
Abstract
The aryl hydrocarbon receptor (AHR) is widely expressed in immune and non-immune cells of the gut and its activation has been correlated to the outcome of inflammatory bowel diseases (IBD). In ulcerative colitis and Crohn’s disease, there is an excessive chronic inflammation with massive accumulation of leukocytes in the gut, in an attempt to constrain the invasion of pathogenic microorganisms on the damaged organ. Accordingly, it is known that dietary components, xenobiotics, and some chemicals or metabolites can activate AHR and induce the modulation of inflammatory responses. In fact, the AHR triggering by specific ligands during inflammatory conditions results in decreased IFNγ, IL-6, IL-12, TNF, IL-7, and IL-17, along with reduced microbial translocation and fibrosis in the gut. Moreover, upon AHR activation, there are increased regulatory mechanisms such as IL-10, IL-22, prostaglandin E2, and Foxp3, besides the production of anti-microbial peptides and epithelial repair. Most interestingly, commensal bacteria or their metabolites may also activate this receptor, thus contributing to the restoration of gut normobiosis and homeostasis. In line with that, Lactobacillus reuteri, Lactobacillus bulgaricus, or microbial products such as tryptophan metabolites, indole-3-pyruvic acid, urolithin A, short-chain fatty acids, dihydroxyquinoline, and others may regulate the inflammation by mechanisms dependent on AHR activation. Hence, here we discussed the potential modulatory role of AHR on intestinal inflammation, focused on the reestablishment of homeostasis through the receptor triggering by microbial metabolites. Finally, the development of AHR-based therapies derived from bacteria products could represent an important future alternative for controlling IBD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The aryl hydrocarbon receptor (AHR) that belongs to the family of the basic helix-loop-helix/Per-Arnt-Sim proteins [1] is widely expressed on vertebrate cells [2]. It recognizes and metabolizes a wide range of molecules that include mainly xenobiotic compounds, some chemicals such as 6-formylindolo (3, 2-b) carbazole (FICZ), polycyclic aromatic hydrocarbons like 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [3], and the environmental contaminant dioxin, present in cigarette smoking [4]. After ligand binding, the cytoplasmic AHR translocates to the nucleus, where it triggers the transcription of several genes related to the metabolism of the recognized compounds. There is an activation of the enzymes of cytochrome P450 family 1A1 (CYP1A1) which metabolizes the receptor ligands, permitting a feedback modulation of AHR-induced responses. Then, following receptor triggering, the cells express the P450 enzymes to reduce the ligands’ availability, thus terminating the receptor activation [5].
AHR has been implicated in many immune or inflammatory processes, such as those presented in cardiovascular diseases [6], multiple sclerosis [7], rheumatoid arthritis [8], depression, obesity [9], and allergic responses, among others. Then, there have been some attempts to develop novel treatments for constraining inflammation, focused on AHR activation. For example, a new approach has been used based on a nonsteroidal topical anti-inflammatory drug able to modulate AHR. This drug, named Tapinarof, led to great improvement in atopic dermatitis [10, 11]. In addition, gut bacteria or its metabolites may also trigger AHR responses. In line with that, a recent study demonstrated that the administration of AHR agonist or a Lactobacillus strain able to produce AHR ligands ameliorated alterations related to metabolic syndrome, which was shown to be associated with an impaired capacity of the gut microbiota to metabolize tryptophan into AHR agonists in mice and humans [12].
Despite the toxic effects triggered by its major ligands, it is clear that the receptor plays a relevant role in the modulation of inflammatory responses, especially on the intestinal mucosal surfaces. Indeed, many leukocytes such as macrophages [13], dendritic cells [14], T lymphocytes [15], and innate lymphoid cells [16] express AHR and therefore its activation may significantly influence the ongoing immune reactions, since many genes including those related to cytokine expression, have dioxin-response elements [17, 18]. Hence, studies have focused on the role of AHR ligands in the resolution of inflammatory diseases.
Inflammatory Bowel Diseases and AHR
Inflammatory bowel diseases (IBD) such as Crohn’s disease (CD) or ulcerative colitis (UC) are disabling conditions of the gut, developed as a result of the interaction among loss of tolerance, genetic predisposition, and environmental triggers [19]. In this scenario, there is dysregulation of innate and T cell responses with impaired influx or function of regulatory cells [20], together with a significant local dysbiosis. In the intestine, a large number of microorganisms coexist and participate in the organ development, maturation, and modulation of host responses. This complex and dynamic microbial ecosystem may be modified by genetic factors of the host [21], infections, use of antibiotics, diet, and environmental stimuli [19].
The AHR expression influences the establishment of microbial community in the gut [22]. For example, the ingestion of 2,3,7,8-tetrachlorodibenzofuran (TCDF), which activates the receptor signaling, led to disrupted mice metabolism and altered the microbiota–host homeostasis, including the intestinal bacteria composition [23]. Hence, it is plausible to assume that the interaction between this receptor and intestinal microorganisms interferes in the mucosal immunity regulation. Indeed, AHR plays a protective role in gut inflammation, as described in most studies until now.
Since dysbiosis plays a central role in the development of Crohn’s disease and ulcerative colitis, the manipulation of gut microbiota has been exploited for the development of new therapies for these diseases. Accordingly, we proposed that the downregulation of the gut inflammation could occur by the AHR activation with commensal bacteria or its metabolites, with subsequent recovery of the normobiosis and mucosal homeostasis. Indeed, many probiotics are derived from these microorganisms that inhabit the healthy human gut. Thus, species such as Lactobacillus and Bifidobacterium could represent an important adjuvant (but not substitutive) approach to the conventional therapies, especially for UC [24, 25], while the benefits for CD are still not clear [26, 27]. Therefore, here we raise the discussion on the benefits of activating AHR by bacteria derived from the commensal microbiota and on the modulation of human intestinal inflammation. However, while the effectiveness of these agents seems promising and a wide range of data for mouse intervention can be found, more clinical studies are needed to understand not only their mechanisms of action but also their effects on human gut immunity, especially regulatory responses activated by AHR pathways.
AHR in the Gut Barrier and the Initial Inflammatory Triggers
AHR may play different roles in the inflammatory responses, depending on the cells, tissues, or organs where the receptor is expressed, such as in the gut. The intestinal epithelial barrier is one of the first innate protections against pathogen invasion due to its essential role in gut anatomy. Together with the subjacent immune effectors, it permits the interaction with food antigens, besides the discrimination between invaders and commensal microorganisms that inhabit the gastrointestinal tract [28].
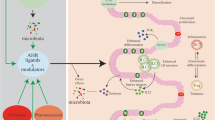
In intestinal epithelial cells (IECs), AHR pathway is required for the development of this population from local stem cells [29]. Also, the lack of this receptor compromises Goblet cells and the mucus production and increases the microbial translocation to other anatomical sites [30]. In experimental colitis, an AHR agonist, β-naphthoflavone, attenuated the disease and reduced the responses of human epithelial colonic cells induced by LPS treatment, indicating that the AHR activation in epithelia may, in fact, represent an important mechanism to regulate gut inflammation [31]. Moreover, in epithelial cells, the AHR ligand FICZ reduced the IL-7 production and ameliorated experimental colitis by decreasing the frequency of activated intraepithelial lymphocytes (IEL) associated with the gut barrier and inflammation development (Fig. 1) [32].

The activation of aryl hydrocarbon receptor (AHR) controls intestinal inflammation. AHR is expressed by leukocytes and non-immune cells of the gut. Its activation by diverse ligands such as xenobiotics and dietary products results in the suppression of inflammatory responses, with reduction of cytokines such as IFNγ, IL-6, IL-12, TNF, IL-7, and Th17 reactions, along with a decrease in microbial translocation and collagen synthesis or fibrosis in the gut. On the contrary, upon AHR triggering, there is an augment in regulatory mechanisms mediated by IL-10, IL-22, prostaglandin E2 (PGE2), and Foxp3 (Tregs), besides anti-microbial peptides and restoration of the epithelial integrity. DC: dendritic cells; ILC: innate lymphoid cells; NK: natural killer; mϴ: macrophages; CD4: CD4 T lymphocytes; CD8: CD8 T lymphocytes; Treg: regulatory T cell; B: B lymphocytes
On the contrary, the lack of AHR or their ligands compromises the epithelial barrier, since this receptor controls the bacterial load, likewise the IEL frequency in the gut [33]. Interestingly, CD8αα+TCRαβ+ IELs become resistant to apoptosis simultaneously to the upregulation of AHR and IL-15 receptor after FICZ treatment in an experimental colitis model. Upon AHR activation, these cells also produced higher amounts of IL-10 and lower IFN-γ [34], indicating a novel pathway to be explored in future development of IBD therapies.
AHR in the Balance of Adaptive Immunity: From Inflammation to Tolerogenic Responses
During experimental gut inflammation, AHR expression is increased in an attempt to constrain the detrimental consequences of colitis. Mice treated with dextran sulfate sodium (DSS) 3% showed augmented expression of AHR than those treated with DSS 2%, which in turn is higher than in healthy mice, like a dose-response expression of the receptor, dependent on the gut inflammation imbalance [31]. Moreover, upon the breakdown of mucosal tolerance, the exacerbated immune response that underlies gut damage is driven by complex interactions between components of the innate and adaptive immune responses including neutrophils, macrophages, T lymphocytes, and inflammatory mediators, such as cytokines and eicosanoids [35], whose production may be altered by AHR activation. Indeed, the mice pretreatment with TCDD was protective against the harmful DSS effects and controlled the intestinal inflammatory reactions by a mechanism dependent on prostaglandin E2 production in the gut [36]. Furthermore, besides lipid mediators, the balance between effector T helper and regulatory cells (Tregs), which usually constrains excessive inflammatory conditions, is essential to determine the homeostasis in the intestinal mucosa and IBD outcome in affected subjects [37].
In general, the AHR activation constrains T cell responses and contributes to the inflammation control [38]. For example, in mice exposed to the colitogenic DSS, the AHR triggering by the dioxin TCDD restored the Th17/Treg ratio by inhibiting Th17 proliferation and inducing Treg differentiation [39]. Similarly, the AHR ligand 3, 3′-diindolylmethane (DIM) alleviated experimental colitis induced by oxazolone through reducing the Th2/Th17 cells and increasing Tregs [3]. In the 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced gut inflammation, the administration of TCDD by gavage, prior to the enema with TNBS, inhibited the response mediated by IL-6, IL-12, IFN-γ, and TNF, besides inducing an increase in Foxp3+ Tregs in the gut (Fig. 1) [18]. Moreover, the AHR agonist 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) was able to suppress effector cells and induce regulation by CD39 and granzyme B in an in vitro approach, along with the amelioration of experimental colitis in a humanized mice model, through reestablishment of immune tolerance in the intestine with Tregs [40]. In accordance, induced Tregs express AHR and this receptor is important to the accumulation of this population as well as its function in the gut (Fig. 1). In fact, the activation of AHR in Tregs protected against experimental T cell-mediated colitis [41]. In line with that, one of the drugs widely used for IBD treatment, mesalamine, plays an anti-inflammatory role in colitis by activating the AHR pathway and inducing Tregs in the colon via activated TGF-β [42]. Thus, a large body of evidences indicates that the AHR activation by ligands could be an alternative approach to the intestinal inflammation control, through the generation of regulatory mechanisms.
The IL-22 cytokine, produced by CD4+ T cells and innate lymphoid cells (ILCs), plays a protective role in gut inflammation as well, mainly by maintaining the integrity of the intestinal epithelium and inducing anti-microbial peptides, which control bacterial translocation in dysbiosis. Considering the relationship between AHR and IL-22, mice injected with the ligand FICZ showed regulation of the excessive inflammation in experimental colitis, by mechanisms dependent on goblet cell differentiation [30] and on protective responses mediated by this cytokine [38]. The AHR activation induced high amounts of IL-22 by CD4+ T cells; meanwhile, the lack of IL-22 producing ILCs and CD4 T cells promoted a more severe colitis in mice exposed to DSS [43, 44]. In addition to the protective role for the epithelium, IL-22 controls Th17 accumulation in the gut, involving AHR activation and local microbiota [45]. Moreover, upon receptor activation with FICZ, there is augmented IL-22 and diminished IFN-γ production by lamina propria cells, thus confirming a counter-regulatory response in IBD, induced by AHR signaling (Fig. 1) [38].
Apart from the Th17-IL-22 axis, the intestinal inflammation in Crohn’s disease develops because of the chronic pathogenic Th1 and IFN-γ responses that mediate excessive inflammation and local tissue injury [46]. This cytokine induces indoleamine 2,3-dioxygenase (IDO1), the enzyme responsible for tryptophan conversion to kynurenine which, in turn, is an endogenous AHR ligand. After the receptor activation by kynurenine, the IL-10R1 is upregulated on IEC. Thus, mice exposed to DSS and treated with exogenous kynurenine presented reduced mucosal inflammation due to an improvement in IL-10 action [47]. When it comes to IBD long-term complications, strictures are one of the main outcomes in some patients presenting fibrosis areas in the gut and requiring surgical intervention, especially in ileal chronic disease. Once AHR activation attenuated the collagen synthesis (Fig. 1), it could be an important target for this chronic inflammatory and fibrotic complication of CD [48]. In a mouse model of intestinal obstruction, AHR triggering by FICZ reduced the intestinal permeability and the epithelial damage, by inhibiting the myosin light chain kinase (MLCK) and the phosphorylated MLC (pMLC) pathway [49]. Likewise, AHR activation by FICZ reduced both the dysfunction of epithelial barrier and the claudin-2 expression, besides maintaining the tissue integrity in cell culture and in vivo studies [50, 51].
On the other hand, some toxins have been linked to the induction of autoimmune diseases, supposedly by DNA epigenetic modifications during developmental exposure (unlike in adult life), resulting in dysregulated immune responses [52]. The correlation between TCDD, an AHR ligand, and autoimmunity is a phenomenon observed mainly in neonatal mice, exposed during mild-gestation or afterbirth [53,54,55,56,57]. This is probably because TCDD induces a disruption in thymic function, which plays the most important activity in early life on T cells selection and on autoreactive clone elimination [53]. Furthermore, TCDD is used in mice experiments as an AHR agonist, for the investigation of the effects of this receptor on immunity. However, in humans, the aim would not be the administration of this dioxin, but the use of protective microbiota and its metabolites, which bind to AHR, for inflammation control.
Therefore, even considering the different roles in inflammation, it is clear that AHR is mainly protective in IBD. Mice deficient for AHR showed a more severe colitis, while those treated with AHR agonist had attenuated disease progression. Nevertheless, though the deficiency of AHR in epithelia usually results in excessive inflammation, the absence of this receptor in T cells may lead to the amelioration of DSS-induced colitis, probably because of the reduced infiltration of Th17 lymphocytes in gut lamina propria [58]. In addition, AHR is essential to the maintenance of the IEL numbers in the intestine, as well as the local bacterial load, which increases in the absence of this receptor, resulting in augmented epithelial damage [33]. Indeed, recent evidences pointed to the importance of AHR ligands and gut bacteria in the modulation of inflammatory responses [59].
AHR Modulation by Intestinal Microbiota
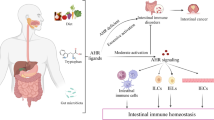
As cited above, some bacteria exert immunoregulatory effects in the intestine, dependent on the activation of the AHR pathway, which culminates in anti-inflammatory responses. Indeed, while the maintenance of gut normobiosis is essential to the regulation of mucosal immunity and homeostasis (Fig. 2) [21, 60, 61], the dysbiosis contributes, undoubtedly, to the dysregulated inflammation. It encompasses the prevalence of pathogenic species which predispose to host diseases frequently caused by effector responses against the altered microbiota, with microorganism translocation in the gut (Fig. 2). However, studies are still necessary to unravel the opposite scenario that involves the direct beneficial effects of bacteria or their metabolites in the regulation of the ongoing immune responses.

Metabolites from gut microbiota may modulate local inflammatory responses and intestinal dysbiosis by AHR activation. In the absence of chronic inflammation, the gut homeostasis is maintained with an equilibrium between commensal and pathogenic microorganisms, in a normobiosis condition (A). Upon local dysbiosis and breakdown of epithelial barrier, there is microbial translocation and triggering of innate and adaptive reactions that amplify the local tissue damage and inflammation (B). Bacteria such as Lactobacillus reuteri, Lactobacillus bulgaricus, or microbial products (tryptophan metabolites, IPA, UroA, SCFA, dihydroxyquinoline and others) may regulate the excessive inflammatory reactions after AHR activation (C) and represent an alternative for future studies aimed at developing novel therapies for Crohn’s disease or ulcerative colitis. IPA: indole-3-pyruvic acid; UroA: urolithin A; SCFA: short-chain fatty acids
The intestinal bacteria represent an important source of products able to trigger AHR [62]. However, the quality of the microbiota is essential to generate AHR ligands. For example, a high-fat diet alters the bacteria components and their capacity to produce some metabolites, such as tryptamine and indole-3-acetate (I3A), both AHR agonists [63]. Moreover, genetic polymorphisms also interfere in the microbial products. CARD9 (caspase recruitment domain family member 9) is necessary to the tryptophan metabolism and IL-22 induction, which protects the intestinal epithelium in colitis, associated with AHR activation. Yet the microbiota (and metabolites) of IBD patients presenting the risk allele of CARD9 does not trigger adequately the AHR molecule, indicating a relationship among intestinal inflammation, CARD9, and AHR activation or the production of its agonists by the host microbiota [64].
The tryptophan metabolites, which ameliorate intestinal inflammation [65], may also be derived from the microbiota and activate the AHR target genes on mouse colonocytes (Fig. 2). In this scenario, the modulation depends on specific agonist or antagonist activities of ligands such as indole-3-acetate, indole-3-aldehyde, indole, and tryptamine. These stimuli may induce different patterns of gene expression after receptor triggering, suggesting a selective AHR modulation by such microbial metabolites [66]. For instance, Lactobacillus reuteri originates indole derivatives from tryptophan metabolism and then activates AHR in CD4+ T cells, which in turn downregulate the transcription factor ThPOK to the induction of CD4+CD8αα+ intraepithelial regulatory lymphocytes [67]. These data indicated that in the presence of tryptophan, a probiotic bacterium is able to mediate the induction of a regulatory profile to control intestinal inflammation through AHR (Fig. 2). In addition, the supplementation of pigs’ diet with tryptophan resulted in increased diversity of the animals’ microbiome, activation of AHR and CYP1A1 in the gut, with IL-8 reduction and improvement of epithelial barrier in the gut. These findings suggested once more an important relationship among tryptophan, microbial metabolism, and gut immunity regulation [68]. However, a novel mechanism of action of Lactobacillus reuteri was recently described in the R2lc and 2010 strains, involving novel identified polyketide synthase (PKS) clusters on the strains’ genome, which are able to trigger AHR in a tryptophan-independent pathway [69]. Regarding other species, Lactobacillus bulgaricus strain OLL1181 ameliorated DSS-colitis by activating AHR signaling (Fig. 2) and inducing the gene expression of the AHR target cytochrome CYP1A1, not only in the gut of treated mice but also in samples of human colon cells [70].
The indole-3-pyruvic acid (IPA), which is a precursor of AHR agonists produced by gut microbiota, can itself activate AHR and control experimental intestinal inflammation (Fig. 2). The oral administration of IPA to SCID mice in the T cell transfer colitis model reduced gut inflammation by inhibiting the expression of IL-12, IFN-γ, TNF, and IL-1β in the intestine, together with an increase in IL-10. Moreover, IPA induced the differentiation and augmented suppressive potential of Tr1 cells, as well as the accumulation of anti-inflammatory dendritic cells in the mesenteric lymph nodes, which was abolished by treatment with an AHR antagonist. These data indicated a relevant modulatory potential for this metabolite on colon inflammation through AHR activation [71]. Likewise, the oral treatment with 1,4-dihydroxy-2-naphthoic acid—DHNA, which is an AHR activator obtained from the cheese bacteria Propionibacterium freudenreichii ET-3, induced anti-microbial peptides such as RegIIIβ and γ in the intestine, and led to the control of inflammation in DSS-colitis [72].
In accordance, several studies have proposed the use of probiotics and prebiotics in the treatment of intestinal inflammation, to reestablish the equilibrium between microbial populations and their products in the gut. Besides a few older studies (ClinicalTrials.gov Identifier: NCT02093767), a recent clinical trial recruiting children and young adults with IBD is being conducted in order to evaluate the effects of the prebiotic inulin in gut bacteria and disease activity (ClinicalTrials.gov Identifier: NCT03653481). The ingestion of fibers is related to a reduced risk for Crohn’s disease [73] and the short-chain fatty acids (SCFAs) butyrate, acetate or propionate, which are derived from bacteria metabolism of ingested fibers, play anti-inflammatory and immunomodulatory activities, by inducing regulatory T cells and constraining cytokines responses in the gut [74, 75]. Moreover, SCFAs act together with AHR ligands to increase the responsiveness and activation of this receptor in gut epithelial cells, a fact that could in theory, potentiate its anti-inflammatory role (Fig. 2) [76]. Importantly, butyrate triggers AHR in human intestinal epithelial cells, indicating once more that metabolites produced by gut microbiota may be an important source of immune-modulatory molecules able to control intestinal inflammation [62].
Apart from the ligands described above, the intestinal microbiota can also metabolize dietary compounds and generate other products that bind AHR, such as urolithin A (UroA). This metabolite has the capacity to reduce IL-6 and TNF production by macrophages and to bind AHR on IECs with further induction of tight junction proteins such as claudin 4, occludin, and zona occludens 1 (ZO1). In fact, mice with experimental colitis have an attenuated disease when treated with UroA (Fig. 2) and, considering that microbial translocation is a hallmark of IBD, the induction of tight junction proteins would be a great advantage on the control of patients’ intestinal inflammation [77]. Furthermore, another microbial derivative such as 2,8 dihydroxyquinoline also plays a role in the AHR activation in human cells [78], thus pointing to an additional important bacteria product with the ability to regulate gut immunity (Fig. 2).
Beyond the potential of individual bacteria species and their metabolites to modulate AHR, the fecal microbiota transplantation (FMT) could also represent a novel interesting approach to achieve intestinal homeostasis, though not fully established yet. It consists of the transference of the intestinal content from a healthy donor to a receptor that is often inflamed, aiming at controlling the gut dysbiosis and restoring the local tolerance with beneficial microbiota. The FMT of normal mice donors to animals with experimental colitis ameliorated the intestinal inflammation, with augmented AHR expression as well as anti-inflammatory cytokines such as IL-10 and TGF-β. There was a later increase in Lactobacillus and Bifidobacterium bacteria in the receptors, together with elevated indole-3-acetic acid (IAA) levels, indicating a link between the AHR activation and microorganisms able to restore the gut normobiosis [79]. Otherwise, the gut microbiota depletion by wide range antibiotic-therapy resulted in the atrophy of intestinal mucosa and decreased production of antimicrobial molecules, which were restored after FMT. The reduction of AHR activation was associated with the diminished antimicrobial peptides, which was rescued by mice treatment with FICZ. These data pointed again to an interplay between gut microbiota and AHR pathway that seemed to be involved in the production of microbicidal molecules relevant to the maintenance of mucosal homeostasis [80].
Conclusions and Future Perspectives
In summary, AHR interacts with endogenous ligands produced by the host, besides a wide range of molecules. Since the signaling through this receptor is altered in IBD patients who usually present intestinal dysbiosis, the putative activation of this pathway could envisage a novel alternative treatment for such pathologies, particularly considering the beneficial effects of certain bacteria metabolites in the gut homeostasis (Fig. 2). Therefore, the future development of AHR-based therapies focused on prebiotics or metabolites derived from probiotic bacteria could represent a novel approach for achieving intestinal health in Crohn’s disease or colitis patients. However, further clinical studies are still necessary to establish the safety and effectiveness of this proposed therapy.
References
Dever DP, Adham ZO, Thompson B, Genestine M, Cherry J, Olschowka JA et al (2016) Aryl hydrocarbon receptor deletion in cerebellar granule neuron precursors impairs neurogenesis. Dev Neurobiol 76(5):533–550. https://doi.org/10.1002/dneu.22330
Kewley RJ, Whitelaw ML, Chapman-Smith A (2004) The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol 36(2):189–204
Huang Z, Jiang Y, Yang Y, Shao J, Sun X, Chen J, Dong L, Zhang J (2013) 3,3′-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Mol Immunol 53(4):335–344. https://doi.org/10.1016/j.molimm.2012.09.007
Rannug U, Rannug A, Sjoberg U, Li H, Westerholm R, Bergman J (1995) Structure elucidation of two tryptophan-derived, high affinity Ah receptor ligands. Chem Biol 2(12):841–845
Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ et al (2017) Feedback control of AHR signalling regulates intestinal immunity. Nature 542(7640):242–245. https://doi.org/10.1038/nature21080
Yi T, Wang J, Zhu K, Tang Y, Huang S, Shui X et al (2018) Aryl hydrocarbon receptor: a new player of pathogenesis and therapy in cardiovascular diseases. Biomed Res Int 2018:6058784. https://doi.org/10.1155/2018/6058784
Zorlu N, Hoffjan S, Haghikia A, Deyneko IV, Epplen JT (2019) Evaluation of variation in genes of the arylhydrocarbon receptor pathway for an association with multiple sclerosis. J Neuroimmunol 334:576979. https://doi.org/10.1016/j.jneuroim.2019.576979
Hui W, Dai Y (2019) Therapeutic potential of aryl hydrocarbon receptor ligands derived from natural products in rheumatoid arthritis. Basic Clin Pharmacol Toxicol. https://doi.org/10.1111/bcpt.13372
Chaves Filho AJM, Lima CNC, Vasconcelos SMM, de Lucena DF, Maes M, Macedo D (2018) IDO chronic immune activation and tryptophan metabolic pathway: a potential pathophysiological link between depression and obesity. Prog Neuro-Psychopharmacol Biol Psychiatry 80(Pt C):234–249. https://doi.org/10.1016/j.pnpbp.2017.04.035
Peppers J, Paller AS, Maeda-Chubachi T, Wu S, Robbins K, Gallagher K, Kraus JE (2019) A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J Am Acad Dermatol 80(1):89–98 e3. https://doi.org/10.1016/j.jaad.2018.06.047
Smith SH, Jayawickreme C, Rickard DJ, Nicodeme E, Bui T, Simmons C, Coquery CM, Neil J, Pryor WM, Mayhew D, Rajpal DK, Creech K, Furst S, Lee J, Wu D, Rastinejad F, Willson TM, Viviani F, Morris DC, Moore JT, Cote-Sierra J (2017) Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J Invest Dermatol 137(10):2110–2119. https://doi.org/10.1016/j.jid.2017.05.004
Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, Michel ML, Chong-Nguyen C, Roussel R, Straube M, Jegou S, McQuitty C, le Gall M, da Costa G, Lecornet E, Michaudel C, Modoux M, Glodt J, Bridonneau C, Sovran B, Dupraz L, Bado A, Richard ML, Langella P, Hansel B, Launay JM, Xavier RJ, Duboc H, Sokol H (2018) Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab 28(5):737–749 e4. https://doi.org/10.1016/j.cmet.2018.07.001
Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, Gutiérrez-Vázquez C, Kenison J, Tjon EC, Barroso A, Vandeventer T, de Lima KA, Rothweiler S, Mayo L, Ghannam S, Zandee S, Healy L, Sherr D, Farez MF, Prat A, Antel J, Reardon DA, Zhang H, Robson SC, Getz G, Weiner HL, Quintana FJ (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 22(5):729–740. https://doi.org/10.1038/s41593-019-0370-y
Meyers JL, Winans B, Kelsaw E, Murthy A, Gerber S, Lawrence BP (2018) Environmental cues received during development shape dendritic cell responses later in life. PLoS One 13(11):e0207007. https://doi.org/10.1371/journal.pone.0207007
Yu H, Jiang L, Liu R, Yang A, Yang X, Wang L, Zhang W, Che T (2019) Association between the ratio of aryl hydrocarbon receptor (AhR) in Th17 cells to AhR in Treg cells and SLE skin lesions. Int Immunopharmacol 69:257–262. https://doi.org/10.1016/j.intimp.2019.01.039
Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF Jr et al (2018) Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity 49(5):915–28 e5. https://doi.org/10.1016/j.immuni.2018.09.015
Mohinta S, Kannan AK, Gowda K, Amin SG, Perdew GH, August A (2015) Differential regulation of Th17 and T regulatory cell differentiation by aryl hydrocarbon receptor dependent xenobiotic response element dependent and independent pathways. Toxicol Sci 145(2):233–243. https://doi.org/10.1093/toxsci/kfv046
Benson JM, Shepherd DM (2011) Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol Sci 120(1):68–78. https://doi.org/10.1093/toxsci/kfq360
Ho SM, Lewis JD, Mayer EA, Plevy SE, Chuang E, Rappaport SM et al (2019) Challenges in IBD research: environmental triggers. Inflamm Bowel Dis 25(Supplement_2):S13–S23. https://doi.org/10.1093/ibd/izz076
Giuffrida P, Cococcia S, Delliponti M, Lenti MV, Di Sabatino A (2019) Controlling gut inflammation by restoring anti-inflammatory pathways in inflammatory bowel disease. Cells 8(5). https://doi.org/10.3390/cells8050397
de Souza PR, Guimaraes FR, Sales-Campos H, Bonfa G, Nardini V, Chica JEL et al (2018) Absence of NOD2 receptor predisposes to intestinal inflammation by a deregulation in the immune response in hosts that are unable to control gut dysbiosis. Immunobiology 223(10):577–585. https://doi.org/10.1016/j.imbio.2018.07.003
Murray IA, Nichols RG, Zhang L, Patterson AD, Perdew GH (2016) Expression of the aryl hydrocarbon receptor contributes to the establishment of intestinal microbial community structure in mice. Sci Rep 6:33969. https://doi.org/10.1038/srep33969
Zhang L, Nichols RG, Correll J, Murray IA, Tanaka N, Smith PB et al (2015) Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ Health Perspect 123(7):679–688. https://doi.org/10.1289/ehp.1409055
Coqueiro AY, Raizel R, Bonvini A, Tirapegui J, Rogero MM (2019) Probiotics for inflammatory bowel diseases: a promising adjuvant treatment. Int J Food Sci Nutr 70(1):20–29. https://doi.org/10.1080/09637486.2018.1477123
Saez-Lara MJ, Gomez-Llorente C, Plaza-Diaz J, Gil A (2015) The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: a systematic review of randomized human clinical trials. Biomed Res Int (2015):505878. https://doi.org/10.1155/2015/505878
Abraham BP, Quigley EMM (2017) Probiotics in inflammatory bowel disease. Gastroenterol Clin N Am 46(4):769–782. https://doi.org/10.1016/j.gtc.2017.08.003
Lichtenstein L, Avni-Biron I, Ben-Bassat O (2016) Probiotics and prebiotics in Crohn’s disease therapies. Best Pract Res Clin Gastroenterol 30(1):81–88. https://doi.org/10.1016/j.bpg.2016.02.002
Lechuga S, Ivanov AI (2017) Disruption of the epithelial barrier during intestinal inflammation: quest for new molecules and mechanisms. Biochim Biophys Acta, Mol Cell Res 1864(7):1183–1194. https://doi.org/10.1016/j.bbamcr.2017.03.007
Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E et al (2018) The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49(2):353–362 e5. https://doi.org/10.1016/j.immuni.2018.07.010
Yin J, Yang K, Zhou C, Xu P, Xiao W, Yang H (2019) Aryl hydrocarbon receptor activation alleviates dextran sodium sulfate-induced colitis through enhancing the differentiation of goblet cells. Biochem Biophys Res Commun 514(1):180–186. https://doi.org/10.1016/j.bbrc.2019.04.136
Furumatsu K, Nishiumi S, Kawano Y, Ooi M, Yoshie T, Shiomi Y, Kutsumi H, Ashida H, Fujii-Kuriyama Y, Azuma T, Yoshida M (2011) A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig Dis Sci 56(9):2532–2544. https://doi.org/10.1007/s10620-011-1643-9
Ji T, Xu C, Sun L, Yu M, Peng K, Qiu Y, Xiao W, Yang H (2015) Aryl hydrocarbon receptor activation down-regulates IL-7 and reduces inflammation in a mouse model of DSS-induced colitis. Dig Dis Sci 60(7):1958–1966. https://doi.org/10.1007/s10620-015-3632-x
Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M (2011) Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147(3):629–640. https://doi.org/10.1016/j.cell.2011.09.025
Chen W, Pu A, Sheng B, Zhang Z, Li L, Liu Z, Wang Q, Li X, Ma Y, Yu M, Sun L, Qiu Y, Yang H (2017) Aryl hydrocarbon receptor activation modulates CD8alphaalpha(+)TCRalphabeta(+) IELs and suppression of colitis manifestations in mice. Biomed Pharmacother 87:127–134. https://doi.org/10.1016/j.biopha.2016.12.061
Hamabata T, Nakamura T, Masuko S, Maeda S, Murata T (2018) Production of lipid mediators across different disease stages of dextran sodium sulfate-induced colitis in mice. J Lipid Res 59(4):586–595. https://doi.org/10.1194/jlr.M079095
Takamura T, Harama D, Matsuoka S, Shimokawa N, Nakamura Y, Okumura K, Ogawa H, Kitamura M, Nakao A (2010) Activation of the aryl hydrocarbon receptor pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunol Cell Biol 88(6):685–689. https://doi.org/10.1038/icb.2010.35
Li J, Ueno A, Iacucci M, Fort Gasia M, Jijon HB, Panaccione R, Kaplan GG, Beck PL, Luider J, Barkema HW, Qian J, Gui X, Ghosh S (2017) Crossover subsets of CD4(+) T lymphocytes in the intestinal lamina propria of patients with Crohn’s disease and ulcerative colitis. Dig Dis Sci 62(9):2357–2368. https://doi.org/10.1007/s10620-017-4596-9
Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L et al (2011) Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 141(1):237–248, 48 e1. https://doi.org/10.1053/j.gastro.2011.04.007
Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS (2011) Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One 6(8):e23522. https://doi.org/10.1371/journal.pone.0023522
Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S et al (2016) AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep 17(5):1318–1329. https://doi.org/10.1016/j.celrep.2016.09.082
Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S et al (2017) The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep 21(8):2277–2290. https://doi.org/10.1016/j.celrep.2017.10.114
Oh-Oka K, Kojima Y, Uchida K, Yoda K, Ishimaru K, Nakajima S et al (2017) Induction of colonic regulatory T cells by mesalamine by activating the aryl hydrocarbon receptor. Cell Mol Gastroenterol Hepatol 4(1):135–151. https://doi.org/10.1016/j.jcmgh.2017.03.010
Parks OB, Pociask DA, Hodzic Z, Kolls JK, Good M (2015) Interleukin-22 signaling in the regulation of intestinal health and disease. Front Cell Dev Biol 3:85. https://doi.org/10.3389/fcell.2015.00085
Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A et al (2014) IL-21 induces IL-22 production in CD4+ T cells. Nat Commun 5:3753. https://doi.org/10.1038/ncomms4753
Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D et al (2013) Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39(2):386–399. https://doi.org/10.1016/j.immuni.2013.08.002
Nizzoli G, Burrello C, Cribiu FM, Lovati G, Ercoli G, Botti F et al (2018) Pathogenicity of in vivo generated intestinal Th17 lymphocytes is IFNgamma dependent. J Crohns Colitis 12(8):981–992. https://doi.org/10.1093/ecco-jcc/jjy051
Lanis JM, Alexeev EE, Curtis VF, Kitzenberg DA, Kao DJ, Battista KD et al (2017) Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol 10(5):1133–1144. https://doi.org/10.1038/mi.2016.133
Monteleone I, Zorzi F, Marafini I, Di Fusco D, Dinallo V, Caruso R et al (2016) Aryl hydrocarbon receptor-driven signals inhibit collagen synthesis in the gut. Eur J Immunol 46(4):1047–1057. https://doi.org/10.1002/eji.201445228
Han B, Sheng B, Zhang Z, Pu A, Yin J, Wang Q, Yang K, Sun L, Yu M, Qiu Y, Xiao W, Yang H (2016) Aryl hydrocarbon receptor activation in intestinal obstruction ameliorates intestinal barrier dysfunction via suppression of MLCK-MLC phosphorylation pathway. Shock 46(3):319–328. https://doi.org/10.1097/SHK.0000000000000594
Ma Y, Wang Q, Yu K, Fan X, Xiao W, Cai Y, Xu P, Yu M, Yang H (2018) 6-Formylindolo(3,2-b)carbazole induced aryl hydrocarbon receptor activation prevents intestinal barrier dysfunction through regulation of claudin-2 expression. Chem Biol Interact 288:83–90. https://doi.org/10.1016/j.cbi.2018.04.020
Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z et al (2018) Aryl hydrocarbon receptor activation modulates intestinal epithelial barrier function by maintaining tight junction integrity. Int J Biol Sci 14(1):69–77. https://doi.org/10.7150/ijbs.22259
Blossom SJ, Gilbert KM (2018) Epigenetic underpinnings of developmental immunotoxicity and autoimmune disease. Curr Opin Toxicol 10:23–30. https://doi.org/10.1016/j.cotox.2017.11.013
Ishimaru N, Takagi A, Kohashi M, Yamada A, Arakaki R, Kanno J et al (2009) Neonatal exposure to low-dose 2,3,7,8-tetrachlorodibenzo-p-dioxin causes autoimmunity due to the disruption of T cell tolerance. J Immunol 182(10):6576–6586. https://doi.org/10.4049/jimmunol.0802289
Mustafa A, Holladay SD, Witonsky S, Sponenberg DP, Karpuzoglu E, Gogal RM Jr (2011) A single mid-gestation exposure to TCDD yields a postnatal autoimmune signature, differing by sex, in early geriatric C57BL/6 mice. Toxicology 290(2–3):156–168. https://doi.org/10.1016/j.tox.2011.08.021
Mustafa A, Holladay S, Witonsky S, Zimmerman K, Manari A, Countermarsh S, Karpuzoglu E, Gogal R (2011) Prenatal TCDD causes persistent modulation of the postnatal immune response, and exacerbates inflammatory disease, in 36-week-old lupus-like autoimmune SNF1 mice. Birth Defects Res B Dev Reprod Toxicol 92(1):82–94. https://doi.org/10.1002/bdrb.20285
Holladay SD, Mustafa A, Gogal RM Jr (2011) Prenatal TCDD in mice increases adult autoimmunity. Reprod Toxicol 31(3):312–318. https://doi.org/10.1016/j.reprotox.2010.08.001
Mustafa A, Holladay SD, Witonsky S, Zimmerman K, Reilly CM, Sponenberg DP et al (2009) Gestational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin disrupts B-cell lymphopoiesis and exacerbates autoimmune disease in 24-week-old SNF1 mice. Toxicol Sci 112(1):133–143. https://doi.org/10.1093/toxsci/kfp177
Chinen I, Nakahama T, Kimura A, Nguyen NT, Takemori H, Kumagai A, Kayama H, Takeda K, Lee S, Hanieh H, Ripley B, Millrine D, Dubey PK, Nyati KK, Fujii-Kuriyama Y, Chowdhury K, Kishimoto T (2015) The aryl hydrocarbon receptor/microRNA-212/132 axis in T cells regulates IL-10 production to maintain intestinal homeostasis. Int Immunol 27(8):405–415. https://doi.org/10.1093/intimm/dxv015
Stedtfeld RD, Chai B, Crawford RB, Stedtfeld TM, Williams MR, Xiangwen S et al (2017) Modulatory influence of segmented filamentous bacteria on transcriptomic response of gnotobiotic mice exposed to TCDD. Front Microbiol 8:1708. https://doi.org/10.3389/fmicb.2017.01708
Ianiro G, Tilg H, Gasbarrini A (2016) Antibiotics as deep modulators of gut microbiota: between good and evil. Gut 65(11):1906–1915. https://doi.org/10.1136/gutjnl-2016-312297
Silva MJ, Carneiro MB, dos Anjos PB, Pereira Silva D, Lopes ME, dos Santos LM (2015) The multifaceted role of commensal microbiota in homeostasis and gastrointestinal diseases. J Immunol Res 2015:321241. https://doi.org/10.1155/2015/321241
Marinelli L, Martin-Gallausiaux C, Bourhis JM, Beguet-Crespel F, Blottiere HM, Lapaque N (2019) Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci Rep 9(1):643. https://doi.org/10.1038/s41598-018-37019-2
Krishnan S, Ding Y, Saedi N, Choi M, Sridharan GV, Sherr DH et al (2018) Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep 23(4):1099–1111. https://doi.org/10.1016/j.celrep.2018.03.109
Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G et al (2016) CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 22(6):598–605. https://doi.org/10.1038/nm.4102
Islam J, Sato S, Watanabe K, Watanabe T, Ardiansyah HK et al (2017) Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J Nutr Biochem 42:43–50. https://doi.org/10.1016/j.jnutbio.2016.12.019
Cheng Y, Jin UH, Allred CD, Jayaraman A, Chapkin RS, Safe S (2015) Aryl hydrocarbon receptor activity of tryptophan metabolites in young adult mouse colonocytes. Drug Metab Dispos 43(10):1536–1543. https://doi.org/10.1124/dmd.115.063677
Cervantes-Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J et al (2017) Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 357(6353):806–810. https://doi.org/10.1126/science.aah5825
Liang H, Dai Z, Liu N, Ji Y, Chen J, Zhang Y et al (2018) Dietary L-tryptophan modulates the structural and functional composition of the intestinal microbiome in weaned piglets. Front Microbiol 9:1736. https://doi.org/10.3389/fmicb.2018.01736
Ozcam M, Tocmo R, Oh JH, Afrazi A, Mezrich JD, Roos S et al (2019) Gut symbionts Lactobacillus reuteri R2lc and 2010 encode a polyketide synthase cluster that activates the mammalian aryl hydrocarbon receptor. Appl Environ Microbiol 85(10). https://doi.org/10.1128/AEM.01661-18
Takamura T, Harama D, Fukumoto S, Nakamura Y, Shimokawa N, Ishimaru K et al (2011) Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol Cell Biol 89(7):817–822. https://doi.org/10.1038/icb.2010.165
Aoki R, Aoki-Yoshida A, Suzuki C, Takayama Y (2018) Indole-3-pyruvic acid, an aryl hydrocarbon receptor activator, suppresses experimental colitis in mice. J Immunol 201(12):3683–3693. https://doi.org/10.4049/jimmunol.1701734
Fukumoto S, Toshimitsu T, Matsuoka S, Maruyama A, Oh-Oka K, Takamura T, Nakamura Y, Ishimaru K, Fujii-Kuriyama Y, Ikegami S, Itou H, Nakao A (2014) Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol Cell Biol 92(5):460–465. https://doi.org/10.1038/icb.2014.2
Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Korzenik JR et al (2013) A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis. Gastroenterology 145(5):970–977. https://doi.org/10.1053/j.gastro.2013.07.050
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, de Roos P et al (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504(7480):451–455. https://doi.org/10.1038/nature12726
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM et al (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341(6145):569–573. https://doi.org/10.1126/science.1241165
Jin UH, Cheng Y, Park H, Davidson LA, Callaway ES, Chapkin RS et al (2017) Short chain fatty acids enhance aryl hydrocarbon (Ah) responsiveness in mouse colonocytes and Caco-2 human colon cancer cells. Sci Rep 7(1):10163. https://doi.org/10.1038/s41598-017-10824-x
Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG et al (2019) Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun 10(1):89. https://doi.org/10.1038/s41467-018-07859-7
Hubbard TD, Liu Q, Murray IA, Dong F, Miller C 3rd, Smith PB et al (2019) Microbiota metabolism promotes synthesis of the human ah receptor agonist 2,8-dihydroxyquinoline. J Proteome Res 18(4):1715–1724. https://doi.org/10.1021/acs.jproteome.8b00946
Wei YL, Chen YQ, Gong H, Li N, Wu KQ, Hu W et al (2018) Fecal microbiota transplantation ameliorates experimentally induced colitis in mice by upregulating AhR. Front Microbiol 9:1921. https://doi.org/10.3389/fmicb.2018.01921
Wang J, Wang P, Tian H, Tian F, Zhang Y, Zhang L, Gao X, Wang X (2018) Aryl hydrocarbon receptor/IL-22/Stat3 signaling pathway is involved in the modulation of intestinal mucosa antimicrobial molecules by commensal microbiota in mice. Innate Immunol 24(5):297–306. https://doi.org/10.1177/1753425918785016
Acknowledgments
We would like to acknowledge the financial agencies and the Biosciences and Biotechnology Post Graduation Program from the Faculty of Pharmaceutical Sciences of Ribeirão Preto at the University of São Paulo that supported this work.
Funding
This work was supported by CAPES financial code 001, CNPq (310174/2016-3) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2017/08651.1).
Author information
Authors and Affiliations
Contributions
LP designed and wrote the review; MDS wrote and discussed the work; CRBC wrote, discussed, and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pernomian, L., Duarte-Silva, M. & de Barros Cardoso, C.R. The Aryl Hydrocarbon Receptor (AHR) as a Potential Target for the Control of Intestinal Inflammation: Insights from an Immune and Bacteria Sensor Receptor. Clinic Rev Allerg Immunol 59, 382–390 (2020). https://doi.org/10.1007/s12016-020-08789-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-020-08789-3