on ice, it can be transported and stored until further processing. Matsumoto et al. [43] have shown that after 2 days of storage, they could still isolate cells, which were capable of generating organoids. However, in general, preparation of the sample for isolation of crypt or single intestinal epithelial cells (IEC) should be performed as soon as possible. Prolonged storage can lead to a loss of intestinal stem cells (ISC) and negatively impact the viability of isolated cells.
Cryopreservation & Thawing
One of the main limitations in stem cells research is access to patient material. In most cases, there is a need for geographical proximity between the place of material collection and the research laboratory. Additionally, the samples must be processed within the short timeframe give by limited tissue viability [21].
To overcome these limitations, Tsai et al. [72] developed a practical method to cryopreserve live human biopsy tissue. This method allows storage of shipment of frozen material, for future thawing and use in the generation of a new organoids cultures. In a first step, the authors compared two formulations of freezing media; a simple freezing medium (DMEM/F12, 10% FBS, 10% DMSO) with a complex L-WRN medium (conditioned media containing Wnt3a, Rspondin3, and Noggin mixed with DMEM/F12, GlutaMax, HEPES, N2 supplement, B27 supplement, antibiotics, N-acetylcysteine, nicotinamide, FBS, DMSO, Y27632, CHIR99021) [72]. The biopsy fragments were frozen in media at -80 °C overnight. As both media types performed equally well, the authors suggested to use of a simple medium formulation for biopsy preservation. In the next step, Tsai et al. compared four different thawing protocols, followed by the establishment of organoid cultures (details can be found in publication [72]). As a consensus, the authors recommend thawing the tissue rapidly at 37 °C, partially digesting it with dispase, and using next a mechanical force to dissociate the tissue and detach the cells. Thereafter, the isolated epithelium can be embedded in Matrigel and subsequently cultured as organoids. The growth of organoids derived from frozen samples is initially delayed compared to organoids cultured from freshly isolated cells. However, after the first passage, frozen organoids become similar to fresh organoids. Finally, as shown by mRNA sequencing, cryopreservation does not cause any significant differences in gene expression as compared to fresh organoids [72].
The authors show that human biopsy specimens from the small intestine and colon can be cryopreserved and used to establish long-term organoid cultures after thawing. Thanks to this method, the geographic proximity between hospital and research laboratory is no longer essential, as frozen tissue samples can be shipped across the globe. Moreover, it effectively increases collaboration perspectives between clinicians, researchers, and diagnostic laboratories worldwide.
Biopsy Digestion to Generate Intestinal Crypt Suspension
Before digestive enzymes are introduced, washing the biopsies thoroughly can help to avoid potential contamination. For washing, either ice-cold PBS, HBSS, or a culture medium-based wash buffer containing an antibody cocktail can be applied. Centrifugation between the washing steps is unnecessary since the biopsies typically settle to the bottom of the tube after a few seconds. The supernatant can be removed with a vacuum pump or manually with a serological pipette. To increase the surface area and improve the digestion process, the washed tissue should be cut into smaller pieces (2–5 mm) prior to adding digestive enzymes. For mincing the tissue, either sharp, small scissors or two razor blades can be used. The minced tissue pieces are then incubated in a digestion buffer at 37 °C or on ice, depending on the applied digestive enzymes. The composition of commonly used digestion buffers can be found in the Table 2. After the digest, crypts are released into the solution through vigorous agitation of the tissue pieces either by pipetting them up and down, manual shaking, or vortexing the tube. The crypt-containing supernatant is then filtered over a 40–100 um cell strainer to remove the undigested epithelium, villi, mucus, and large debris. It might be necessary to repeat shaking and filtration in order to generate several fractions. Fractions containing the highest number of crypts can afterward be pooled. Optionally, the number of crypts per volume can be counted for calculating the optimized seeding density. The crypt-enriched epithelial preparation can directly be suspended in a basement membrane mix and used for generating intestinal organoids (see chapter). Alternatively, the crypt fraction can be further dissociated into single cells (see chapter).
Dissociation into a Single-cell Suspension
For specific experimental approaches and downstream analysis such as FACS or single-cell RNA sequencing, single-cell suspensions are required. To generate single-cell suspensions, either freshly isolated crypts from human intestinal tissue or disrupted, patient-derived organoids are incubated together with a dissociation reagent that cleaves cell–cell junctions. Commonly used dissociation reagents are summarized in Table 3. To prevent dissociation-induced apoptosis, it is recommended to supplement the dissociation buffer with the Rho kinase inhibitor Y-27632. Released DNA molecules from dying cells can clump neighboring cells together. Therefore, the tissue should additionally be treated with DNase I to reduce cell clumping and subsequent loss of material through filtration. During the incubation, the solution should regularly (every 1–2 min) be checked by microscopy to monitor the dissociation progress and stop the reaction at the earliest time point at which the majority (80–90%) of crypts are dissociated. If there are still cell clumps visible, the dissociation cycle can be repeated. To assist the enzymatic digestion, the solution can be gently triturated with a P1000 pipette tip or a glass Pasteur pipette with a P10 pipette tip attached and connected to a pipette controller. Following the dissociation of single cells into solution, the supernatant should be filtered in order to remove any undigested tissue and cell aggregates. After the dissociation procedure, it is critical to evaluate the quality of the single-cell suspension by assessing cell viability, the absence of debris, and cell aggregates [51].
In vitro Expansion Techniques
3D Cultivation
Monolayer cultures of colon cancer cell lines (e.g., HT-29, Caco-2) often lack tissue-specific cell types and architecture and show high gene mutation rates. The resulting insufficient clinical predictive power of those traditional culture systems has led to the development of 3D culture systems that enable the growth of self-organized, gut-like structures. In contrast to conventional immortalized cell lines, intestinal organoids can recapitulate the intestinal epithelium’s in vivo characteristics and physiological features and even retain their self-renewal and differentiation abilities, their phenotype, and gene expression profile over several passages [19]. Furthermore, since the discovery of stem cell niche factors, which allowed long-term cultivation [3, 58], the organoid technology has become a powerful, novel system to study organogenesis [28, 30, 87], self-organization [61], stem cell biology [67, 68, 90] and human disease modeling [16, 17].
Intestinal organoids can be generated directly from biopsy-derived crypts or dissociated, single IECs (see chapter). Isolated crypts or single IECs need a cell–matrix anchorage and cell–cell contact; otherwise, they undergo anoikis within hours [53]. Therefore, optimized culture matrices that enable attachment of IEC and support 3D growth have been developed [29, 31, 33]. Matrigel, a mix of extracellular matrix (ECM) components consisting of laminin, type IV collagen, entactin, and heparin sulfate proteoglycan perlecan [29], is widely used as a scaffold material for organoid cultures. However, its batch-to-batch variability in mechanical and biochemical properties can negatively affect the reproducibility of experiments. Moreover, it has been shown that the inherent properties of the matrix material might influence stem cell fate decisions. Therefore, alternative scaffolds have emerged on the market ranging from simple mixtures of different collagen types to advanced synthetic materials, which are xenogenic-free, chemically defined, and tailorable for specific experimental designs [1, 31, 33].
For seeding, the isolated crypts or single cells are resuspended in ice-cold Matrigel or an alternative ECM (final concentration should be between 75 to 100%) and plated as drops into wells of a pre-warmed cell culture plate. Due to the special rheological properties of Matrigel, the droplets form a stable gel in the incubator at 37 °C after 10 to 15 min. of incubation. Pleguezuelos et al. [47] have recently published a detailed protocol for the establishment of human intestinal organoid cultures, including a table with recommendations regarding the volumes of ECM that are needed for different multiwall formats. The solidified domes can afterward be overlaid with culture medium. Successful cultivation of intestinal organoids requires an optimized culture media formulation [43, 57, 62, 86] containing all necessary growth factors for promoting intestinal stem cell proliferation and/or differentiation (see Table 4 with recommendations for human organoid growth medium). To improve the survival of IECs and prevent anoikis after dissociation, it is generally recommended to add the Rho kinase inhibitor Y-27632 for the first 2–4 days of culture. The standard WENR medium, containing Wnt-3a, EGF, Noggin, and R-Spondin-1, results in stem-cell enriched organoids [57]. Further enrichment for stem cells can be achieved by simultaneous addition of CHIR990221 (GSK3β-inhibitor, Wnt enhancer) and valproic acid (HDAC inhibitor, Notch activator) to the medium (WENR-CV) [84]. While the standard WENR medium is widely used, Fujii et al. [19] have established a refined human intestinal organoid culture condition by adding IGF-1 and FGF-2 instead of SB202190R as niche factors to the growth medium. This p38 inhibitor-free medium has been shown to better conserve the native cellular diversity in human small intestinal organoids compared to the conventional methods. Since the growth factors Wnt-3a, R-Spondin-1 and Noggin are rather costly; many research groups are taking advantage of the L-Wnt3a (ATCC CRL-2647) and L-WRN (ATCC CRL3276) cell line that produces Wnt3a or Wnt3a + RSpondin + Noggin, respectively, to prepare conditioned media [44, 47, 73, 83]. In order to generate stem-cell depleted and more differentiated organoids, components like Wnt-3a, Noggin, R-Spondin-1, Nicotinamide, SB202190R, CHIR990-21, and valproic acid have to be withdrawn or reduced from the culture media [89]. Additionally, by stimulating or inhibiting the Wnt, Notch, and MAPK signaling pathway, the organoid cultures can be enriched for certain IEC types such as Paneth cells (+ DAPT, + CHIR99021), goblet cells (+ DAPT, + IWP2, or XAV939), enteroendocrine cells (+ DAPT, + IWP2 or XAV939, + PD0325901) or Tuft cells (+ IL-4/13) [8]. For optimal results, Pleguezuelos et al. [47] recommend conducting experimental assays and sample collection as early as possible after terminal differentiation since the viability of fully differentiated organoids tends to drop quite rapidly. During the differentiation process, more granular cells (like Paneth cells) appear, organoid walls start to thicken, and dead cells are accumulating in the organoid lumen. Altogether this leads to a less transparent appearance of the organoids and can be used as an indicator for the progress of differentiation.
The first and all following passages should be performed after 7–12 days in culture (each line must be determined empirically). Passaging organoids repeatedly at a too early timepoint will lead to higher material and reagent costs and is therefore not recommended. Late passaging, on the other hand, might substantially decrease cell viability and consequently recovery after splitting. For passaging, the organoids have to be released from the growth matrix and dissociated into smaller fragments. Commonly used techniques for splitting either involve mechanical disruption by pipetting and/or the addition of a chemical dissociation reagent (e.g., EDTA, Dispase, TrypLE). After dissociation, the fragments are resuspended in fresh ECM and replated as domes. The split ratio for a fully developed culture is typically 1:3 to 1:4. By repeating this process regularly, the organoids can be expanded further for experimental purposes and downstream analysis (see chapters). For cryopreservation (see chapter), ideally, exponentially growing small organoids should be collected (2–3 days after passaging) to optimize the recovery after thawing.
Instead of freshly isolated crypts, intestinal organoids can also serve as a source of IECs to generate 2D epithelial monolayers culture systems (see chapter).
Despite the significant advances in using organoids as intestinal models, the system also comes with several limitations. For example, the inaccessibility of the apical side, due to the orientation of the epithelia into the lumen of the organoid, complicates studies to characterize epithelial transport mechanisms and barrier function after exposure to chemical compounds or microorganisms. Moreover, intestinal organoids lack the interaction with the immune, vascular, and enteric nervous system that are essential components of the living digestive tract. In addition, a great heterogeneity between organoids within one dome is often observed, and the 3D architecture tends to be less regular than in vivo. However, these issues might be solved in the future with more defined growth substrates (e.g., synthetic polymers) that allow for consistent interaction with matrix components and prevent spatiotemporal gradients of essential growth factors within the dome. Besides that, the microfluidic gut-on-chip technology (see chapter) will allow for the development of more complex models, including interactions with the microvascular endothelium, commensal microbes, immune cells, pathogens, and even provide the option to mimic peristalsis-like deformations. Nevertheless, the 3D intestinal organoid system remains a promising tool for a broad range of applications [46] and still harbors a substantial potential for further development and improvement, which might in the future also increase the consistency and reproducibility of the in vitro 3D models.
2D – Enteroid Monolayer Cultivation
Human intestinal organoids have become a powerful tool to study intestinal epithelial physiology and model human diseases. Due to the increasing interest in using patient-derived organoids for disease modeling and drug screening, substantial efforts have been made to advance the 3D-based organoid culture. However, the restricted luminal access of the spheroidal structure to exogenous compounds and pathogens complicates studies on nutrient metabolism and absorption, epithelial barrier function, and gut microbiota [37]. To overcome these limitations, researchers have focused on 2D monolayer culture systems, which also allow for improved live-cell imaging, observation of ISC growth, and cell-to-cell interaction. Rapid stem cell loss and high apoptosis rates made the long-term culture of primary intestinal epithelial cells as monolayer practically impossible in the past. Meanwhile, several 2D monolayer culture systems that support ISC maintenance have been successfully established [2, 36, 38, 39, 52, 55, 60, 70, 78]. Extracellular matrix (ECM)-coated surfaces are commonly used as a base for generating intestinal epithelial monolayers. While initially those ECM were produced by feeder layers of irradiated fibroblasts [69], reproducibility issues with cell batch-to-batch variability and potential risk for pathogens and xenogeneic transmission have led to the development of feeder-cell-free culture systems [74]. In these systems, often type 1 collagen, recombinant human laminin, or Matrigel are used for coating. Alongside biological matrices, synthetic matrices with more defined physiochemical and mechanical properties have been developed [65].
Several groups showed robust epithelial monolayer growth by using transwell systems where the apical part of the growing epithelial monolayer faces towards the culture medium allowing for direct contact with other cell types or compounds. The transwell-based monolayer culture therefore serves as a particularly suitable system to study the crosstalk between IECs and immune cells, myofibroblasts, pathogens, or microbiota [32, 66, 81, 88]. There is also a more complex sandwich culture system described, consisting of a bottom collagen IV coating and collagen I gel overlay with IECs between the two layers [71]. While this approach seems to improve the expansion of human LGR5+ intestinal stem cells, the complexity of the system and the lack of certain mature secretory cell types may limit its application for pathophysiological models. To mimic the architectural organization of the intestine, more sophisticated collagen scaffolds microfabricated with an array of crypt-like invaginations have been developed [27],Y. L. [79]. After seeding and expanding primary, immature IECs, the proliferative monolayer can be converted into fully polarized, mature crypts with the typical basal stem cell niche and luminal differentiated cell zone. Using the micromold-scaffold platforms, a chemical gradient across the crypt array can be generated to study the impact of growth and differentiation factors, inflammatory cues, food, and bacterial metabolites on stemness and differentiation.
Dutton et al. [15] concluded that the proliferation of stem cell monolayers is controlled by a synergistic combination of different factors, including stiffness, porosity, and ECM composition. According to the authors, systems utilizing thick collagen layers are most suitable for long-term monolayer expansion of ISC in vitro. The ability to cultivate and expand ISC has laid the foundation for the development of in vitro 2D models that recapitulate the physiological features of the in vivo intestinal epithelium. Micro-engineered models harbor the potential to become cost-effective and customizable experimental platforms, which can mimic the most relevant physiological properties of the gastrointestinal tract, including tissue architecture, chemical gradients, luminal flow, and mechanical forces, all of which cannot be modeled by a 3D organoid system.
Downstream Analyses
Flow Cytometry / FACS
Jung et al. described a method to isolate stem cells directly from the human colon sample without the need for cell culture [34]. The authors took advantage of the differential expression of EPHB2 (ephrin type-B receptor 2) on the cell surface to isolate different cell types from human colon biopsies. As shown by qRT-PCR, CD45(-)CD11b(-)CD31(-)EpCAM( +)EPHB2(high) sorted cells are characterized by a lower expression of differentiation markers (KRT20, ANPEP, CEACAM7, CA1, FAB2) but higher expression of intestinal stem cells markers (LGR5, ASCL2, OLFM4, EPHB3) and proliferation genes (MKI67, FOXM1, MYC, AURKB) when compared to cells expressing low or medium levels of EPHB2. Furthermore, single EPHB2(high) cells from the human colon could form in vitro spheroids characterized by multilineage differentiation capacity and long-term proliferation [34].
On the other hand, some authors [75]
An alternative approach combines staining for the markers CD44 and CD24 [23] to isolate stem cells from human jejunum-derived patients samples. This method, proposed by Gracz et al., takes advantage of the fact that cells originating from the region between the crypt base to the crypt-villus junctions express CD44, whereas the remaining upper part of the villus does not. A similar expression pattern is observed for CD24 with a main staining distribution along the apical cell membranes. Both markers are also broadly expressed by non-epithelial cells such as the lamina propria, submucosa, and muscle cells, but the staining protocol that combines CD326 (EpCAM) with CD24 and CD44 allows to separate epithelial from non-epithelial cells [23]. The authors demonstrate that the CD24(-)CD44( +) cell population is enriched for the active cycling intestinal stem cells (ISC) markers LGR5 and OLFM4. On the contrary, the cell population expressing both markers CD24( +)CD44( +) is enriched for the reserve/facultative ISC maker HOPX. Subsequently, sorted cells were cultured to assess the functional properties of stemness. CD24(-)CD44(-) and CD24( +)CD44(-) single cells failed to form cystic enterosphere structures while CD24(-)CD44( +) as well as CD24( +)CD44( +) succeeded to do so. Interestingly, after the first week of culture, CD24( +)CD44( +) derived enterospheres exhibited a limited increase in size, but CD24(-)CD44( +) continued to develop. The cell culture efficiency of CD24( +)CD44( +) was even further increased when cells were cocultured with myofibroblasts. Finally, at day 21, enteroids derived from both cell populations contained enteroendocrine cells, goblet cells, Paneth cells, and absorptive enterocytes, demonstrating functional stemness of the originally sorted populations [23].
A more complex FACS staining protocol to identify ISC from human as well as mouse tissue, based on the expression of surface markers, was developed by Wang and colleagues [75]. Analysis of CD44, CD24, CD166, and GRP78/c-kit expression allowed the identification of putative stem cells in the gastrointestinal tract. The authors show that CD44( +)CD24(lo)CD166( +) and GRP78(lo/-) or c-kit identifies putative intestinal (GRP78) or colonic mouse stem cells. In addition, the authors prove that, in principle, a similar gating strategy can be applied to human settings and will have a critical influence on future clinical applications.
OMICS Analysis
The term Omics usually refers to the global profiling of specific molecules in order to extract a meaningful message or pattern by analyzing the large-scale and high dimensional dataset as a whole. In recent years, advances in sequencing technology have allowed the generation of massive omics datasets. This section discusses the most recent technologies to study the multilayer of omics, including genomics, epigenomics, transcriptomics, proteomics, metabolomics, and metagenomics.
High-throughput Sequencing
Next-generation sequencing (NGS) has multiple advantages over traditional sequencing, including multiplexing of samples and parallel sequencing, leading to faster sequencing with lower costs (see Table 5). Nowadays, a complete human genome can be sequenced within a few days for less than $1,000 [59]. Generally, the omics analysis is done in two significant steps, 1) library preparation and 2) sequencing via sequencer platform. Once the biopsy has been digested, as mentioned earlier, the choice of library preparation is based on the used technology for the sequencing. For example, Van et al. used Ion torrent technology to study the genomic alteration in human embryonic stem cells [45]. The Ion Torrent platform is more rapid, direct, and less expensive than other technologies. It is suitable for targeted sequencing since one run could be finished in 2–7 h [42]. Ying et al. used Illumina platforms to compare the differentially expressed genes between embryonic stem cell lines and feed cell lines [85]. Illumina platforms are currently the most used in the NGS field. Also, researchers can use customized panels to detect specific profiles of interest, as Anezeka et al. who studied the allogeneic hematopoietic stem cell transplantation using custom next generation sequencing (NGS) panel to detect the mutations on DNA and these panel sequenced by MiSeq device [26].
The remarkable increase in data output and decrease in cost per base sequenced has been driven primarily by increases in parallelization in short-read sequencing technologies such as the Illumina and Ion Torrent platforms. These short reads usually cause errors and gaps in genome assembly and face many challenges in repetitive regions and structural information of genomic. That was the motivation for the development of third technologies sequencing platforms. Pacific Biosciences single-molecule real-time SMRT technology does not require amplification and offers much longer reads than second-generation sequencing technologies. On the other hand, Oxford Nanopore sequencing technologies do not rely on DNA polymerase like PacBio SMRT, and it can directly sequence RNA and protein. [49] These technologies enabled Wu et al. to study the DNA methylation in mammalian embryonic stem cells [80].
Single-cell RNAseq
For the analysis of tissues, cell dissociation is the most critical step, as the conditions of this step directly affect the molecular profiles of cells, and the impacts of stress and damage depend on the cell type. To measure transcripts in individual cells, reverse transcription (RT) and cDNA amplification must be performed from minimal amounts of RNA [35]. Several methods for the simple procedure of scRNA-seq library construction have been reported [50].
Several protocols based on microdroplet technology have been reported, such as Drop-Seq and DroNc-seq. In these methods, a cell/nucleus, reaction liquid, and a barcoded bead are included in an oil droplet, and RT is conducted with molecular/cell barcoding within each oil droplet. On the other hand, in the microwell-seq approach, a cell and barcoded bead are isolated in a well. Nx1-seq and Seq-Well are reported to be portable, low-cost microwell-based platforms. In addition, this microdroplet- and microwell-based protocols enable the easy handling of thousands of single cells [, 22, 25].
Microdroplet-based systems such as Chromium (10 × Genomics), ddSEQ (Bio-Rad/Illumina), Nadia (Dolomite), and inDrop (1CellBio), and microwell-based systems such as Rhapsody (BD) and ICELL8 (Takara) also exist. Problems such as limited cell capture, low RT efficiency, amplification bias, and the requirement for a large number of sequencing reads remain, depending on the platform. Users should select appropriate methods of scRNA-seq for their sample type and research purpose [24].
Future Directions
Multi-layered Sequencing from the Same Cells
A single omics layer alone cannot sufficiently decipher the complexity of the biological system. The use of individual cells for sequencing prevents the recognition of different omics profiling layers of the same cell. Therefore additional methods, developed to analyze tow or more omics layers from a single cells have been reported e.g., G&T-seq and DR-seq. These methods allowed simultaneous analysis of genomic DNA sequences and mRNA profiles from single cells. They accuracy of the copy number profile and expression profile of these methods are similar to conventional WGA and WTA methods, respectively. ScDam&T-seq also measures both protein–DNA interactions and transcriptome profiles in the same cell and thus couples transcriptional regulation and gene expression analysis in individual cells [, 11, 40].
The Organ on a Chip
The human gut-on-a-chip technology provides an ideal tool to establish a microenvironment that mimics the natural conditions of the human intestine in a small-scale and controllable in vitro platform [6]. This microfluidic-based technology imitates different organs to address the complexity of animal-based models. Organ-on-chips consist of multiple cell types in 3D cell culture, allowing cell interactions similar to tissue. Additionally, peristaltic motion, mechanical forces, and flow of nutrients in fluidic channels recreate the microenvironment seen in the living colon. As a unique feature, the fluidic channels of the chip even offer the possibility to coculture microbes with host cells. As a result, the method can mimic normal organ physiology or, even more importantly, induce disease pathology on the organ level [4, 5].
The development of the first gut-on-a-chip model techniques opens new avenues to study the contributions of a broad range of factors to the pathogenesis of different intestinal diseases and to control these parameters independently. In addition, the ability to co-culture microorganisms [48] with patient-derived gut cells, falls into the field of precision medicine, where patients’ own cells and microbiota are cocultured on the chip. More importantly, in the near future, a patient-derived disease-on-a-chip model will be available to test different therapeutics for the possible identification of individualized treatment strategies.
Data Availability
N/A.
Code Availability
N/A.
References
Aisenbrey, E. A., & Murphy, W. L. (2020). Synthetic alternatives to Matrigel. Nature Reviews Materials, 5(7), 539–551. https://doi.org/10.1038/s41578-020-0199-8
Altay, G., Larranaga, E., Tosi, S., Barriga, F. M., Batlle, E., Fernandez-Majada, V., & Martinez, E. (2019). Self-organized intestinal epithelial monolayers in crypt and villus-like domains show effective barrier function (vol 9, 10140, 2019). Scientific Reports, 9. ARTN 18822. https://doi.org/10.1038/s41598-019-55181-z
Barker, N., van Es, J. H., Kuipers, J., Kujala, P., van den Born, M., Cozijnsen, M., … Clevers, H. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449(7165), 1003-U1001. https://doi.org/10.1038/nature06196
Beaurivage, C., Kanapeckaite, A., Loomans, C., Erdmann, K. S., Stallen, J., & Janssen, R. A. J. (2020). Development of a human primary gut-on-a-chip to model inflammatory processes. Science and Reports, 10(1), 21475. https://doi.org/10.1038/s41598-020-78359-2
Beaurivage, C., Naumovska, E., Chang, Y. X., Elstak, E. D., Nicolas, A., Wouters, H., … Kurek, D. (2019). Development of a Gut-On-A-Chip Model for High Throughput Disease Modeling and Drug Discovery. Int J Mol Sci, 20(22). https://doi.org/10.3390/ijms20225661
Bein, A., Shin, W., Jalili-Firoozinezhad, S., Park, M. H., Sontheimer-Phelps, A., Tovaglieri, A., … Ingber, D. E. (2018). Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell Mol Gastroenterol Hepatol, 5(4), 659-668. https://doi.org/10.1016/j.jcmgh.2017.12.010
Bergenheim, F., Fregni, G., Buchanan, C. F., Riis, L. B., Heulot, M., Touati, J., … Nielsen, O. H. (2020). A fully defined 3D matrix for ex vivo expansion of human colonic organoids from biopsy tissue. Biomaterials, 262. doi:ARTN 120248. https://doi.org/10.1016/j.biomaterials.2020.120248
Boonekamp, K. E., Dayton, T. L., & Clevers, H. (2020). Intestinal organoids as tools for enriching and studying specific and rare cell types: Advances and future directions. Journal of Molecular Cell Biology, 12(8), 562–568. https://doi.org/10.1093/jmcb/mjaa034
Bottcher, A., Buttner, M., Tritschler, S., Sterr, M., Aliluev, A., Oppenlander, L., … Lickert, H. (2021). Non-canonical Wnt/PCP signalling regulates intestinal stem cell lineage priming towards enteroendocrine and Paneth cell fates (vol 23, pg 23, 2021). Nature Cell Biology, 23(5), 566-576. https://doi.org/10.1038/s41556-021-00667-0
Capdevila, C., Calderon, R. I., Bush, E. C., Sheldon-Collins, K., Sims, P. A., & Yan, K. S. (2020). Single-Cell Transcriptional Profiling of the Intestinal Epithelium. Methods in Molecular Biology, 2171, 129–153. https://doi.org/10.1007/978-1-0716-0747-3_8
Dey, S. S., et al. (2015). Integrated genome and transcriptome sequencing of the same cell. Nature Biotechnology, 33(3), 285–289.
Dieterich, W., Neurath, M. F., & Zopf, Y. (2020). Intestinal ex vivo organoid culture reveals altered programmed crypt stem cells in patients with celiac disease. Scientific Reports, 10(1). doi:ARTN 3535. https://doi.org/10.1038/s41598-020-60521-5
Dokladny, K., In, J. G., Kaper, J., & Kovbasnjuk, O. (2021). Human Epithelial Stem Cell-Derived Colonoid Monolayers as a Model to Study Shiga Toxin-Producing Escherichia coli-Host Interactions. Methods in Molecular Biology, 2291, 285–296. https://doi.org/10.1007/978-1-0716-1339-9_13
Dotti, I., Mora-Buch, R., Ferrer-Picon, E., Planell, N., Jung, P., Masamunt, M. C., … Salas, A. (2017). Alterations in the epithelial stem cell compartment could contribute to permanent changes in the mucosa of patients with ulcerative colitis. Gut, 66(12), 2069-2079. https://doi.org/10.1136/gutjnl-2016-312609
Dutton, J. S., Hinman, S. S., Kim, R., Wang, Y. L., & Allbritton, N. L. (2019). Primary Cell-Derived Intestinal Models: Recapitulating Physiology. Trends in Biotechnology, 37(7), 744–760. https://doi.org/10.1016/j.tibtech.2018.12.001
Fatehullah, A., Tan, S. H., & Barker, N. (2016). Organoids as an in vitro model of human development and disease. Nature Cell Biology, 18(3), 246–254. https://doi.org/10.1038/ncb3312
Freire, R., Ingano, L., Serena, G., Cetinbas, M., Anselmo, A., Sapone, A.,… Senger, S. (2019). Human gut derived-organoids provide model to study gluten response and effects of microbiota-derived molecules in celiac disease. Sci Rep, 9(1), 7029. https://doi.org/10.1038/s41598-019-43426-w
Fujii, M., Matano, M., Nanki, K., & Sato, T. (2015). Efficient genetic engineering of human intestinal organoids using electroporation. Nature Protocols, 10(10), 1474–1485. https://doi.org/10.1038/nprot.2015.088
Fujii, M., Matano, M., Toshimitsu, K., Takano, A., Mikami, Y., Nishikori, S., … Sato, T. (2018). Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell, 23(6), 787-+. https://doi.org/10.1016/j.stem.2018.11.016
Fujimichi, Y., Otsuka, K., Tomita, M., & Iwasaki, T. (2019). An Efficient Intestinal Organoid System of Direct Sorting to Evaluate Stem Cell Competition in Vitro. Scientific Reports, 9. doi:ARTN 20297. https://doi.org/10.1038/s41598-019-55824-1
Fuller, M. K., Faulk, D. M., Sundaram, N., Mahe, M. M., Stout, K. M., von Furstenberg, R. J., … Henning, S. J. (2013). Intestinal stem cells remain viable after prolonged tissue storage. Cell Tissue Res, 354(2), 441-450. https://doi.org/10.1007/s00441-013-1674-y
Gierahn, T. M., et al. (2017). Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nature Methods, 14(4), 395–398.
Gracz, A. D., Fuller, M. K., Wang, F., Li, L., Stelzner, M., Dunn, J. C., … Magness, S. T. (2013a). Brief report: CD24 and CD44 mark human intestinal epithelial cell populations with characteristics of active and facultative stem cells. Stem Cells, 31(9), 2024-2030. https://doi.org/10.1002/stem.1391
Han, X., et al. (2018) Mapping the Mouse Cell Atlas by Microwell-Seq. Cell, 172(5), 1091-1107 e17.
Hashimoto, S. (2019). Nx1-Seq (Well Based Single-Cell Analysis System). Advances in Experimental Medicine and Biology, 1129, 51–61.
Heumuller, A., et al. (2020). Clonal hematopoiesis of indeterminate potential in older patients having received an allogeneic stem cell transplantation from young donors. Bone Marrow Transplant, 55(3), 665–668.
Hinman, S. S., Wang, Y. L., Kim, R., & Allbritton, N. L. (2021). In vitro generation of self-renewing human intestinal epithelia over planar and shaped collagen hydrogels. Nature Protocols, 16(1), 352–382. https://doi.org/10.1038/s41596-020-00419-8
Holloway, E. M., Czerwinski, M., Tsai, Y. H., Wu, J. H., Wu, A., Childs, C. J., … Spence, J. R. (2021). Mapping Development of the Human Intestinal Niche at Single-Cell Resolution. Cell Stem Cell, 28(3), 568-+. https://doi.org/10.1016/j.stem.2020.11.008
Hughes, C. S., Postovit, L. M., & Lajoie, G. A. (2010). Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics, 10(9), 1886–1890. https://doi.org/10.1002/pmic.200900758
Hung, Y. H., Huang, S., Dame, M. K., Yu, Q. H., Yu, Q. C., Zeng, Y. A., … Sethupathy, P. (2021). Chromatin regulatory dynamics of early human small intestinal development using a directed differentiation model. Nucleic Acids Research, 49(2). https://doi.org/10.1093/nar/gkaa1204
Hunt, D. R., Klett, K. C., Mascharak, S., Wang, H. Y. Y., Gong, D. N., Lou, J. Z., … Heilshorn, S. C. (2021). Engineered Matrices Enable the Culture of Human Patient-Derived Intestinal Organoids. Advanced Science, 8(10). ARTN 2004705. https://doi.org/10.1002/advs.202004705
In, J. G., Foulke-Abel, J., Clarke, E., & Kovbasnjuk, O. (2019). Human Colonoid Monolayers to Study Interactions Between Pathogens, Commensals, and Host Intestinal Epithelium. Jove-Journal of Visualized Experiments (146). ARTN e59357. https://doi.org/10.3791/59357
Jabaji, Z., Brinkley, G. J., Khalil, H. A., Sears, C. M., Lei, N. Y., Lewis, M., … Dunn, J. C. (2014). Type I collagen as an extracellular matrix for the in vitro growth of human small intestinal epithelium. PLoS One, 9(9), e107814. https://doi.org/10.1371/journal.pone.0107814
Jung, P., Sato, T., Merlos-Suarez, A., Barriga, F. M., Iglesias, M., Rossell, D., … Batlle, E. (2011). Isolation and in vitro expansion of human colonic stem cells. Nat Med, 17(10), 1225-1227. https://doi.org/10.1038/nm.2470
Kashima, Y., Suzuki, A., & Suzuki, Y. (2019). An Informative Approach to Single-Cell Sequencing Analysis. Advances in Experimental Medicine and Biology, 1129, 81–96.
Kozuka, K., He, Y., Koo-Mccoy, S., Kumaraswamy, P., Nie, B., Shaw, K., … Siegel, M. (2017). Development and Characterization of a Human and Mouse Intestinal Epithelial Cell Monolayer Platform. Stem Cell Reports, 9(6), 1976-1990. https://doi.org/10.1016/j.stemcr.2017.10.013
Liu, Y., & Chen, Y. G. (2018). 2D-and 3D-Based Intestinal Stem Cell Cultures for Personalized Medicine. Cells, 7(12). ARTN 225. https://doi.org/10.3390/cells7120225
Liu, Y., Qi, Z., Li, X. T., Du, Y. N., & Chen, Y. G. (2018). Monolayer culture of intestinal epithelium sustains Lgr5(+) intestinal stem cells. Cell Discovery, 4. ARTN 32. https://doi.org/10.1038/s41421-018-0036-z
Luu, L., Johnston, L. J., Derricott, H., Armstrong, S. D., Randle, N., Hartley, C. S., … Coombes, J. L. (2019). An Open-Format Enteroid Culture System for Interrogation of Interactions Between Toxoplasma gondii and the Intestinal Epithelium. Frontiers in Cellular and Infection Microbiology, 9. ARTN 300. https://doi.org/10.3389/fcimb.2019.00300
Macaulay, I. C., et al. (2015). G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nature Methods, 12(6), 519–22.
Mahe, M. M., Sundaram, N., Watson, C. L., Shroyer, N. F., & Helmrath, M. A. (2015). Establishment of Human Epithelial Enteroids and Colonoids from Whole Tissue and Biopsy. Jove-Journal of Visualized Experiments(97). ARTN e52483. https://doi.org/10.3791/52483
Mason, C. E., & Elemento, O. (2012). Faster sequencers, larger datasets, new challenges. Genome Biology, 13(3), 314.
Matsumoto, Y., Koga, H., Takahashi, M., Suda, K., Ochi, T., Seo, S., … Nakamura, T. (2021). Defined serum-free culture of human infant small intestinal organoids with predetermined doses of Wnt3a and R-spondin1 from surgical specimens. Pediatric Surgery International. https://doi.org/10.1007/s00383-021-04957-4
Miyoshi, H., & Stappenbeck, T. S. (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nature Protocols, 8(12), 2471–2482. https://doi.org/10.1038/nprot.2013.153
Nguyen, V., et al. (2016). A Genomic Study of DNA Alteration Events Caused by Ionizing Radiation in Human Embryonic Stem Cells via Next-Generation Sequencing. Stem Cells International, 2016, 1346521.
Ostrop, J., Zwiggelaar, R. T., Pedersen, M. T., Gerbe, F., Bosl, K., Lindholm, H. T., … Oudhoff, M. J. (2021). A Semi-automated Organoid Screening Method Demonstrates Epigenetic Control of Intestinal Epithelial Differentiation. Frontiers in Cell and Developmental Biology, 8. ARTN 618552. https://doi.org/10.3389/fcell.2020.618552
Pleguezuelos-Manzano, C., Puschhof, J., van den Brink, S., Geurts, V., Beumer, J., & Clevers, H. (2020). Establishment and Culture of Human Intestinal Organoids Derived from Adult Stem Cells. 130(1), e106. https://doi.org/10.1002/cpim.106
Poceviciute, R., & Ismagilov, R. F. (2019). Human-gut-microbiome on a chip. Nat Biomed Eng, 3(7), 500–501. https://doi.org/10.1038/s41551-019-0425-0
Quail, M. A., et al. (2012). A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics, 13, 341.
Ramskold, D., et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology, 30(8), 777–82.
Reichard, A., & Asosingh, K. (2019). Best Practices for Preparing a Single Cell Suspension from Solid Tissues for Flow Cytometry. Cytometry Part A, 95a(2), 219–226. https://doi.org/10.1002/cyto.a.23690
Roodsant, T., Navis, M., Aknouch, I., Renes, I. B., van Elburg, R. M., Pajkrt, D., … Muncan, V. (2020). A Human 2D Primary Organoid-Derived Epithelial Monolayer Model to Study Host-Pathogen Interaction in the Small Intestine. Frontiers in Cellular and Infection Microbiology, 10. ARTN 272. https://doi.org/10.3389/fcimb.2020.00272
Rosen, K., Shi, W., Calabretta, B., & Filmus, J. (2002). Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of Fas ligand - A novel mechanism of anoikis of intestinal epithelial cells. Journal of Biological Chemistry, 277(48), 46123–46130. https://doi.org/10.1074/jbc.M207883200
Ross, A. D. B., Perrone, F., Elmentaite, R., Teichmann, S. A., & Zilbauer, M. (2021). Obtaining purified human intestinal epithelia for single-cell analysis and organoid culture. STAR Protoc, 2(2), 100597. https://doi.org/10.1016/j.xpro.2021.100597
Sanman, L. E., Chen, I. W., Bieber, J. M., Thorne, C. A., Wu, L. F., & Altschuler, S. J. (2020). Generation and Quantitative Imaging of Enteroid Monolayers. In P. Ordóñez-Morán (Ed.), Intestinal Stem Cells: Methods and Protocols (pp. 99–113). Springer, US.
Sasaki, N., Sachs, N., Wiebrands, K., Ellenbroek, S. I. J., Fumagalli, A., Lyubimova, A., … Clevers, H. (2016). Reg4(+) deep crypt secretory cells function as epithelial niche for Lgr5(+) stem cells in colon. Proceedings of the National Academy of Sciences of the United States of America, 113(37), E5399-E5407. https://doi.org/10.1073/pnas.1607327113
Sato, T., Stange, D. E., Ferrante, M., Vries, R. G. J., van Es, J. H., van den Brink, S., … Clevers, H. (2011). Long-term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology, 141(5), 1762-1772. https://doi.org/10.1053/j.gastro.2011.07.050
Sato, T., Vries, R. G., Snippert, H. J., van de Wetering, M., Barker, N., Stange, D. E., … Clevers, H. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 459(7244), 262-U147. https://doi.org/10.1038/nature07935
Schwarze, K., et al. (2020). The complete costs of genome sequencing: a microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet Med, 22(1), 85–94.
Scott, A., Rouch, J. D., Jabaji, Z., Khalil, H. A., Solorzano, S., Lewis, M., … Dunn, J. C. Y. (2016). Long-term renewable human intestinal epithelial stem cells as monolayers: A potential for clinical use. Journal of Pediatric Surgery, 51(6), 995-1000. https://doi.org/10.1016/j.jpedsurg.2016.02.074
Serra, D., Mayr, U., Boni, A., Lukonin, I., Rempfler, M., Meylan, L. C., … Liberali, P. (2019). Self-organization and symmetry breaking in intestinal organoid development. Nature, 569(7754), 66-+. https://doi.org/10.1038/s41586-019-1146-y
Shin, W., Wu, A., Min, S., Shin, Y. C., Fleming, R. Y. D., Eckhardt, S. G., & Kim, H. J. (2020). Spatiotemporal Gradient and Instability of Wnt Induce Heterogeneous Growth and Differentiation of Human Intestinal Organoids. iScience, 23(8). ARTN 101372. https://doi.org/10.1016/j.isci.2020.101372
Shokralla, S., Spall, J. L., Gibson, J. F., & Hajibabaei, M. (2012). Next-generation sequencing technologies for environmental DNA research. Molecular Ecology, 21(8), 1794–1805. https://doi.org/10.1111/j.1365-294X.2012.05538.x
Smillie, C. S., Biton, M., Ordovas-Montanes, J., Sullivan, K. M., Burgin, G., Graham, D. B., … Regev, A. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell, 178(3), 714-+. https://doi.org/10.1016/j.cell.2019.06.029
Snyder, J., Wang, C. M., Zhang, A. Q., Li, Y., Luchan, J., Hosic, S., … Koppes, A. (2020). Materials and Microenvironments for Engineering the Intestinal Epithelium. Annals of Biomedical Engineering, 48(7), 1916-1940. https://doi.org/10.1007/s10439-020-02470-8
Staab, J. F., Lemme-Dumit, J. M., Latanich, R., Pasetti, M. F., & Zachos, N. C. (2020). Co-Culture System of Human Enteroids/Colonoids with Innate Immune Cells. Current Protocols in Immunology, 131(1), e113. https://doi.org/10.1002/cpim.113
Sugimoto, S., Fujii, M., & Sato, T. (2020). Organoid Derivation and Orthotopic Xenotransplantation for Studying Human Intestinal Stem Cell Dynamics. In P. Ordóñez-Morán (Ed.), Intestinal Stem Cells: Methods and Protocols (pp. 303–320). Springer, US.
Suzuki, K., Murano, T., Shimizu, H., Ito, G., Nakata, T., Fujii, S., … Okamoto, R. (2018). Single cell analysis of Crohn’s disease patient-derived small intestinal organoids reveals disease activity-dependent modification of stem cell properties. J Gastroenterol, 53(9), 1035-1047. https://doi.org/10.1007/s00535-018-1437-3
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., & Jones, J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science, 282(5391), 1145–1147. https://doi.org/10.1126/science.282.5391.1145
Thorne, C. A., Chen, I. W., Sanman, L. E., Cobb, M. H., Wu, L. F., & Altschuler, S. J. (2018). Enteroid Monolayers Reveal an Autonomous WNT and BMP Circuit Controlling Intestinal Epithelial Growth and Organization. Developmental Cell, 44(5), 624-+. https://doi.org/10.1016/j.devcel.2018.01.024
Tong, Z. X., Martyn, K., Yang, A., Yin, X. L., Mead, B. E., Joshi, N., … Karp, J. M. (2018). Towards a defined ECM and small molecule based monolayer culture system for the expansion of mouse and human intestinal stem cells. Biomaterials, 154, 60-73. https://doi.org/10.1016/j.biomaterials.2017.10.038
Tsai, Y. H., Czerwinski, M., Wu, A., Dame, M. K., Attili, D., Hill, E., … Spence, J. R. (2018). A Method for Cryogenic Preservation of Human Biopsy Specimens and Subsequent Organoid Culture. Cell Mol Gastroenterol Hepatol, 6(2), 218–222 e217. https://doi.org/10.1016/j.jcmgh.2018.04.008
VanDussen, K. L., Sonnek, N. M., & Stappenbeck, T. S. (2019). L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Research, 37. ARTN 101430. https://doi.org/10.1016/j.scr.2019.101430
Villa-Diaz, L. G., Ross, A. M., Lahann, J., & Krebsbach, P. H. (2013). Concise Review: The Evolution of Human Pluripotent Stem Cell Culture: From Feeder Cells to Synthetic Coatings. Stem Cells, 31(1), 1–7. https://doi.org/10.1002/stem.1260
Wang, F., Scoville, D., He, X. C., Mahe, M. M., Box, A., Perry, J. M., … Li, L. (2013). Isolation and characterization of intestinal stem cells based on surface marker combinations and colony-formation assay. Gastroenterology, 145(2), 383–395 e381–321. https://doi.org/10.1053/j.gastro.2013.04.050
Wang, F. C., Scoville, D., He, X. C., Mahe, M. M., Box, A., Perry, J. M., … Li, L. H. (2013). Isolation and Characterization of Intestinal Stem Cells Based on Surface Marker Combinations and Colony-Formation Assay. Gastroenterology, 145(2), 383-+. https://doi.org/10.1053/j.gastro.2013.04.050
Wang, X., Yamamoto, Y., Wilson, L. H., Zhang, T., Howitt, B. E., Farrow, M. A., … Xian, W. (2015). Cloning and variation of ground state intestinal stem cells. Nature, 522(7555), 173-+. https://doi.org/10.1038/nature14484
Wang, Y. L., DiSalvo, M., Gunasekara, D. B., Dutton, J., Proctor, A., Lebhar, M. S., … Allbritton, N. L. (2017). Self-renewing Monolayer of Primary Colonic or Rectal Epithelial Cells. Cellular and Molecular Gastroenterology and Hepatology, 4(1), 165-+. https://doi.org/10.1016/j.jcmgh.2017.02.011
Wang, Y. L., Kim, R., Gunasekara, D. B., Reed, M. I., DiSalvo, M., Nguyen, D. L., … Allbritton, N. L. (2018). Formation of Human Colonic Crypt Array by Application of Chemical Gradients Across a Shaped Epithelial Monolayer. Cellular and Molecular Gastroenterology and Hepatology, 5(2), 113-130. https://doi.org/10.1016/j.jcmgh.2017.10.007
Wu, T. P., et al. (2016). DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature, 532(7599), 329–33.
Yl, Wang, Kim, R., Hwang, S. H. J., Dutton, J., Sims, C. E., & Allbritton, N. L. (2018). Analysis of Interleukin 8 Secretion by a Stem-Cell-Derived Human-Intestinal-Epithelial-Monolayer Platform. Analytical Chemistry, 90(19), 11523–11530. https://doi.org/10.1021/acs.analchem.8b02835
Wang, Y. L., Song, W. L., Wang, J. L., Wang, T., Xiong, X. C., Qi, Z., … Chen, Y. G. (2020). Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. Journal of Experimental Medicine, 217(2). https://doi.org/10.1084/jem.20191130
Wilson, S. S., Mayo, M., Melim, T., Knight, H., Patnaude, L., Wu, X. M., … Terrillon, S. (2021). Optimized Culture Conditions for Improved Growth and Functional Differentiation of Mouse and Human Colon Organoids. Frontiers in Immunology, 11. ARTN 547102. https://doi.org/10.3389/fimmu.2020.547102
Yin, X. L., Farin, H. F., van Es, J. H., Clevers, H., Langer, R., & Karp, J. M. (2014). Niche-independent high-purity cultures of Lgr5(+) intestinal stem cells and their progeny. Nature Methods, 11(1), 106-+. https://doi.org/10.1038/nmeth.2737
Ying, Q. L., et al. (2008). The ground state of embryonic stem cell self-renewal. Nature, 453(7194), 519–23.
Yokota, J., Yamashita, T., Inui, T., Nomoto, R., Kishimoto, W., Nakase, H., & Mizuguchi, H. (2021). Comparison of culture media for human intestinal organoids from the viewpoint of pharmacokinetic studies. Biochemical and Biophysical Research Communications, 566, 115–122. https://doi.org/10.1016/j.bbrc.2021.06.007
Yu, Q. H., Kilik, U., Holloway, E. M., Tsai, Y. H., Harmel, C., Wu, A., … Camp, J. G. (2021). Charting human development using a multi-endodermal organ atlas and organoid models. Cell, 184(12), 3281-+. https://doi.org/10.1016/j.cell.2021.04.028
Zhang, J. B., Hernandez-Gordillo, V., Trapecar, M., Wright, C., Taketani, M., Schneider, K., … Griffith, L. G. (2021). Coculture of primary human colon monolayer with human gut bacteria. Nature Protocols. https://doi.org/10.1038/s41596-021-00562-w
Zhao, X. Y., Li, C., Liu, X. J., Chiu, M. C., Wang, D., Wei, Y. X., …Zhou, J. (2021). Human Intestinal Organoids Recapitulate Enteric Infections of Enterovirus and Coronavirus. Stem Cell Reports, 16(3), 493-504. https://doi.org/10.1016/j.stemcr.2021.02.009
Zwarycz, B., Gracz, A. D., Rivera, K. R., Williamson, I. A., Samsa, L. A., Starmer, J., … Magness, S. T. (2019). IL22 Inhibits Epithelial Stem Cell Expansion in an Ileal Organoid Model. Cellular and Molecular Gastroenterology and Hepatology, 7(1), 1-17. https://doi.org/10.1016/j.jcmgh.2018.06.008
Author information
Authors and Affiliations
Contributions
J.H., Y.M., M.W: made substantial contributions to the conception and design of the work, J.H., Y.M., M.W.: drafted the work, M.W., M.S.: revised the work critically for important intellectual content, all authors.: approved the version to be published;
Corresponding author
Ethics declarations
Ethical Approval
N/A
Consent to Participate
All authors agreed with the content and that all gave explicit consent to submit.
Consent for Publication
Obtained.
Conflicts of Interest
The authors have no financial or proprietary interests in any material discussed in this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Häfliger, J., Morsy, Y., Scharl, M. et al. From Patient Material to New Discoveries: a Methodological Review and Guide for Intestinal Stem Cell Researchers. Stem Cell Rev and Rep 18, 1309–1321 (2022). https://doi.org/10.1007/s12015-021-10307-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-021-10307-7