
Abstract
There is an ongoing shift in demographics such that older persons will outnumber young persons in the coming years, and with it age-associated tissue attrition and increased diseases and disorders. There has been increased information on the association of the aging process with dysregulation of hematopoietic stem (HSC) and progenitor (HPC) cells, and hematopoiesis. This review provides an extensive up-to date summary on the literature of aged hematopoiesis and HSCs placed in context of potential artifacts of the collection and processing procedure, that may not be totally representative of the status of HSCs in their in vivo bone marrow microenvironment, and what the implications of this are for understanding aged hematopoiesis. This review covers a number of interactive areas, many of which have not been adequately explored. There are still many unknowns and mechanistic insights to be elucidated to better understand effects of aging on the hematopoietic system, efforts that will take multidisciplinary approaches, and that could lead to means to ameliorate at least some of the dysregulation of HSCs and HPCs associated with the aging process.

Graphical Abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
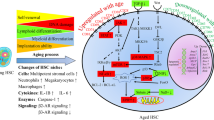
Aging is an inevitable process if one lives long enough. There is an ongoing shift in demographics such that older persons will out number young persons in the coming years, and with it age-associated tissue attrition and increased diseases and disorders. There has been an increased influx in literature on the association of the aging process with dysregulation of hematopoietic stem (HSC) and progenitor (HPC) cells, and hematopoiesis. Most such hematopoietic aging studies have been carried out in mice, where it has been reported that the aging process (in this case mice in the range of 2 years old) compared to that of younger mice is associated with increased absolute numbers of phenotypically defined HSCs identified by cell surface antigens in the bone marrow (BM). Yet, the functional capacities of these increased numbers of HSCs are grossly deficient in their engrafting capability in competitive and non-competitive HSC transplants in lethally irradiated mice. Moreover, the differentiation capacity of the engrafted donor BM cells from the old mice is different from that of young donor BM cells; there is a shift in the lymphoid/myeloid ratio of engrafting cells such that the older donor BM cells manifest greater numbers of myeloid to lymphoid cell output. This is the opposite of that of young engrafting mouse BM HSCs. How informative this and other information on aged hematopoiesis is remains to be determined by further investigation. Recent work from our laboratory [1] has observed that at least some of the abnormalities of HSCs from old mice may be more of an artifact of the collection and processing of mouse BM cells, rather than how they manifest their numbers and functional capacities in vivo.
This review provides an extensive, although not necessarily complete, summary on the literature of aged hematopoiesis, HSCs and HPCs. When placed in context of potential artifacts of the collection and processing procedure, that may not be totally representative of the status of HSCs in the in vivo microenvironment of BM, the site in which HSCs, HPCs, and hematopoiesis are nurtured for self-renewal, proliferation, survival, and differentiation some of what we know may have to be re-evaluated. This review encompasses the following sections: A) Aging and Stem Cells in General, B) Age-Related Changes in HSCs/HPCs and Hematopoiesis, C) Age-Related Clonal Hematopoiesis of Indeterminant Potential (CHIP) and Inflammation, D) DNA Damage, Transcriptional and Epigenetic Changes During Aging, E) Metabolic Processes, Mitochondria and Reactive Oxygen Species (ROS) During Aging, F) Apoptosis, Autophagy, Radiation and a Role for the Sirtuin Family of Proteins During Aging, G) The Microbiome, Hematopoiesis, and Aging, H) Additional Age-Related Information, The Microenvironment, Exosomes, Leptin (Lep) and Leptin Receptors (R), and Means to Better Evaluate and Understand Hematopoiesis During Aging in part in context of our recent studies [1], I) COVID-19, SARS-CoV-2, Aging and Hematopoiesis, and J) Conclusions in Context of Potential Future Interventions for Better Health of the Hematopoietic System During Aging. One of the authors (HEB) of this review had an interest in Gerontology, the study of aging, over 50 years ago, but it is only most recently, that he, his lab members, and collaborators have been involved in actual experiments in this area, having previously focused on the regulation of hematopoiesis in the young.Footnote 1
A) Aging and Stem Cells in General
It has been suggested that aging is not caused by active gene programming, but that it rather evolved through limitations in maintenance of somatic cells in which there was a build up of damage [2], which in fact is associated with gene mutations that affect endocrine signaling, stress responses, metabolism and telomere length [3]. Thus, aging is believed to entail “damage” due to multiple mechanisms, some information of which may possibly be used to slow some of the “damage” during aging for healthier outcome. Covered in these papers [2, 3] are the areas of: why aging occurs, is it programmed, how does evolutionary genetics and physiology fit into these processes, how aging is “caused” in terms of molecular mechanisms, mitochondria, and network themes. Whether we yet know enough about the aging process of cells, their organelles, and organisms is still open for debate, although more insight into problems and causes associated with aging could provide the means to potentially intervene at least partially in the future.
Human aging is associated with a number of diseases and defects including the heart, muscle wasting, osteoporosis, and in some cases mental deterioration [4]. Senescent cells and their accumulating damage can contribute to aging, through a number of intracellular signaling pathways including the p53 and RB tumor suppressors, and the influences of neighboring cells in the environment [5]. These, and a number of other genetic pathways have been implicated in aging [6], including nutrient sensing pathways. Over thirty genetic mutations have been reported to extend the lifespan of mice, and a number of genes have been associated in genome wide association studies with longevity of humans [6]. Ames Dwarf mice which harbor a spontaneous mutation in the Prop1df gene resulting in the lack of growth hormone, prolactin, and thyroid-stimulating hormone are known to live close to two times the lifespan of other mouse strains [7,8,9,10], a situation mimicking certain conditions in humans. Why we age has been commented on from an evolutionary point of view [11]. While somatic cells have a limited lifespan, the lifespan of stem cells, which have the property of making more of themselves (self-renewal) and being able under the appropriate stimuli for differentiation to more mature cell types has not yet been conclusively defined, although transcriptional fingerprinting and other pathway analyses suggest that stem cells do themselves age as one gets older [12, 13].
B) Age-Related Changes in HSCs/HPCs and Hematopoiesis
A number of articles and reviews appeared in this area of research since 2005 [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37] which will be mainly described in chronological order so that the reader can see what research was reported first and then subsequently, as not all research findings may agree. It was reported that the aging of long-term (LT) HSCs was associated with autonomous changes that increased the self-renewal of these cells, but that these HSCs manifested decreased potential for lymphoid cell differentiation and production [14]. This was associated with down-modulation of genes that mediate lymphoid specification, and up-modulation of myeloid fate decisions and functions [14]. Competitive transplants were done using the congenic CD45.1/CD45.2 mouse system with relatively purified populations of donor HSCs to assess engraftment, and self-renewal was estimated by secondary transplants. This paper [14] did not note the decreased engrafting capacity of HSCs from BM of older mice that most of the other numerous publications in this area have reported, and lacked detailed month by month chimerism data comparing engraftment of old vs. young mice; nor did it quantitate numbers of functional HSCs using limiting dilution analysis to calculate competitive repopulating units (CRUs, a measure of the numbers of functional HSCs [38]. Age-related defects in lymphoid-bias [15] and B-lymphopoiesis [16] have been reported by others, and have been suggested to underlie the dominance of myeloid cells in adult leukemia [17]. In order for HSCs to engraft, they must first home to the BM after IV injection. This process of homing for BM cells of old mouse CRUs was about 3-fold lower than the homing efficiency of CRUs from young mice [18], hence one potential reason for decreased engrafting capability noted by others. Of some interest, although not completely understood, the ability to mobilize HSCs from old mice with G-CSF to the blood was increased compared to that of G-CSF mobilization of HSCs from younger mice [19]. This correlated with a reduced adhesion capacity of an immature cell population (not a purified HSC population) to stromal cells, and with increased activation of Cdc42, a small RhoGTPase. This work has not yet been reproduced to the knowledge of the authors of this review, and more rigorous analysis is needed to fully understand this interesting phenomenon. What has not been defined yet is the mobilization of lymphoid vs. myeloid-biased HSCs in old vs. young mice. It will also be of interest to assess the mobilizing capacity of bonified HSCs to the combination of G-CSF plus AMD3100 (Plerixafor), as G-CSF and AMD3100 synergize to mobilize HSCs and HPCs from young mice [39].
Other reviews have noted age-related changes in hematopoiesis of old vs. young mice [20, 21], with one short report [22] not seeing differences in engraftment of sorted populations of HPCs from elderly (>70 years old vs. young) human BM in immune deficient NSG mice. This clearly needs more rigorous investigation in terms of numbers and engrafting capability of rigorously purified populations of functional human BM HSCs (not HPCs) from old and young donors.
While an earlier report [14] suggested increased self-renewal of HSCs from BM of old mice, a later report by others with more in-depth analysis demonstrated that HSCs from the BM of old mice manifested significantly reduced self-renewal in secondary transplants using highly purified populations of LT-HSCs [23] for both primary and secondary engraftment. They [23] as did others [18] showed decreased homing efficiency of HSCs from the BM of old mice. Moreover, they [23] showed significantly delayed proliferative responses of old vs. young BM HSCs.
What is clear is that all studies thus far that have assessed old vs. young BM engrafting HSCs have shown a bias of the myeloid vs. lymphoid production capability of HSCs from old mice [14, 16,17,18, 20,21,22,23,24]. Whether this apparent bias of donor HSCs from old mice might be due to potential artifacts in how donor cells were collected, processed, and injected into recipient mice [1] will be discussed in Section H.
The impact of hematopoiesis in aging primates was investigated by clonal tracking in which clonal output of thousands of genetically barcoded HSCs and HPCs was determined in old vs. young macaques after autologous transplantation [25, 26]. Delayed output from multipotent clones was observed in old macaques with persistence of lineage biased clones noted; in contrast to aging studies in mice which showed persistence of myeloid-biased clones with old age, there was persistent output from both B-lymphoid- and myeloid-biased clones. Whether or not macaque vs. mouse differences were due to aging differences between species requires further investigation as these studies [25] were based on only two old macaques 18 and 25 years of age, which were considered “aged” on their lifespans of captivity of 20-30 years.
The multipotential progenitor (MPP) cell compartment is a composite of 4 different cell types, with the MPP4 compartment being considered to be lymphoid-primed [27]. A yet to be understood observation in context of lymphoid-biased aging studies is the progressive loss and increased cycling of the MPP4 population with aging; other cells and factors may be involved in lymphoid-biased output from engrafted aged HSCs.
Two intriguing reviews on HSC aging are entitled: “The slippery slope of hematopoietic stem cell aging” [30], and “Age-related clonal hematopoiesis: Stem cells tempting the devil” [29]. The latter review touches on clonal hematopoiesis of indeterminate potential (CHIP), an area that will be covered in detail in Section C, and is associated with increased risk of hematological cancers, as well as that of the mortality associated with cardiovascular problems. A number of other more recent reviews are worth noting including: “Aging of hematopoietic stem cells” [31], “Anemia at older age: etiologies, clinical implications and management” [32], “Aged murine stem cells drive aging-associated immune remodeling” [33], “The global complexity of the murine blood system declines throughout life and after serial transplantation” [34], “Hematopoietic stem cells aging, life span and transplantation” [35], “The ageing hematopoietic stem cell compartment” [36], and “Relationships between aging and hematopoietic cell transplantation” [37]. All these reviews suggest that intervention in age-related dysfunction of HSCs may be possible, in part by targeting selected intracellular regulatory pathways. We suggest in Section H, the potential use based on studies in mice of HSCs and HPCs from older individuals for efficient hematopoietic cell transplantation (HCT), if the cells are more appropriately collected and processed under conditions that maintain their in vivo numbers and functional characteristics [40,41,42]. How these cells are collected may be crucial, as cells are currently collected in almost all mouse studies, except those noted by us [40,41,42], and in all human studies, in ambient air (~21% O2 tension). Collection of cells in ambient air subjects them to a phenomenon which we termed Extra Physiological Oxygen Shock Stress (EPHOSS) [40, 41]. Ambient air causes the very rapid loss of HSCs and a concomitant increase in numbers of HPCs due to EPHOSS-induced differentiation of HSCs [40]. This differentiation process during collection of cells in air occurs within minutes, and may likely be needed to be considered in interpretation of at least some of the published information presented. This may require some re-evaluation of past studies to ensure that studies accurately describe the situation of numbers and functions of these cells as when they are present in their BM microenvironment, before removal for collection and analysis, as we reported in [1] and discuss in Section H.
C) Age-Related Clonal Hematopoiesis of Indeterminant Potential (CHIP) and Inflammation
Age-Related Clonal Hematopoiesis
CHIP, also known as age-related clonal hematopoiesis (ARCH), is characterized by expansion of somatic mutations in various hematopoietic lineages of older persons and is associated with risks of developing leukemia [43], as well as other age-associated disorders including cardiovascular disease [44]. Human aging is associated with an exponential increase in the occurrence of CHIP in aged individuals. It is an emerging public health issue that affects at least 15-20% of individuals aged 70 or above [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. A number of reviews and reports on clonal hematopoiesis have been published [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. This is currently a heavily researched area of investigation, with the causes still relatively unknown. Clarity is needed on why some cells with mutations, likely involving and caused by several factors [57] noted in the below Sections, persist and/or expand with resultant disorders such as leukemias, myelodysplasias, and cancers associated with aging individuals.
The vast majority of the mutations identified in CHIP are dispersed across the genome. However, five genes, including DNMT3A, TET2, ASXL1, JAK2, and TP53, have high numbers of somatic mutations [54,55,56,57,58,59]. The most common base-pair change in the somatic variants identified in CHIP was a cytosine to thymine (C to T) transition, a somatic mutational signature of aging [54,55,56]. CHIP is an age-dependent risk factor for both hematological malignancies and cardiovascular disease [53,54,55,56,57,58,59]. Thus, preventing CHIP progression may prove to be beneficial for human health. However, mechanisms by which somatic mutations in HSCs and other blood cells contribute to the pathogenesis of age-related diseases are largely unknown.
Clinical studies revealed that hematopoietic clones harboring specific mutations in individuals with CHIP may expand over time [54,55,56,57,58]. However, how different cellular stressors affect clonal expansion is largely unknown. Recently, three different stressors, including hematopoietic transplantation, cytotoxic therapy and inflammation, have been shown to expand hematopoietic clones. TP53 mutations identified in CHIP confer a competitive advantage to HSCs and HPCs following transplantation through modulating epigenetic pathways [52]. Considering that common mutations identified in CHIP affect epigenetic modulators, including DNMT3A, ASXL1, and TET2, these findings underscore the importance of dysregulated epigenetic control in CHIP development.
PPM1D is a phosphatase that negatively regulates p53 and several proteins involved in the DNA damage response (DDR) pathway [60]. Recently, PPM1D mutations were found in CHIP [54,55,56,57,58]. PPM1D mutations result in the expansion of PPM1D-mutant hematopoietic cells following chemotherapy treatment. However, they do not confer competitive advantage to HSCs and HPCs following bone marrow transplantation [61, 62]. TP53 mutations are associated with prior exposure to chemotherapy [63]. Genotoxic stresses selectively expand TP53-mutant HSPCs [50, 64]. While both p53 and PPMID are involved in the DDR pathway, they appear to play distinct roles in promoting of HSCs and HPCs expansion.
The Effects of Chronic, Low-Grade Inflammation Associated with Aging
During aging, chronic and low-grade inflammation - inflammaging - develops, which contributes to the pathogenesis of age-related diseases [65, 66]. Aberrant innate immune activation and pro-inflammatory signaling within the malignant clone and the BM microenvironment have been identified as key pathogenic drivers of myelodysplastic syndrome (MDS), an age-related disease [67]. Mutations identified in CHIP may utilize cell extrinsic mechanisms to promote clonal hematopoiesis. For example, TET2-deficient macrophages exhibit an increased in NLRP3 inflammasome-mediated interleukin-1β secretion [68]. Inflammasomes are multiprotein complexes that activate Caspase-1 and increase the release of pro-inflammatory cytokines such as IL-1β, leading to caspase-1-dependent death, known as pyroptosis [69]. HSCs and HPCs from low to high-risk human patients with MDS manifest activated NLRP3 inflammasome [70]. NLRP1 inflammasome activation increases IL-1β secretion that inhibits wild-type HSPC function through inducing pyroptosis [71]. The NLRP1 inflammasome, but not the NLRP3 inflammasome, is specifically activated in p53 mutant HSPCs, leading to increased secretion of IL-1β, which induces pyroptosis of wild-type HSPCs in a paracrine fashion (YL and HEB, unpublished data). Tet2-deficient hematopoietic stem and progenitor cells manifest a hyperactive IL-6 pathway, which promotes cell survival under basal conditions and in response to inflammatory stress. Inhibiting inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis [72].
Splicing of pre-mRNAs by the spliceosome plays a key role in tissue development [73, 74]. Genome wide splicing analysis revealed an increased number of spliced genes during aging [75, 76]. Changes in spliceosome gene expression and alterations in pre-mRNA splicing are associated with lifespan in mice and humans [77]. Notably, both human and mouse HSPCs display dysregulated pre-mRNA splicing with age [78, 79]. Further, spliceosome gene mutations, including SF3B1, SRSF2 and U2AF1, were frequently found in CHIP and MDS [54,55,56,57, 80,81,82], implicating that aberrant splicing in hematopoietic cells may contribute to CHIP and pathogenesis of MDS. Although both SRSF2 and SF3B1 mutations alter mRNA splicing, these mutations functionally converge with hyperactivation of NF-κB, a key mediator of the inflammatory response [83]. These findings underscore the importance of chronic inflammation in promoting CHIP development during aging.
Inflammation
Inflammation is a double-edged sword in hematopoiesis and disease. The hematopoietic system gives rise to the immune cells of the body and is, therefore, closely linked to inflammation. Even at early stages of hematopoietic development, inflammatory cytokines, such as interleukin-1 (IL-1), interferon-γ (IFN-γ), tumor necrosis factor (TNF), and granulocyte colony stimulating factor (G-CSF), play critical roles in the specification of hematopoietic stem cells (HSCs) [84]. Remarkably, different levels and combinations of inflammatory signaling molecules can elicit opposing responses, suggesting that context may be significant. For example, while IFN-γ and TNF are linked to bone marrow failure and decreased self-renewal capacity in adults, they enable hematopoiesis during development [84]. Under normal conditions in the mature hematopoietic system, cytokines influence HSC proliferation, differentiation, and self-renewal [84]. IFN-γ, IL-3, and IL-1 can influence the differentiation of HSCs toward myeloid lineages by activating myeloid transcription factors [84]. These same inflammatory factors stimulate hematopoiesis to support the immune system during infection and injury in a process called emergency granulopoiesis [85]. This process leads to the expansion of myeloid cells, which serve as the first line of defense against foreign pathogens [85]. These mechanisms rely on inflammatory mediators and are essential for maintaining homeostasis.
While inflammation enables hematopoietic development and stimulation of HSCs during illness and injury, it also contributes to pathogenesis and disease progression. During infection, inflammatory signals, such as IFN-γ and IL-27, trigger the proliferation and differentiation of HSCs to bolster the immune response either by acting directly on HSCs or indirectly via mature hematopoietic cells, endothelial cells, or the bone marrow microenvironment [86,87,88,89,90]. However, prolonged bacterial or viral infection can hinder self-renewal and competitive repopulation capacity, leading to HSC depletion [91, 92]. This exhaustion of HSCs during chronic inflammation may be attributed to increased myeloid differentiation [93]. Notably, dysregulation of the myeloid cell compartment also occurs in patients with severe COVID-19 [94]. This inflammation-mediated HSC dysfunction may occur via the TLR4-TRIF-ROS-p38 signaling pathway rather than Myd88 signaling, suggesting that the mechanism underlying chronic inflammation may be distinct from that of emergency granulopoiesis [91]. In the context of sepsis, activation of Myd88 caused myelosuppression without significant effects on HSCs, whereas activation of TRIF strongly inhibited HSC self-renewal without direct effects on myeloid cells, inferring cell type-specific effects of these inflammatory mechanisms [95]. Targeting these two pathways may have therapeutic value. While foreign infections can alter hematopoiesis by triggering inflammation, the normal microbiome can also influence the hematopoietic system. See more on this in Section G. Antibiotic-treated mice exhibit depletion of HSCs and progenitor cells as well as anemia, thrombocytosis, pan-lymphopenia, and leukopenia [96]. The complexity of the intestinal microbiome regulates the size of the myeloid cell population in the bone marrow via Myd88 signaling [97]. The disruption of these interactions may have implications for the potential contribution of infection to the progression of preleukemic conditions to hematological disease. For example, disruption of the intestinal barrier promotes myeloproliferation in mice lacking the preleukemic gene Tet2, whereas germ-free Tet2-deficient mice do not exhibit myeloproliferation [98]. Germline Tet2 loss of function is associated with immunodeficiency and lymphoma in children [99, 100]. Importantly, myeloproliferation was alleviated in Tet2-deficient mice with loss of intestinal integrity by treatment with antibiotics [98, 101]. Similarly, bacterial signals cause the expansion of HSCs lacking Tet2 and induce the production of IL-6 from HSCs, bolstering the role of infection and inflammation in the pathogenesis of hematological malignancy [102]. A link to these effects in aged animals remains to be better elucidated, with effects of bacteria on tumor progression and metastasis covered in Section I.
Remarkably, many of the same inflammatory pathways that guide hematopoietic development and strengthen the immune system under normal conditions can also drive leukemia in the context of infection or other inflammatory conditions. For example, IL-6 facilitates the development of chronic myelogenous leukemia in mice, and IL-33 contributes to myeloproliferative neoplasms by altering myelopoiesis [103, 104]. In addition, inflammation can cause genotoxic stress, the accumulation of mutations, and the progression of preleukemic conditions to leukemia [105, 106]. An unexplored area is the elucidation of the factors and events responsible for the transition from the normal inflammatory response to the deleterious inflammation that can promote hematological disease. As during hematopoietic development, the context of the inflammatory response may be important in shaping disease outcomes. For example, chronic IL-1 signaling can reduce HSC self-renewal, limit hematopoietic lineages, and impair the response of HSCs to replicative challenges [107]. A recent study of MDS indicates that inflammation acts as a selective pressure that specifically fosters expansion of preleukemic or malignant HSCs compared to normal HSCs [108]. Distinct hematopoietic cell subtypes may exhibit differential responses to specific inflammatory signaling molecules, further supporting heterogeneous HSC populations that can drive leukomogenesis [84, 109].
The effect of inflammation on HSCs has valuable implications for age-associated diseases, as older individuals exhibit elevated levels of inflammation [110]. Inflammation plays a significant role in expansion of HSC clones carrying preleukemic mutations in CHIP. Characterized by acquisition of somatic mutations in hematopoietic lineages with age, CHIP is associated with both hematological malignancy and cardiovascular disease (CVD), broadening the role of preleukemic mutations in disease states [111, 112]. The pro-inflammatory response observed in HSCs carrying preleukemic mutations indicates the existence of intrinsic mechanisms of inflammation for HSCs [102, 113, 114]. The addition of a pro-inflammatory preleukemic mutation can alter the presentation of hematological malignancy [115]. It has been proposed that HSCs and mature hematopoietic lineages propagate the inflammatory response via a feedforward mechanism, in which inflammatory signals from one cell type amplifies the other [116]. Pro-inflammatory macrophages secreting IL-1α enable CHIP-associated CVD, underscoring that some of the same pathways involved in hematopoietic homeostasis and leukemogenesis also contribute to the non-hematopoietic manifestations of CHIP [68]. In addition, as in emergency granulopoiesis, aging promotes myeloid expansion, further emphasizing potential mechanistic overlap [109]. While intrinsic factors appear to be important to the inflammatory response involved in the pathogenesis of CHIP-associated diseases, extrinsic mechanisms may also influence this process as the conditioned media from aged mesenchymal stromal cells impairs the function of young HSCs [117]. In addition to acting directly on HSCs, inflammation can also remodel the bone marrow microenvironment, which regulates hematopoiesis [118]. Additional studies are needed to understand the ways in which these pathways are altered during aging and pathogenesis, and how they may be modified for health benefit.
Elderly populations may be uniquely susceptible to the effects of inflammation and CHIP as comorbidities and CHIP are more common in aging individuals. Several comorbidities with inflammatory components, such as ulcerative colitis, rheumatoid arthritis, and systemic sclerosis, have been linked to increased clonal hematopoiesis [119,120,121]. Pro-inflammatory features of CHIP-associated HSCs/HPCs may influence outcomes of hematopoietic stem cell transplants, leading to cytopenias, chronic graft vs. host disease, and/or donor-derived leukemia (DDL) [122]. These patient populations may especially be vulnerable to development of hematological malignancies and CHIP-associated diseases following infection, as infection may exacerbate the existing inflammatory response in these patients. Inflammation may be a key factor contributing to the heterogeneity observed in hematological malignancies and CHIP-associated diseases and should be considered in the clinical management of patients with these conditions. In particular, differences in the clinical presentation between donors and recipients that develop DDL highlight the potential role of inflammation and comorbidities in promoting leukemogenesis. It is yet not known if these different sources of inflammation influence hematological malignancies and CHIP-associated diseases via common mechanisms.
Intrinsic and extrinsic sources of inflammation may represent potential therapeutic targets for hematological malignancies and CHIP-associated diseases. Inhibition of inflammation can impede clonal expansion in response to inflammatory stimuli [102]. Blocking inflammation may also be a valuable therapeutic approach in CVD, as inhibiting IL-1 receptor signaling from pro-inflammatory macrophages can prevent CHIP-associated CVD in mice [68]. It is currently unclear whether suppressing inflammatory HSCs or mature hematopoietic lineages is more effective. Inflammatory signals from the bone marrow microenvironment may also be viable therapeutic targets but have not yet been investigated. A more thorough understanding of the timing of the inflammatory response and the cell types involved will facilitate the effective inhibition of inflammation in hematological malignancies and CHIP-associated diseases. Strategies to both prevent pathogenesis and to treat existing disease will be valuable. A critical aspect of targeting inflammation in these disease contexts will be to maintain the normal inflammatory response necessary for responses to infection and injury while targeting aberrant inflammatory pathways that promote disease; however, additional studies are needed to elucidate factors governing these processes. Thus, Inflammation acts as a dysregulator of tissue maintenance and regeneration during aging as evidenced by the fact that HSC do not regenerate well after inflammatory challenge [123].
D) DNA Damage, Transcriptional, and Epigenetic Changes During Aging
DNA damage accumulates with age, and defects in DNA repair can cause cellular changes that resemble a premature aging phenomenon [124,125,126,127]. Tables on selected models of premature aging in mice and their common features have been summarized in a review [126] and p53 implicated in DNA damage [87]. While DNA damage to HSCs and HPCs during aging is clearly impacted, such damage to the microenvironmental niche cannot be overlooked [124]. Transcriptional changes in stem cell populations have been profiled for HSCs and other stem cell types, but it is not clear yet if a common age-related signature has been identified [12]. A role for epigenetics in the aging process is also considered [128]. Epigenetic hallmarks of aging and senescence have been diagrammed, as have been the pros and cons of using model systems to study aging and senescence in a variety of species, along with a short listing of repositories and tools for evaluating a role for research in the aging process [128].
Repair of damage has been shown to offset deficient HSC function during aging [129]. This was especially apparent under stress conditions, in which DNA damage led to loss of the potential of HSC reconstitution, proliferative capacity, self-renewal activity, enhanced apoptosis, and then exhaustion of function [129]. It was suggested that the accrual of DNA damage may be a means contributing to HSC functional defects of these cells to respond to acute stress or injury [129].
A shift from canonical to non-canonical signaling by Wnt, in response to elevated expression of Wnt5a was associated with the process of HSC aging [130]. Treatment of cells from young mice with Wnt5a induced aging associated HSC apolarity, reduced their capacity for regeneration, and resulted in a age-related shift in myeloid/lymphoid differentiation, that was associated with activation of the GTPase Cdc42 [130]. Moreover, haploinsufficiency of Wnt5a resulted in the attenuation of the aging phenotype of HSCs [130]. Other studies defined replication stress as a driver of functional declines in HSCs during aging [131]. This was associated with decreased expression of mini-chromosomal maintenance helicase components and altered DNA replication fork dynamics [132].
There are reports on a role for epigenetics in abnormalities associated with HSCs in old mice [132,133,134,135]. While the decline of HSC function seemed to be dependent on their proliferative history, it was noted to be independent of the length of their telomeres [133]. HSCs from old mice manifested reduced signaling of transforming growth factor-beta with changes in genes involved in proliferation and differentiation of HSCs [135]. HSCs from old mice had broader peaks of H3K4me3 with increased methylation of DNA at the transcription factor binding sites that were associated with genes involved in promotion of differentiation, and a reduction of genes associated with maintenance of HSCs [135]. Ribosomal biogenesis was found to be a particular target of this age-related HSC phenotype; there was increased transcription of ribosomal protein and RNA genes, and the hypomethylation of genes for ribosomal RNA [135].
Proteosome analysis [136] and single-cell RNA sequencing [137] have been performed on HSCs from old mice. How much these analyses really inform us about the prime drivers in HSC dysfunction remains to be determined, especially since the cells were collected in ambient air prior to analysis, which may not be optimal for assessing physioxia associated effects [1]. Of some interest, deletion of inhibition of DNA binding1 (Id1), a helix-loop-helix transcription faction protected HSCs from both the effects of stress-induced exhaustion and that of aging [138].
E) Metabolic Processes, Mitochondria and Reactive Oxygen Species (ROS) During Aging
Metabolism, mitochondria and ROS are important aspects of HSC function [40, 41], as well as for other stem and progenitor cell types [139,140,141]. Aging is associated with extensive changes in metabolism [75,76,77]. A short report questioned whether or not metabolic mechanisms of stem cell maintenance might explain aging and its associated impact on stem cells [142]. Another review concentrated on mitochondrial contributions to dysfunction of somatic stem cells in general and in context of aging [143] and a review on mitochondrial metabolic checkpoints and aging of HSCs implicated mitochondrial maintenance mechanisms including mitophagy and asymmetric segregation of “aged” mitochondria [144]. This is an area that clearly requires more detailed investigation, although it has been suggested that mutations in mitochondrial DNA are not a primary driver of stem cell aging [145].
ROS has been implicated in various stem cell functions [40, 41, 146,147,148], and STAT3, mitochondrial dysfunction, and overproduction of ROS has been associated with a rapid aging-like phenotype [149]. Symmetric divisions of stem cells, including HSCs, results in increased stem cell numbers with maintenance of stem cell characteristics of the original “mother” cell. However, asymmetric division of stem cells can result in one daughter cell maintaining the original stem cell characteristics of the “mother” stem cell, while the other cell can be a more differentiated progenitor cell. To assess selective apportioning of subcellular contents between “daughter” cells using mammary stem like cells, it was found that “daughter” cells that received fewer “old” mitochondria were associated with maintenance of stem cell traits; inhibition of mitochondrial fission disrupted age-dependent subcellular localization and segregation of mitochondria with resultant loss of stem cell properties in the progeny [150]. It is not clear if such studies with mitochondria apportuning between HSCs undergoing symmetric or asymmetric divisions have yet been done, but it is certainly an area of interest if done in context of HSC from young and old bone marrow HSCs, and their collection and processing under physioxia conditions as noted [1].
There is still much to be learned regarding how stem cells maintain metabolic homeostasis. The unfolded protein response has been implicated as modulating the HSC pool during stress [151], but has apparently not yet been evaluated in HSCs from aged mice. However, a regulatory branch of the mitochondrial unfolded protein response, mediated by the interplay of the sirtuin, SIRT7 (more on sirtuins in Section F), and nuclear respiratory factor 1 (NRF1) which is a master regulator of mitochondria, was interrogated in HSCs [152]. It was noted that inactivation of SIRT7 resulted in reduced quiescence, increased mitochondrial protein folding stress, and decreased regenerative capacity of HSCs. Moreover, expression of SIRT7 was decreased in HSCs from old mice, and up-regulation of SIRT7 in the aged HSCs improved their regenerative capacity. This implicated the mitochondrial unfolded protein response-mediated metabolic checkpoint as a contributor to HSCs in old mice [152]. In addition, mitochondrial DNA polymerase, when defective, has been associated with premature aging in mice [153], but how, if at all, this relates to HSC function from old mice remains to be determined.
Thioredoxin-interacting protein (TXNIP) is a 397 amino acid residue, belonging to the arrestin family of proteins. It has been reported to regulate HSC quiescence and mobilization after stress [154,155,156], and is likely to be involved in HSC function, but has not to our knowledge been extensively investigated. Reasons to evaluate this during aging is that Txnip-/- mice have decreased HSC reconstitution resulting in HSC exhaustion, effects associated with hyperactive signaling of Wnt, an active cell cycle, and reduced expression of p21cip1. These stresses also affect the BM microenvironment resulting in decreased expression of CXCL12 (a chemotactic and homing chemokine)- and osteopontin-mediated interactions between HSCs and the BM [154]. TXNIP helps to maintain the pool of HSCs by functional switching of p53 after oxidative stress, effects that have been reviewed [155].
There is much to be learned regarding metabolic influences in aging, and molecular mechanisms underlying aging effects on HSCs still remain unclear. Elevated activity of the small RhoGTPase cdc42, previously noted by the investigators in another paper was linked casually to effects on HSCs in old mice [152], with a correlation of the loss of polarity in these cells. Moreover, by inhibiting cdc42 activity by pharmacological means, it “rejuvenated” the aged populations of HSCs by increasing the percent of polarized cells and restoring the level and spatial distribution of histone H4 lysine16 acetylation such that it was similar to that in HSCs isolated from young mice [152, 157]. This information further identified epigenetic regulatory changes in functional effects of HSCs from old mice, and may relate to metabolic changes.
Other studies linked the interaction of ROS dependent DNA damage, mitochondria, and p38 MAPK with senescence of adult mesenchymal stem/stroma cells (MSCs) from humans, with pharmacological inhibition of p38 MAPK partially recovered the senescence phenotype by partial prevention of hydrogen peroxide-induced senescence [158]. How linked senescence phenotypes are to the function of HSCs in aged persons remains to be determined. Somatic cell mitochondrial DNA (mtDNA) mutations contribute to such age-related disorders as those associated with myelodysplasia (MDS), and it was noted that the mito-protective effect of autophagy was impaired in erythroid cells of old mice [159]. mtDNA-mutated mice had somatic mtDNA mutations that were a targeted defect in the function of proofreading mtDNA polymerase, PolgA, and developed macrocytic anemia similar that seen in MDS patients. Mechanistic insight into these processes was reported [159], but whether or not these processes reflected changes in HSCs from old mice was not explored.
F) Apoptosis, Autophagy, Radiation, and a Role for the Sirtuin Family of Proteins During Aging
Aging-Related Apoptosis and Autophagy
Apoptosis, the phenomenon of programmed cell death, and autophagy, a self-degrative process responsible for eliminating cytosolic constituents such as long-lived proteins, aggregated proteins, and damaged organelles (mitochondria, ribosomes, peroxysomes) [160] have been linked to functional changes noted during aging [161, 162] and HCT [163]. Autophagy is associated with repair pathways that can protect hematopoiesis from injury due to nuclear radiation [164, 165]. Inhibition of autophagy by genetic manipulation was associated with normal and pathological aging, with its inhibition compromising the “longevity-promoting” effects of restriction of calories, the activation of SIRT1, inhibition of insulin and insulin growth factor signaling, and the administration of rapamycin, resveratrol, or spermine [161]. Autophagy was shown to maintain the metabolism of HSCs from both young and old mice [164]. These influences were not noted in all HSCs from old mice, with about a third of HSCs from aged mice demonstrating high autophagy levels being associated with a low metabolic state and high potential for regeneration [165]. This suggests that not all HSCs in aged mice are functionally compromised, an important point in aging HSC research that can be overlooked when studying HSCs from old mice at a total HSC population level. It is known that there are subsets of rigorously purified HSC populations that differ in mitotic history [166], and intracellular characteristics [1]. FOXO4 was suggested as a pivotal agent in the area of cellular senescence [167]. Using a FOXO4 peptide that disrupted the FOXO4 interactions with p53 in vivo where it was tolerated, restored certain functions in naturally aged and in fast aging XpdTTD/TTD mice. How this relates to HSCs in old mice remains to be evaluated.
Mitophagy is a process that is evolutionary conserved involving autophagic targeting and clearance of mitochondria that are destined for removal [168]. It is induced by short ubiquitin chains on the mitochondria [169]. Reviews on this process have been reported [168, 169] and discuss how metformin, an oral diabetes medication, both enhances and normalizes mitochondrial function that leads to alleviation of inflammation associated with aging [170]. What remains to be determined is if there is a role for mitophagy in HSC and HPC during aging, and if this can be modulated for health benefit.
Radiation Effects and Aging
Like aging, exposure to radiation is an additional stressor to the hematopoietic (H) system, the most sensitive tissue in the body to radiation damage. Therapeutic radiation, nuclear accidents, and malicious exposure from radiologic-warfare put mankind at risk for life-threatening acute radiation syndromes (ARS) and the delayed effects of acute radiation exposure (DEARE) in those fortunate to survive ARS. H-ARS, due to direct and indirect effects of radiation exposure on all classes of hematopoietic cells, leads to death within weeks if untreated [171, 172].
Hematopoietic DEARE, also known as residual bone marrow damage (RBMD), is characterized by diminished immunity and decreased production of blood cells persisting for years after radiation exposure [173,174,175,176,177,178,179]. Survivors of H-ARS exhibit severe lifelong damage to HSC, characterized by significantly decreased complete blood count, loss of HSC repopulating potential, loss of HSC quiescence, decreased numbers of HPC, and dramatic myeloid skewing, all most evident under stress [173,174,175,176,177,178,179,180,181,182,183]. An increased incidence of lymphoid malignancies, shortened life span, decreased mesenchymal stem/progenitor cell (MSC) number, and aberrant levels of endothelial cell-derived HSC niche proteins in aged H-ARS survivors have also been documented (Orschell, unpublished data). Long-term damage to the HSC-supportive niche also likely contributes to HSC dysfunction and RBMD. As enhanced cycling of HSC is believed to lead to loss of self-renewal potential and is detrimental to engraftment potential [184], it seems likely that the enhanced cycling of HSC from H-ARS survivors is a major contributor to RBMD. These data illustrate an unrecoverable loss of HSC self-renewal and differentiation potential, the two hallmarks of HSC [185,186,187,188,189,190,191], in survivors of H-ARS and suggest that compensatory mechanisms of hematopoietic support cannot overcome the “second hit” imparted by aging [14, 124, 129, 192, 193].
The DEARE are generally thought to result from persistent inflammation and chronic oxidative stress [194,195,196,197,198,199,200,201], leading to fibrosis [202] and loss of stem cell self-renewal functions. Indeed, elevated levels of pro-inflammatory cytokines associated with oxidative stress [203] have been reported in Japanese atomic bomb survivors [198, 202]. Other studies in atomic bomb survivors have shown possible reductions in self renewal capability of HSC secondary to dose-dependent DNA damage [204], as well as detriments in immune function [205], corroborating mouse H-ARS data. NAD(P)H oxidase, xanthine oxidase, and mitochondria have all been implicated as primary oxidant sources in various models and conditions [203, 206,207,208], and ROS has been documented in HSC post-irradiation as well [181]. NF-kB, one of several transcription factors activated by ionizing radiation [209], plays a central role in inflammation [210], is activated by oxidative stress and induces oxidative stress through interactions with cytokines [211], creating a potential feed-forward mechanisms to maintain chronic inflammation and oxidative stress.
Cellular senescence, and its associated oxidative/pro-inflammatory phenotype, has recently emerged as a causative mechanism of DEARE [211,212,213,214], making senescent cells a new therapeutic target for RBMD and other DEARE. Importantly, senolytic drugs have the potential to be used as an effective treatment for DEARE even after DEARE becomes a progressive disease [212], but it is not yet clear how this might be used in and for elderly exposed individuals.
As mice age, like humans, health issues and phenotypic changes begin to manifest and variability in experimental endpoints increases, necessitating the need for larger group sizes for sufficient statistical power. For example, mice of similar strains have been shown to exhibit significantly different life spans [215, 216] and radiation sensitivities when aged (Orschell, unpublished data), as well as differing susceptibilities to radiation-induced swollen muzzle syndrome [217], all depending on the vendor from which they were sourced. Mice from different vendors have also been shown to possess different fecal microbiota [218], which may contribute to vendor-specific phenotypic differences. For these reasons, investigators should consider stringent control of the vendor and barrier of their mice for aging studies to ensure optimal stability of their experimental models.
It is noted that more profound effects of aging may be produced not by life-threatening ARS (where the majority of those are exposed to high dose radiation), but rather by moderate or even low dose exposure.
The Role of Sirtuins in Regulating Aged HSC Function
Sirtuins are part of a large family of molecules, some of which have been linked in longevity/aging studies. The role of the sirtuin SIRT1 in stem cell biology, the aging process and in HSC function in old mice had been reviewed [219]. In this review it was noted that although the role of SIRT1 in teleomere maintenance was not resolved, its role in mitochondria and generation of ROS was highly implicated. It was observed that the genetic, hormonal, or drug manipulation of stem cell mitochondria may be useful as an intervening tool for manipulating HSCs from old mice. It was later reported that deficiency of SIRT1 compromised mouse embryonic stem cell hematopoietic cell differentiation in addition to embryonic and adult mouse hematopoiesis [220]. SIRT1 was reported to be required for maintenance of HSCs and lineage specification, in part by the transcription factor FOXO3 [221]. These investigators also suggested that SIRT1 may be involved in HSC function during aging by “delaying” HSC functional abnormalities [221], but this has not been rigorously studied. Although the role of SIRT1 and other sirtuins in the caloric restriction of modifying the aging process have been extensively reviewed [222,223,224], such studies do not always take into account the sex and mouse strains utilized [225] which could influence the reported results. SIRT3, while found to be dispensable for maintenance of HSCs and homeostasis of tissues during young age, was reported to be essential following stress and with the aging process [226]. SIRT3 expression was decreased with aging and upregulation of SIRT3 expression in HSCs of old mice improved their regenerative capacity, effects involving a role for mitochondrial homeostasis [226]. As noted above in Section E, SIRT7 in the mitochondrial unfolded protein response and aging-associated changes in HSCs in the old mice were linked [152]. This was discussed more thoroughly in a short commentary [227].
G) The Microbiome, Hematopoiesis, and Aging
The microbiome describes microorganisms such as bacteria, viruses, and fungi that colonize the human and animal body and influence various biological processes. Most studies that explored microbiome–hematopoiesis interactions are based on characterizing HSC and HPC populations in germ-free (GF) or broad-spectrum antibiotic-treated mice and in human subjects under prolonged antibiotic regimens or diagnosed with gut dysbiosis such as inflammatory bowel syndrome [228,229,230]. GF mice demonstrated myelosuppression, smaller HSC, MPP, and common lymphoid progenitor (CLP) populations, and impaired neutrophils, monocytes, and T-cell functions. Recolonization of GF mice restored immune response to infection [96, 228, 231]. However, a closer evaluation of HSCs and HPCs in oral antibiotic-treated mice revealed normal HSC and HPC populations but reduced mature T cell, B cell, and granulocyte populations, suggesting impaired differentiation of mature immune cells in microbiota-depleted mice after oral antibiotics treatment and introduced some discrepancies between animal models used to study the microbiome [232]. Following HCT, microbiota-depleted recipient mice immune reconstitution was significantly lower than their control counterparts [232] supporting the conclusion that the microbiome plays a role in regulating mature immune cell development. Several studies have linked the human gut microbiome imbalance or dysbiosis in conditions such as inflammatory bowel syndrome, malnutrition, and obesity to altered hematopoiesis [229].
The microbiome and hematopoiesis have been intimately linked [233,234,235,236,237,238]. Gut microbiota are known to sustain hematopoiesis [233], microbiota can regulate HSC differentiation by altering the BM niche [234], and CX3CR1+ mononuclear cells influence HPCs [235]. Reduced mPB is noted in mice receiving antibiotics [236] and microbiota modification has been discussed in context of hematology [237]. Moreover, gut microbiota are known to control bacterial infection by promoting hematopoiesis [238], but definitive and rigorous comparative studies on a role of the microbiome on hematopoiesis in the young and old are yet to be done.
Bacteria and the microbiome present a not uncomplicated scenario that has not been adequately addressed in context of aged hematopoiesis and this needs adequate attention. Certain bacteria, using that of Fusobacterium nucleatum as an example, have been implicated in enhancing metastasis of cancer cells [239,240,241,242,243,244,245,246,247,248]. If such bacteria have this capacity for cancer cell metastasis, then why not for HSC and HPC, migration and/or homing an area worthy of investigation.
Up to 15% of patients with a history of prolonged antibiotic use have suffered hematological adverse effects in the form of neutropenia, anemia, and pancytopenia [249, 250]. Although associations between the hematopoietic system and microbiota imbalance is apparent in mice and humans, mechanistic understanding of this interaction is limited. In the signal transducer and activator of transcription protein 1 (STAT1) knockout mouse, the antibiotic effects on HSC and HPC numbers were abrogated [96]. In another report, administrating a ligand of the pathogen recognition receptor 1 (NOD1) restored HSC and HPC numbers in GF mice [251]. To the authors' knowledge, aged-HSC and HPC function and phenotyping in relation to the microbiome have not yet been reported in mice or humans. Aging is associated with perturbation of intestinal epithelial integrity and upregulation of permeability, allowing microbiota entrance to the circulation and induces a chronic inflammatory state in the aged subject. Aging results in microbiota-associated increases in pro-inflammatory cytokine levels (e.g., TNF-α, TGF-β, IL-6, etc), changes in T-cell numbers (e.g., Treg, Th1, and Th2 T-cell subsets), and activation of TLR2, NF-κB and mTOR [252]. Considering how the well-characterized low-grade, chronic inflammation associated with aging affects the hematopoietic system, a role for microbiota promoting such inflammation is a strong possibility. How this might regulate hematopoiesis in the young and old remains an unexplored area to be better studied in steady- and stressed-states.
H) Additional Age-Related Information and Means to Better Evaluate/Understand Hematopoiesis During Aging
Role of Collection/Processing of Cells
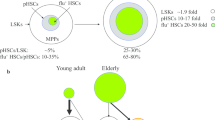
Many studies have acknowledged the probable effects of oxidative stress on functional changes in stem cells during aging [253]. This oxidative stress is associated with damage to macromolecules including that of nucleic acids, proteins, lipids, and carbohydrates that could contribute to changes in HSC function. This however, did not consider how even the mere removing of HSC-containing populations of cells from mouse BM [40], mouse mobilized peripheral blood (mPB) [42], or human cord blood [40] could so quickly change HSC numbers and impinge on the function of the removed HSCs. As previously mentioned, collecting and processing cells under ambient air conditions for as little as 15 minutes exposes the cells to extra physiological oxygen stress/shock or EPHOSS. EPHOSS is associated with increased differentiation of HSCs to HPCs through a sequence of events involving p53, the opening of the mitochondrial permeability transition pore, cyclophilin D, hypoxia inducing factor (HIF)1-alpha and the hypoxamir, miR210 [40, 41]. By collecting and processing BM and mPB from mice and cord blood from humans in a hypoxic chamber set at 3% O2 and taking care to make sure that the cells are never exposed to ambient air conditions, it is possible to obtain many more phenotyped and functional HSCs [40,41,42]. Increased numbers of collected HSCs under hypoxia has also been reported for BM cells from Fanca-/- and Fancc-/- mice [254]. It is possible that some of the EPHOSS-related effects on HSCs exposed to ambient air can be compensated for by collection and processing of these cells at ~4°C [255]. Such cold collections and processing of human cord blood and mouse BM cells mimic at least some of the effects seen during hypoxia collection/processing of cells including increased numbers of collected HSCs. However, the mechanisms involved with preserving HSC numbers/function following cold collection/processing of cord blood cells have not yet been worked out and may differ somewhat from that of physioxia/hypoxia collected/processed cell populations.
Collecting/processing BM cells from old vs. young mice under different oxygen tensions [1], allowed us to demonstrate that functional engrafting HSCs from old mouse BM collected/processed at 3% O2 were equal in number to that of ambient air (~21% O2) collected/processed young mouse BM HSCs. Perhaps more importantly, the abnormal myeloid to lymphoid ratio seen when aged BM cells were engrafted into lethally irradiated recipient mice in a competitive transplant setting was not noticeable and completely resembled that seen after engraftment of young mouse BM. This was consistent with increased CLP numbers and decreased CMP and GMP numbers in the donor BM from old mice following their collection in hypoxia. These phenomena seen with old BM HSCs collected/processed in hypoxia, were associated with decreased total and mitochondria ROS, and decreased expression of stress-induced proteins [1]. Hence, aged mouse BM HSC function may not be as dysregulated as many others have reported, with differences perhaps due to their increased response to EPHOSS following ex vivo analysis in ambient air. A corollary of this may suggest that with the more physiological collection and processing of HSCs (a.k.a. in hypoxia conditions, or by means of a physiological strategy (e.g. collecting cells in the cold [255]) from BM of older individuals may be more functional in context of clinical HCT than if they were collected/processed as usual in ambient air O2. It is known that there are distinct populations of even rigorously purified populations of HSCs [1, 256]. Which of these HSC populations survive EPHOSS effects when cells are collected/processed in ambient air from young and old mouse BM remains to be better elucidated. While much more obviously needs to be done to more mechanistically and physiologically understand the true state of HSCs isolated from older human individuals or mice, it may be that much of the literature on aged HSCs needs to be re-evaluated for their functional status as they are within their in vivo physioxia microenvironment. Studies to be done with hypoxia vs. ambient collected HSCs include more in-depth intracellular events and signaling pathways that include gene and protein expression profiles, as well as epigenetic changes, work currently ongoing.
Molecular chaperones and heat shock responses have been postulated to play a role in longevity and aging [257], but rigorous studies in the area of aged HSCs are lacking. Age and organ specific differences in bioenergistics in Brown Norway rats have been noted [258], and p53 deficiency induces diverse dysregulated processes under physiological oxygen/physioxia [259], but we know that such events are influenced by modes of cell collection and processing [40, 41].
The Microenvironment
Not to be ignored in studies of aged HSCs is the role that their microenvironment in vivo plays in the functional cellular and intracellular abnormalities/defects associated with their engrafting deficiencies and biased differentiation patterns. The microenvironment niche for HSCs has been studied for young mice [260], but more in-depth studies of the aged BM microenvironment is warranted, especially in context of oxygen content [260], as we noted [1]. There is a report of the rejuvenation of progenitors from old mice when placed into and exposed to a young BM environment [261], and a more recent report demonstrated that degeneration of adrenergic nerves in BM affects aging of the HSC niche [262], and that aging in humans alters the special organization between populations of CD34+ cells (contains mainly HPCs, but also a small percentage of HSCs) and adipocytes in the BM [263]; this is of potential relevance as increased adiposity is associated with the aged BM microenvironment and can alter the functionality of surrounding cells. MSCs showed aging related expression of cxcr4 [264], a “homing” HSC/HPC receptor, but this has not yet been evaluated under conditions of hypoxia collection and processing such as in [40, 41].
Leptin (Lep) and Lep Receptors (R)
Metabolic activities of cells are adaptively regulated by systemic signals that reflect the nutritional status of an organism throughout its life [265, 266]. This is particularly crucial in the case of HSCs as they are rare, and they maintain the integrity of the entire hematopoietic organ system. One way that the body communicates nutritional cues to HSCs is via systemic hormonal regulations. Various metabolic hormones have been documented to influence hematopoiesis, and aging can significantly alter these metabolic messengers, hence indirectly affecting HSC functional behavior [267,268,269].
Among them, leptin controls the body energy expenditure and storage through both central and peripheral mechanisms [270]. As an adipokine, it is recognized to have broad spectrum effects on numbers and functions of different immune cells under homeostasis [271]. Aging is known to be associated with multiple dysregulations of the immune system, including a declined adaptive immune response [272]. Lep induces gene expression of p16, a marker of cellular immunosenescence in human B cells from young lean adults. These cells also exhibited lowered class switching activity and ability to produce influenza-specific IgG [273, 274]. Unfortunately, the study was limited to in vitro treatment only and did not provide full mechanistic insights. In line with this finding, it was demonstrated that lep induced significantly higher levels of IL-10, TNF-α, and IL-6 from aged human B cells as compared to young controls, and this effect was mediated through the STAT3 signaling pathway downstream of lepR activation [275]. Another group reported that sustainably higher lep levels were found in LPS-treated older (24 month) compared to young (2 month) rats, and the old rats showed delayed but longer febrile response. Elevated lep concentrations were accompanied by increased levels of pro-inflammatory cytokines including IL-6 and IL-1Rα; however, it was not determined whether the increase in lep level was causative and how [276]. Although lep signaling was consistently reported to be altered in aged animals, more rigorously designed studies are needed to help us understand how this well-known proinflammatory neuroendocrine adipokine may play any roles in aging-associated immunological changes [277, 278].
Beyond its role in immunity, lepR-expressing MSCs in murine hematopoiesis have been well-characterized as an indispensable source of stem cell factor for maintenance of both HSCs and more differentiated progenitor cells [279,280,281,282]. In addition, it has been demonstrated that BM adipose tissue possessed brown fat properties that declined in old or diabetic mice [283]. It was demonstrated that during the process of aging or in obesity, MSCs preferentially differentiated into adipocytes, which impaired hematopoietic recovery [284]. Since BM adipocytes could be a potential source of lep, it is important to know how BM adipocyte-derived lep can directly or indirectly alter hematopoiesis as the animal ages. In the context of HSC biology, we recently discovered that Lepr marks a small subset of robustly repopulating and self-renewing long-term HSCs in adult murine BM; Lepr+ HSCs (defined as LSKCD150+CD48-) were found to generate equal myeloid-lymphoid outputs as compared to Lepr-HSCs [285]. Given that lepr (OB-R) was also reported to be expressed by different types of both myeloid and lymphoid leukemic cells [286,287,288,289], it will be intriguing and important to determine whether aging has a differential selection pressure on Lepr+HSCs as compared to the rest of total BM HSCs, particularly in the context of clonal hematopoiesis. Although RNA-seq data suggested that Lepr+HSCs and Lepr+MPPs (defined as LSKCD150-CD48-) predominantly expressed the truncated short isoforms OB-Ra and OB-Rc, it remains to be determined whether aged HSCs or leukemic cells can express the long functional isoform OB-Rb (LepR). Future studies with more mechanistic insight should be able to address these questions, perhaps providing potential meaningful clinical implications.
More on Inflammation
Aging-related inflammation promoted aging characteristics of HSCs through a tumor necrosis (TNF)-alpha, ERK, ETS1, IL-27 receptor (R) pathway [290]. TNF-alpha increases during aging and induced expression of HSC IL-27R-alpha by ERK-ETS-1 intracelluar signaling, with deletion of IL-27R-alpha rescuing functional decline and myeloid bias of HSCs. Old IL-27R-alpha knockout mice had reduced proportions of myeloid-biased HSCs. Thus, this is another report implicating factors external to HSCs that effect the functional capacity of HSCs from old mice. Somatotrophic/Insulin-Insulin-Like Growth Factor has also been implicated in aging effects on stem cells as well, in these cases on a population of very small embryonic like stem cells [291, 292].
Exosomes
Exosomes are a subset of small extracellular vesicles that range in size from 30-150nm, that are produced by normal and malignant cells [293, 294], originate from the endocytic compartment of producer cells [295], and have emerged as a universal intercellular communication system [296, 297]. Exosomes are in a protective protein/lipid bilayer, are delivered to recipient cells without degradation, and freely cross biological barriers [296, 297]. Exosomes [298] influence proliferation of HPCs [299], but there is no information on effects of exosomes on HSCs and HPCs during aging.
Biological Time Keeping and Circadian Rhythms
Biological time keeping [300] and circadian variations are known to influence numbers of HSCs and HPCs in BM and blood, but how this might occur in old mice has not yet been explored. Such studies may be of interest, based on when it is best to collect HSCs and HPCs from older individuals from BM and mPB for use in HCT. Studies of circadian deep sequencing revealed stress-response genes that adapted to high rhythmic expression during aging [301], and daily onset of light and darkness have been reported to manifest control over HSC maintenance and differentiation [302]. Moreover, circadian host and microbiome interactions have been suggested to play relevant roles [303]. There is a noted sexual dimorphism in body clock regulation [304] which has not been adequately addressed, especially in context of aging, and aged HSCs and HPCs.
HSC homing to the BM plays an important role in the engrafting capability of the HSCs [305], and more consideration needs to be given to this capacity of aged HSCs. There are a number of means to enhance the homing of HSCs for enhanced engraftment [306,307,308,309], but these have not yet been evaluated in context of HSCs from old mice, or when cells are collected/processed in hypoxia vs. ambient air. Notably, there are a number of animal models to study aging, with an example being the Ames hypopituitary dwarf mice [7,8,9,10], that live approximately two times longer than that of most other mice. Imbalanced myelopoiesis was noted in myelopoiesis between BM and spleen in Ames dwarf mice [310].
I) COVID-19/SARS-CoV-2, Aging, and Hematopoiesis
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the pathogen responsible for causing Coronavirus Disease 2019 (COVID-19), a disease which has spread worldwide, infected over 32 million people and claimed the lives of nearly 1 million people as of September 2020 [311]. SARS-CoV-2 is a virus which infects cells by binding to cell surface proteins via its Spike protein that extends out from the viral envelope, facilitating entry to the host cell and allowing for viral replication within the cell [312,313,314]. The most well-studied presentation of COVID-19 is an infection of the lungs, with symptoms ranging from a mild cough and fever to a severe pneumonia [315, 316].
The prognosis and severity of COVID-19 in patients appears strongly tied to the age of the patient [317]. The risk for SARS-CoV-2 infection leading to symptomatic disease rises dramatically with age. According to the Center for Disease Control (CDC), as age increases the likelihood of being hospitalized for COVID-19 increases, with adults over 65 having 5-13x higher hospitalization rate [318]. Older people are, as presently known, also more likely to die from the disease, with a 90-630x higher death rate in patients over 65 [318], although it is likely that the full story on this is not yet known. This is not likely due to older people being more susceptible to infection, as RT-PCR tests for the presence of SARS-CoV-2 in mild to moderate cases of COVID-19 demonstrate that the host cells of younger patients contain more viral RNA than older patients [319]. A potential interpretation of this is that a higher viral load is necessary for younger people to display symptoms compared to older people. This suggests that children are equally likely to be infected, but may be less likely to display symptoms of the disease, likely due to a general lack of additional contributing factors, possibly including a predisposition to hematologic disorders.
Understandably, many studies of SARS-CoV-2 have focused on its infection of and impact on the lungs. However, it has become increasingly evident that COVID-19 is systemic in nature [320], affecting many different systems including primitive and mature hematopoietic cells [317, 321,322,323,324]. The impact that the disease has on the hematopoietic system is evident in the hematological manifestations of COVID-19. A review looking at hematological factors in COVID-19 patients found that both lymphocytopenia and thrombocytopenia are common symptoms with hospitalized patients [322]. It is also evident that more severe cases of COVID-19 were more frequently associated with these hematological factors [322]. Further, one of the more devastating effects of SARS-CoV-2 infection is the induction of a "cytokine storm" [325, 326]. Cytokine storm refers to a toxic excessive release of immune cytokines leading to an autoimmune response. Thus, there is a strong need to address the mechanisms and effects of SARS-CoV-2 exposure on primitive and mature hematopoietic cells and the impact it may have for COVID-19 patients, especially in older individuals.
Recently it was demonstrated that ACE2 is expressed on the cell surface of small numbers of HPCs and mature immune cells [324] and moderate to large numbers of HSCs [323, 324]. Importantly, exposure of human HSCs to SARS-CoV-2 Spike protein induces increases in expression of inflammatory molecules such as NLRP3 and IL-1beta [323], indicating that the cytokine storm may be mediated in part through primitive hematopoietic cells. Further, human HSCs and HPCs exposed to Spike protein ex vivo have decreased capacity for functional HPC colony formation, exhibit decreased cell growth, and decreased expansion of HSCs, HPCs, and functional HPC colony forming units compared to cells that were unexposed to the viral protein [324]. These effects can be neutralized by co-treatment with Angiotensin1-7 [323, 324], a peptide linked to ACE2 regulation of hypertension [327]. The effects of the SARS-CoV-2 Spike protein on colony forming capacity and expansion can also be neutralized by treatment with an antibody targeting SARS-CoV-2 Spike protein or by treatment with soluble human ACE2 [324]. Human peripheral blood mature immune cells also exhibit a response to exposure to Spike protein ex vivo, with monocytes upregulating CD14 and undergoing aberrant changes in morphology [324]. It is clear that SARS-CoV-2 does not have to infect HSCs and HPCs to cause some of the above noted effects of the SARS-CoV-2 Spike protein [323, 324]. These, and that yet to be reported, data are important because they may help to explain the origin of hematological manifestations of COVID-19 such as lymphocytopenia and thrombocytopenia and provide insight into neutralizing these effects on the hematopoietic system.
It is very possible that one of the contributing factors to COVID-19 disease severity in the aged population is due to the impact of the disease on a hematopoietic system that has already been accumulating alterations and damage for many years. Additionally, effects of cytokine storm associated inflammation may further damage aged hematopoietic cells, possibly making them even more vulnerable to the development of hematological disorders even after recovery from COVID-19. The relationships between hematologic manifestations of COVID-19 and age should be further studied, as should the effects of SARS-CoV-2 exposure on HSC/HPC and mature immune cells from the aged versus young. It will also be important to determine whether the hematological manifestations of COVID-19 may be neutralized by specifically targeting the effects on the hematopoietic system, thus potentially relieving some of the disease burden on more severely affected patients, including older patients.
J) Conclusions in Context of Potential Future Intervention for Better Health of the Hematopoietic System During Aging
A number of studies have reviewed the aging process in general including protein sequestration at the nuclear periphery, and pathways of cellular proteostasis, the effects of aging on stem cell populations, and potential therapeutic interventions including that for aged HSCs [328,329,330,331,332,333,334]. Organoids have been suggested as experimental means to study the process of cellular aging [335], but much more rigorous work is needed in this area, especially with analysis ex vivo of HSCs in a relevant physioxia microenvironment BM niche model.
It has been noted that there are molecules in aged blood that promote the spread of cancer [336] and the accumulation of methylmalonic acid promotes tumor progression in the aged [337]. How these phenomenon relate to HSC, HPC, and hematopoiesis and pre-leukemia and leukemia, and to effects on cells collected in hypoxia/physioxia remains to be determined. A recent book [338] has described the aging process from the perspective of a long-time investigator in this field and noted the mTOR, AMPK and sirtuin pathways as main longevity signaling pathways.
Other considerations in context of aging and hematopoiesis to be elucidated are: how mitochondrial ROS acts as a double-edged conundrum for that of host defense in contrast to infection associated pathological inflammation [339] and its effects on HSC and HPC [340]. More in-depth insights can be gained from approaches incorporating single cell multiomics [341] and what role the mechanoregulation [342] of hematopoiesis might play during aging and disorders associated with aging. There is also the question of a role for mitochondrial transfer from Cx43-expressing HPC to stroma [343] during aging of the hematopoietic system and its interaction with the bone marrow microenvironment during regeneration. As noted in more recent reviews on CHIP [344,345,346], there is still much we do not understand about this phenomenon and its relationship to aging and aged hematopoiesis.
Cytokines, Chemokines and Intracellular Signaling
HSCs, HPCs, and hematopoiesis are regulated by numerous interacting cytokines and chemokines [347], effects mediated by receptor-induced intracellular signaling (348; Broxmeyer; submitted solicited review on Cytokines/Chemokines/Other Growth Regulators and their Receptors, 8th Edition, Hematology: Basic Principles and Practice, Eds. R. Hoffman, et. al., 2020). Some recent intracellular players involved in HSC and HPC function have been reported [307,308,309, 349, 350]. However, all such intracellular signaling events have been carried out with HSCs, HPCs, and immune cells of young mice, or human CB or human BM or mPB from younger individuals. Whether or not such regulatory intracellular signaling is similar in cells from old vs. the young remains to be determined, and such studies need to be assessed with purified populations of HSCs and HPCs, and in context of such cells isolated and processed under physioxia conditions, so that they are not exposed to ambient air oxygen which will likely modify the signaling events.
Dipeptidylpeptidase (DPP)4
We had noted that it may be feasible to use HSCs from older individuals for hematopoietic cell transplantation (HCT) if the cells are collected so that they are not induced by EPHOSS to decrease HSC numbers [1] when stressed by ambient air collection [40,41,42], but are there means to decrease the acute GVHD associated with allogeneic HCT, which perhaps has the potential to be more aggressive when donor cells are from more aged individuals? Inhibition of the enzyme dipeptidylpeptidase (DPP)4/CD26 has been shown to enhance mouse BM HCT and to accelerate recovery after radiation and selected chemotherapeutic drugs [351, 352] and to enhance time to engraftment of cord blood (CB) HCT [353,354,355]. This enhancement in time to neutrophil engraftment was also associated with a decrease in the already low acute GVHD noted for CB HCT [356]. It is now clear that the orally active DPP4 inhibitor, sitagliptin used in the CB Trials also greatly dampens acute GVHD in the setting of clinical mPB HCT [357]. Hence, there may be a role for DPP4 inhibitors such as sitagliptin in context of aged hematopoiesis and HCT. DPP4 has also been implicated in exosomes from patients with acute myeloid leukemia [299]. More in depth information on DPP4 during the aging process is clearly warranted. There are many proteins that have purported DPP4-truncation sites [358, 359]. This is of relevance as DPP4-trucated proteins can have decreased activity and block the effects of the full length molecules [352, 360]. Hence, a better undertaking of DPP4 on hematopoiesis in the old, as well as young, could uncover additional means to enhance hematopoiesis during health, aging and disease.
Concluding Thoughts
There are still many unknowns, and still to be elucidated mechanistic insights to understand changes in hematopoiesis during aging. Enhanced genomics may provide additional clues to age-related hematopoiesis [361]. Some of these areas for further investigation are noted in Table 1. It will likely take a multidisciplinary approach to fully understand the causes of abnormalities associated with the aging process, not only of the hematopoietic system. This information might at least partially control what might not be the inevitable consequences of the time- and disease-induced aging process. Future studies of hematopoiesis and HSCs during the aging process that investigate these processes in context of their status in an in vivo/physioxia environment, and ex-vivo under conditions that more closely mimic their in vivo condition of O2 tension and with microenvironmental niche cell interactions [362] will bring us closer to better understanding aged HSCs and what their true functional capacities and abnormalities are. Only with this enhanced understanding can we truly know how this information can best be used and better modified, if necessary and possible, for health benefit.
Notes
While HEB was pursuing a Masters degree in microbiology, prior to his PhD studies in blood cell development and regulation, he was especially interested in reading the literature on Gerontology. In 1969, he attended as an on-looker an International Conference on Gerontology, the first conference he ever attended. The conference was held in Washington, DC, where he heard talks by all the leading experts in this area of research, including Leonard Hayflick, the original proponent on identifying the limited life-span of normal cells. On a whim, HEB registered to attend a closed discussion group of 20 individuals in the conference to discuss the ramifications of what was then currently known about the biology of aging. While he was the only one in the discussion group without an advanced degree, the others in the group were understanding when the Discussion participants were asked to describe their academic background and interest in the field of Gerontology. When it came time for him to talk he apologized, as he had not worked in the area, but noted his interest in the field, and told them his very limited academic training to that time. To his surprise, he was warmly welcomed into the discussion group, which included amongst those present, Dr. Hayflick. The other participants made it clear that the field of Gerontology was still in its infancy, and their words of advice to him, in not so many words, was to find a field to work in that was more advanced and then when he became an expert in that field to integrate the knowledge from that field into the studies of aging. This he did, a half century later.
References
Capitano, M. L., Mohamad, S. F., Cooper, S., Guo, B., Huang, X., Gunawan, A. M., Sampson, C., Ropa, J., Srour, E. F., Orschell, C. M., & Broxmeyer, H.E. (2020). Mitigating oxygen stress enhances aged mouse hematopoietic stem cell numbers and function. Journal of Clinical Investigation. In Press.
Kirkwood, T. B. (2005). Understanding the odd science of aging. Cell, 120(4), 437–447. https://doi.org/10.1016/j.cell.2005.01.027.
Kenyon, C. (2005). The plasticity of aging: insights from long-lived mutants. Cell, 120(4), 449–460. https://doi.org/10.1016/j.cell.2005.02.002.
Chien, K. R., & Karsenty, G. (2005). Longevity and lineages: toward the integrative biology of degenerative diseases in heart, muscle, and bone. Cell, 120(4), 533–544. https://doi.org/10.1016/j.cell.2005.02.006.
Campisi, J. (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell, 120(4), 513–522. https://doi.org/10.1016/j.cell.2005.02.003.
Singh, P. P., Demmitt, B. A., Nath, R. D., & Brunet, A. (2019). The genetics of aging: a vertebrate perspective. Cell, 177(1), 200–220. https://doi.org/10.1016/j.cell.2019.02.038.
Bartke A. (2000). Delayed aging in Ames dwarf mice. Relationships to endocrine function and body size. Results and problems in cell differentiation, 29, 181–202. https://doi.org/10.1007/978-3-540-48003-7_10.
Bartke, A., Coschigano, K., Kopchick, J., Chandrashekar, V., Mattison, J., Kinney, B., & Hauck, S. (2001). Genes that prolong life: relationships of growth hormone and growth to aging and life span. The Journals of gerontology. Series A, Biological Sciences and Medical Sciences, 56(8), B340–B349. https://doi.org/10.1093/gerona/56.8.b340.
Brown-Borg, H. M., Borg, K. E., Meliska, C. J., & Bartke, A. (1996). Dwarf mice and the ageing process. Nature, 384(6604), 33. https://doi.org/10.1038/384033a0.
Bartke, A. (1964). Histology of the anterior hypophysis, thyroid and gonads of two types of dwarf mice. The Anatomical Record, 149, 225–235. https://doi.org/10.1002/ar.1091490206.
Shahrestani, P. (2012). Aging: an evolutionarily derived condition. Frontiers in Genetics, 3, 187. https://doi.org/10.3389/fgene.2012.00187.
Keyes, B. E., & Fuchs, E. (2018). Stem cells: aging and transcriptional fingerprints. The Journal of Cell Biology, 217(1), 79–92. https://doi.org/10.1083/jcb.201708099.
Sharpless, N. E., & DePinho, R. A. (2007). How stem cells age and why this makes us grow old. Nature reviews. Molecular Cell Biology, 8(9), 703–713. https://doi.org/10.1038/nrm2241.
Rossi, D. J., Bryder, D., Zahn, J. M., Ahlenius, H., Sonu, R., Wagers, A. J., & Weissman, I. L. (2005). Cell intrinsic alterations underlie hematopoietic stem cell aging. Proceedings of the National Academy of Sciences of the United States of America, 102(26), 9194–9199. https://doi.org/10.1073/pnas.0503280102.
Cho, R. H., Sieburg, H. B., & Muller-Sieburg, C. E. (2008). A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood, 111(12), 5553–5561. https://doi.org/10.1182/blood-2007-11-123547.
Signer, R. A., Montecino-Rodriguez, E., Witte, O. N., McLaughlin, J., & Dorshkind, K. (2007). Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood, 110(6), 1831–1839. https://doi.org/10.1182/blood-2007-01-069401.
Miller, J. P., & Allman, D. (2005). Linking age-related defects in B lymphopoiesis to the aging of hematopoietic stem cells. Seminars in Immunology, 17(5), 321–329. https://doi.org/10.1016/j.smim.2005.05.003.
Liang, Y., Van Zant, G., & Szilvassy, S. J. (2005). Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood, 106(4), 1479–1487. https://doi.org/10.1182/blood-2004-11-4282.
Xing, Z., Ryan, M. A., Daria, D., Nattamai, K. J., Van Zant, G., Wang, L., Zheng, Y., & Geiger, H. (2006). Increased hematopoietic stem cell mobilization in aged mice. Blood, 108(7), 2190–2197. https://doi.org/10.1182/blood-2005-12-010272.
Warren, L. A., & Rossi, D. J. (2009). Stem cells and aging in the hematopoietic system. Mechanisms of Ageing and Development, 130(1-2), 46–53. https://doi.org/10.1016/j.mad.2008.03.010.
Beerman, I., Maloney, W. J., Weissmann, I. L., & Rossi, D. J. (2010). Stem cells and the aging hematopoietic system. Current Opinion in Immunology, 22(4), 500–506. https://doi.org/10.1016/j.coi.2010.06.007.
Kuranda, K., Vargaftig, J., de la Rochere, P., Dosquet, C., Charron, D., Bardin, F., Tonnelle, C., Bonnet, D., & Goodhardt, M. (2011). Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell, 10(3), 542–546. https://doi.org/10.1111/j.1474-9726.2011.00675.x.
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M., & de Haan, G. (2011). Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. The Journal of Experimental Medicine, 208(13), 2691–2703. https://doi.org/10.1084/jem.20111490.
Pang, W. W., Price, E. A., Sahoo, D., Beerman, I., Maloney, W. J., Rossi, D. J., Schrier, S. L., & Weissman, I. L. (2011). Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proceedings of the National Academy of Sciences of the United States of America, 108(50), 20012–20017. https://doi.org/10.1073/pnas.1116110108.
Yu, K. R., Espinoza, D. A., Wu, C., Truitt, L., Shin, T. H., Chen, S., Fan, X., Yabe, I. M., Panch, S., Hong, S. G., Koelle, S., Lu, R., Bonifacino, A., Krouse, A., Metzger, M., Donahue, R. E., & Dunbar, C. E. (2018). The impact of aging on primate hematopoiesis as interrogated by clonal tracking. Blood, 131(11), 1195–1205. https://doi.org/10.1182/blood-2017-08-802033.
Di Micco, R., & Montini, E. (2018). De(bar)coding aged hematopoiesis in primates. Blood, 131(11), 1157–1159. https://doi.org/10.1182/blood-2018-02-826412.
Pietras, E. M., Reynaud, D., Kang, Y. A., Carlin, D., Calero-Nieto, F. J., Leavitt, A. D., Stuart, J. M., Göttgens, B., & Passegué, E. (2015). Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell, 17(1), 35–46. https://doi.org/10.1016/j.stem.2015.05.003.
Young, K., Borikar, S., Bell, R., Kuffler, L., Philip, V., & Trowbridge, J. J. (2016). Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. The Journal of Experimental Medicine, 213(11), 2259–2267. https://doi.org/10.1084/jem.20160168.
Busque, L., Buscarlet, M., Mollica, L., & Levine, R. L. (2018). Concise review: age-related clonal hematopoiesis: stem cells tempting the devil. Stem Cells (Dayton, Ohio), 36(9), 1287–1294. https://doi.org/10.1002/stem.2845.
Wahlestedt, M., & Bryder, D. (2017). The slippery slope of hematopoietic stem cell aging. Experimental Hematology, 56, 1–6. https://doi.org/10.1016/j.exphem.2017.09.008.
de Haan, G., & Lazare, S. S. (2018). Aging of hematopoietic stem cells. Blood, 131(5), 479–487. https://doi.org/10.1182/blood-2017-06-746412.
Stauder, R., Valent, P., & Theurl, I. (2018). Anemia at older age: etiologies, clinical implications, and management. Blood, 131(5), 505–514. https://doi.org/10.1182/blood-2017-07-746446.
Leins, H., Mulaw, M., Eiwen, K., Sakk, V., Liang, Y., Denkinger, M., Geiger, H., & Schirmbeck, R. (2018). Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood, 132(6), 565–576. https://doi.org/10.1182/blood-2018-02-831065.
Ganuza, M., Hall, T., Finkelstein, D., Wang, Y. D., Chabot, A., Kang, G., Bi, W., Wu, G., & McKinney-Freeman, S. (2019). The global clonal complexity of the murine blood system declines throughout life and after serial transplantation. Blood, 133(18), 1927–1942. https://doi.org/10.1182/blood-2018-09-873059.
Van Zant, G., & Liang, Y. (2012). Concise review: hematopoietic stem cell aging, life span, and transplantation. Stem Cells Translational Medicine, 1(9), 651–657. https://doi.org/10.5966/sctm.2012-0033.
Geiger, H., de Haan, G., & Florian, M. C. (2013). The ageing haematopoietic stem cell compartment. Nature Reviews Immunology, 13(5), 376–389. https://doi.org/10.1038/nri3433.
Cupit-Link, M. C., Arora, M., Wood, W. A., & Hashmi, S. K. (2018). Relationship between aging and hematopoietic cell transplantation. Biology of Blood and Marrow Transplantation : Journal of the American Society for Blood and Marrow Transplantation, 24(10), 1965–1970. https://doi.org/10.1016/j.bbmt.2018.08.015.
Cooper, S. H., Broxmeyer, H. E., & Capitano, M. L. (2020). Experimental mouse models of mouse and human hematopoietic stem cell transplantation. In L. M. Pelus & J. Hoggatt (Eds.), Methods in molecular biology: Hematopoietic stem cells. New York: Springer Nature In Press.
Broxmeyer, H. E., Orschell, C. M., Clapp, D. W., Hangoc, G., Cooper, S., Plett, P. A., Liles, W. C., Li, X., Graham-Evans, B., Campbell, T. B., Calandra, G., Bridger, G., Dale, D. C., & Srour, E. F. (2005). Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. The Journal of Experimental Medicine, 201(8), 1307–1318. https://doi.org/10.1084/jem.20041385.
Mantel, C. R., O'Leary, H. A., Chitteti, B. R., Huang, X., Cooper, S., Hangoc, G., Brustovetsky, N., Srour, E. F., Lee, M. R., Messina-Graham, S., Haas, D. M., Falah, N., Kapur, R., Pelus, L. M., Bardeesy, N., Fitamant, J., Ivan, M., Kim, K. S., & Broxmeyer, H. E. (2015). Enhancing hematopoietic stem cell transplantation efficacy by mitigating oxygen shock. Cell, 161(7), 1553–1565. https://doi.org/10.1016/j.cell.2015.04.054.
Broxmeyer, H. E., O'Leary, H. A., Huang, X., & Mantel, C. (2015). The importance of hypoxia and extra physiologic oxygen shock/stress for collection and processing of stem and progenitor cells to understand true physiology/pathology of these cells ex vivo. Current Opinion in Hematology, 22(4), 273–278. https://doi.org/10.1097/MOH.0000000000000144.
Aljoufi, A., Cooper, S., & Broxmeyer, H.E. (2020). Collection and processing of mobilized mouse peripheral blood at lowered oxygen tension yields enhanced numbers of hematopoietic stem cells. Stem Cell Reviews and Reports. In Press.
Burns, S. S., & Kapur, R. (2020). Clonal hematopoiesis of indeterminate potential as a novel risk factor for donor-derived leukemia. Stem Cell Reports, 15(2), 279–291. https://doi.org/10.1016/j.stemcr.2020.07.008.
Burns, S. S., & Kapur, R. (2020). Putative mechanisms underlying cardiovascular disease associated with clonal hematopoiesis of indeterminate potential. Stem Cell Reports, 15(2), 292–306. https://doi.org/10.1016/j.stemcr.2020.06.021.
Shlush, L. I. (2018). Age-related clonal hematopoiesis. Blood, 131(5), 496–504. https://doi.org/10.1182/blood-2017-07-746453.
Fabre, M. A., McKerrell, T., Zwiebel, M., Vijayabaskar, M. S., Park, N., Wells, P. M., Rad, R., Deloukas, P., Small, K., Steves, C. J., & Vassiliou, G. S. (2020). Concordance for clonal hematopoiesis is limited in elderly twins. Blood, 135(4), 269–273. https://doi.org/10.1182/blood.2019001807.
Hansen, J. W., Pedersen, D. A., Larsen, L. A., Husby, S., Clemmensen, S. B., Hjelmborg, J., Favero, F., Weischenfeldt, J., Christensen, K., & Grønbæk, K. (2020). Clonal hematopoiesis in elderly twins: concordance, discordance, and mortality. Blood, 135(4), 261–268. https://doi.org/10.1182/blood.2019001793.
Steensma, D. P., Bejar, R., Jaiswal, S., Lindsley, R. C., Sekeres, M. A., Hasserjian, R. P., & Ebert, B. L. (2015). Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood, 126(1), 9–16. https://doi.org/10.1182/blood-2015-03-631747.
Pardali, E., Dimmeler, S., Zeiher, A. M., & Rieger, M. A. (2020). Clonal hematopoiesis, aging, and cardiovascular diseases. Experimental Hematology, 83, 95–104. https://doi.org/10.1016/j.exphem.2019.12.006.
Chen, S., Gao, R., Yao, C., Kobayashi, M., Liu, S. Z., Yoder, M. C., Broxmeyer, H., Kapur, R., Boswell, H. S., Mayo, L. D., & Liu, Y. (2018). Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia, 32(3), 850–854. https://doi.org/10.1038/leu.2017.325.
Nabinger, S. C., Chen, S., Gao, R., Yao, C., Kobayashi, M., Vemula, S., Fahey, A. C., Wang, C., Daniels, C., Boswell, H. S., Sandusky, G. E., Mayo, L. D., Kapur, R., & Liu, Y. (2019). Mutant p53 enhances leukemia-initiating cell self-renewal to promote leukemia development. Leukemia, 33(6), 1535–1539. https://doi.org/10.1038/s41375-019-0377-0.
Chen, S., Wang, Q., Yu, H., Capitano, M. L., Vemula, S., Nabinger, S. C., Gao, R., Yao, C., Kobayashi, M., Geng, Z., Fahey, A., Henley, D., Liu, S. Z., Barajas, S., Cai, W., Wolf, E. R., Ramdas, B., Cai, Z., Gao, H., Luo, N., et al. (2019). Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nature Communications, 10(1), 5649. https://doi.org/10.1038/s41467-019-13542-2.
Sperling, A. S., Gibson, C. J., & Ebert, B. L. (2017). The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nature Reviews Cancer, 17(1), 5–19. https://doi.org/10.1038/nrc.2016.112.
Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., Lindsley, R. C., Mermel, C. H., Burtt, N., Chavez, A., Higgins, J. M., Moltchanov, V., Kuo, F. C., Kluk, M. J., Henderson, B., Kinnunen, L., Koistinen, H. A., Ladenvall, C., Getz, G., Correa, A., et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. The New England Journal of Medicine, 371(26), 2488–2498. https://doi.org/10.1056/NEJMoa1408617.
Genovese, G., Kähler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., Chambert, K., Mick, E., Neale, B. M., Fromer, M., Purcell, S. M., Svantesson, O., Landén, M., Höglund, M., Lehmann, S., Gabriel, S. B., Moran, J. L., Lander, E. S., Sullivan, P. F., Sklar, P., et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England Journal of Medicine, 371(26), 2477–2487. https://doi.org/10.1056/NEJMoa1409405.
Xie, M., Lu, C., Wang, J., McLellan, M. D., Johnson, K. J., Wendl, M. C., McMichael, J. F., Schmidt, H. K., Yellapantula, V., Miller, C. A., Ozenberger, B. A., Welch, J. S., Link, D. C., Walter, M. J., Mardis, E. R., Dipersio, J. F., Chen, F., Wilson, R. K., Ley, T. J., & Ding, L. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine, 20(12), 1472–1478. https://doi.org/10.1038/nm.3733.
Young, A. L., Challen, G. A., Birmann, B. M., & Druley, T. E. (2016). Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nature Communications, 7, 12484. https://doi.org/10.1038/ncomms12484.
Zink, F., Stacey, S. N., Norddahl, G. L., Frigge, M. L., Magnusson, O. T., Jonsdottir, I., Thorgeirsson, T. E., Sigurdsson, A., Gudjonsson, S. A., Gudmundsson, J., Jonasson, J. G., Tryggvadottir, L., Jonsson, T., Helgason, A., Gylfason, A., Sulem, P., Rafnar, T., Thorsteinsdottir, U., Gudbjartsson, D. F., Masson, G., et al. (2017). Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood, 130(6), 742–752. https://doi.org/10.1182/blood-2017-02-769869.
Xia, J., Miller, C. A., Baty, J., Ramesh, A., Jotte, M., Fulton, R. S., Vogel, T. P., Cooper, M. A., Walkovich, K. J., Makaryan, V., Bolyard, A. A., Dinauer, M. C., Wilson, D. B., Vlachos, A., Myers, K. C., Rothbaum, R. J., Bertuch, A. A., Dale, D. C., Shimamura, A., Boxer, L. A., et al. (2018). Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood, 131(4), 408–416. https://doi.org/10.1182/blood-2017-08-801985.
Fiscella, M., Zhang, H., Fan, S., Sakaguchi, K., Shen, S., Mercer, W. E., Vande Woude, G. F., O'Connor, P. M., & Appella, E. (1997). Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proceedings of the National Academy of Sciences of the United States of America, 94(12), 6048–6053. https://doi.org/10.1073/pnas.94.12.6048.
Hsu, J. I., Dayaram, T., Tovy, A., De Braekeleer, E., Jeong, M., Wang, F., Zhang, J., Heffernan, T. P., Gera, S., Kovacs, J. J., Marszalek, J. R., Bristow, C., Yan, Y., Garcia-Manero, G., Kantarjian, H., Vassiliou, G., Futreal, P. A., Donehower, L. A., Takahashi, K., & Goodell, M. A. (2018). PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell, 23(5), 700–713.e6. https://doi.org/10.1016/j.stem.2018.10.004.
Kahn, J. D., Miller, P. G., Silver, A. J., Sellar, R. S., Bhatt, S., Gibson, C., McConkey, M., Adams, D., Mar, B., Mertins, P., Fereshetian, S., Krug, K., Zhu, H., Letai, A., Carr, S. A., Doench, J., Jaiswal, S., & Ebert, B. L. (2018). PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood, 132(11), 1095–1105. https://doi.org/10.1182/blood-2018-05-850339.
Coombs, C. C., Zehir, A., Devlin, S. M., Kishtagari, A., Syed, A., Jonsson, P., Hyman, D. M., Solit, D. B., Robson, M. E., Baselga, J., Arcila, M. E., Ladanyi, M., Tallman, M. S., Levine, R. L., & Berger, M. F. (2017). Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell, 21(3), 374–382.e4. https://doi.org/10.1016/j.stem.2017.07.010.
Wong, T. N., Miller, C. A., Jotte, M., Bagegni, N., Baty, J. D., Schmidt, A. P., Cashen, A. F., Duncavage, E. J., Helton, N. M., Fiala, M., Fulton, R. S., Heath, S. E., Janke, M., Luber, K., Westervelt, P., Vij, R., DiPersio, J. F., Welch, J. S., Graubert, T. A., Walter, M. J., et al. (2018). Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nature Communications, 9(1), 455. https://doi.org/10.1038/s41467-018-02858-0.
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C., & Santoro, A. (2018). Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology, 14(10), 576–590. https://doi.org/10.1038/s41574-018-0059-4.
Franceschi, C., Garagnani, P., Vitale, G., Capri, M., & Salvioli, S. (2017). Inflammaging and ‘Garb-aging’. Trends in Endocrinology and Metabolism: TEM, 28(3), 199–212. https://doi.org/10.1016/j.tem.2016.09.005.
Sallman, D. A., & List, A. (2019). The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood, 133(10), 1039–1048. https://doi.org/10.1182/blood-2018-10-844654.
Fuster, J. J., MacLauchlan, S., Zuriaga, M. A., Polackal, M. N., Ostriker, A. C., Chakraborty, R., Wu, C. L., Sano, S., Muralidharan, S., Rius, C., Vuong, J., Jacob, S., Muralidhar, V., Robertson, A. A., Cooper, M. A., Andrés, V., Hirschi, K. K., Martin, K. A., & Walsh, K. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, N.Y.), 355(6327), 842–847. https://doi.org/10.1126/science.aag1381.
Guo, H., Callaway, J. B., & Ting, J. P. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature Medicine, 21(7), 677–687. https://doi.org/10.1038/nm.3893.
Basiorka, A. A., McGraw, K. L., Eksioglu, E. A., Chen, X., Johnson, J., Zhang, L., Zhang, Q., Irvine, B. A., Cluzeau, T., Sallman, D. A., Padron, E., Komrokji, R., Sokol, L., Coll, R. C., Robertson, A. A., Cooper, M. A., Cleveland, J. L., O'Neill, L. A., Wei, S., & List, A. F. (2016). The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood, 128(25), 2960–2975. https://doi.org/10.1182/blood-2016-07-730556.
Masters, S. L., Gerlic, M., Metcalf, D., Preston, S., Pellegrini, M., O'Donnell, J. A., McArthur, K., Baldwin, T. M., Chevrier, S., Nowell, C. J., Cengia, L. H., Henley, K. J., Collinge, J. E., Kastner, D. L., Feigenbaum, L., Hilton, D. J., Alexander, W. S., Kile, B. T., & Croker, B. A. (2012). NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity, 37(6), 1009–1023. https://doi.org/10.1016/j.immuni.2012.08.027.
Cai, Z., Kotzin, J. J., Ramdas, B., Chen, S., Nelanuthala, S., Palam, L. R., Pandey, R., Mali, R. S., Liu, Y., Kelley, M. R., Sandusky, G., Mohseni, M., Williams, A., Henao-Mejia, J., & Kapur, R. (2018). Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell, 23(6), 833–849.e5. https://doi.org/10.1016/j.stem.2018.10.013.
Wahl, M. C., Will, C. L., & Lührmann, R. (2009). The spliceosome: design principles of a dynamic RNP machine. Cell, 136(4), 701–718. https://doi.org/10.1016/j.cell.2009.02.009.
Saltzman, A. L., Pan, Q., & Blencowe, B. J. (2011). Regulation of alternative splicing by the core spliceosomal machinery. Genes & Development, 25(4), 373–384. https://doi.org/10.1101/gad.2004811.
Harries, L. W., Hernandez, D., Henley, W., Wood, A. R., Holly, A. C., Bradley-Smith, R. M., Yaghootkar, H., Dutta, A., Murray, A., Frayling, T. M., Guralnik, J. M., Bandinelli, S., Singleton, A., Ferrucci, L., & Melzer, D. (2011). Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell, 10(5), 868–878. https://doi.org/10.1111/j.1474-9726.2011.00726.x.
Deschênes, M., & Chabot, B. (2017). The emerging role of alternative splicing in senescence and aging. Aging Cell, 16(5), 918–933. https://doi.org/10.1111/acel.12646.
Lee, B. P., Pilling, L. C., Emond, F., Flurkey, K., Harrison, D. E., Yuan, R., Peters, L. L., Kuchel, G. A., Ferrucci, L., Melzer, D., & Harries, L. W. (2016). Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell, 15(5), 903–913. https://doi.org/10.1111/acel.12499.
Crews, L. A., Balaian, L., Delos Santos, N. P., Leu, H. S., Court, A. C., Lazzari, E., Sadarangani, A., Zipeto, M. A., La Clair, J. J., Villa, R., Kulidjian, A., Storb, R., Morris, S. R., Ball, E. D., Burkart, M. D., & Jamieson, C. (2016). RNA splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell, 19(5), 599–612. https://doi.org/10.1016/j.stem.2016.08.003.
Sun, D., Luo, M., Jeong, M., Rodriguez, B., Xia, Z., Hannah, R., Wang, H., Le, T., Faull, K. F., Chen, R., Gu, H., Bock, C., Meissner, A., Göttgens, B., Darlington, G. J., Li, W., & Goodell, M. A. (2014). Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell, 14(5), 673–688. https://doi.org/10.1016/j.stem.2014.03.002.
McKerrell, T., Park, N., Moreno, T., Grove, C. S., Ponstingl, H., Stephens, J., Understanding Society Scientific Group, Crawley, C., Craig, J., Scott, M. A., Hodkinson, C., Baxter, J., Rad, R., Forsyth, D. R., Quail, M. A., Zeggini, E., Ouwehand, W., Varela, I., & Vassiliou, G. S. (2015). Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Reports, 10(8), 1239–1245. https://doi.org/10.1016/j.celrep.2015.02.005.
Yoshida, K., Sanada, M., Shiraishi, Y., Nowak, D., Nagata, Y., Yamamoto, R., Sato, Y., Sato-Otsubo, A., Kon, A., Nagasaki, M., Chalkidis, G., Suzuki, Y., Shiosaka, M., Kawahata, R., Yamaguchi, T., Otsu, M., Obara, N., Sakata-Yanagimoto, M., Ishiyama, K., Mori, H., et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature, 478(7367), 64–69. https://doi.org/10.1038/nature10496.
Papaemmanuil, E., Cazzola, M., Boultwood, J., Malcovati, L., Vyas, P., Bowen, D., Pellagatti, A., Wainscoat, J. S., Hellstrom-Lindberg, E., Gambacorti-Passerini, C., Godfrey, A. L., Rapado, I., Cvejic, A., Rance, R., McGee, C., Ellis, P., Mudie, L. J., Stephens, P. J., McLaren, S., Massie, C. E., et al. (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. The New England Journal of Medicine, 365(15), 1384–1395. https://doi.org/10.1056/NEJMoa1103283.
Lee, S. C., North, K., Kim, E., Jang, E., Obeng, E., Lu, S. X., Liu, B., Inoue, D., Yoshimi, A., Ki, M., Yeo, M., Zhang, X. J., Kim, M. K., Cho, H., Chung, Y. R., Taylor, J., Durham, B. H., Kim, Y. J., Pastore, A., Monette, S., et al. (2018). Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell, 34(2), 225–241.e8. https://doi.org/10.1016/j.ccell.2018.07.003.
Pietras, E. M. (2017). Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood, 130(15), 1693–1698. https://doi.org/10.1182/blood-2017-06-780882.
Boettcher, S., & Manz, M. G. (2017). Regulation of inflammation- and infection-driven hematopoiesis. Trends in Immunology, 38(5), 345–357. https://doi.org/10.1016/j.it.2017.01.004.
Boettcher, S., Gerosa, R. C., Radpour, R., Bauer, J., Ampenberger, F., Heikenwalder, M., Kopf, M., & Manz, M. G. (2014). Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood, 124(9), 1393–1403. https://doi.org/10.1182/blood-2014-04-570762.
Ziegler, P., Boettcher, S., Takizawa, H., Manz, M. G., & Brümmendorf, T. H. (2016). LPS-stimulated human bone marrow stroma cells support myeloid cell development and progenitor cell maintenance. Annals of Hematology, 95(2), 173–178. https://doi.org/10.1007/s00277-015-2550-5.
Fernandez, L., Rodriguez, S., Huang, H., Chora, A., Fernandes, J., Mumaw, C., Cruz, E., Pollok, K., Cristina, F., Price, J. E., Ferkowicz, M. J., Scadden, D. T., Clauss, M., Cardoso, A. A., & Carlesso, N. (2008). Tumor necrosis factor-alpha and endothelial cells modulate Notch signaling in the bone marrow microenvironment during inflammation. Experimental Hematology, 36(5), 545–558. https://doi.org/10.1016/j.exphem.2007.12.012.
Furusawa, J., Mizoguchi, I., Chiba, Y., Hisada, M., Kobayashi, F., Yoshida, H., Nakae, S., Tsuchida, A., Matsumoto, T., Ema, H., Mizuguchi, J., & Yoshimoto, T. (2016). Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathogens, 12(3), e1005507. https://doi.org/10.1371/journal.ppat.1005507.
Baldridge, M. T., King, K. Y., Boles, N. C., Weksberg, D. C., & Goodell, M. A. (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature, 465(7299), 793–797. https://doi.org/10.1038/nature09135.
Takizawa, H., Fritsch, K., Kovtonyuk, L. V., Saito, Y., Yakkala, C., Jacobs, K., Ahuja, A. K., Lopes, M., Hausmann, A., Hardt, W. D., Gomariz, Á., Nombela-Arrieta, C., & Manz, M. G. (2017). Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell, 21(2), 225–240.e5. https://doi.org/10.1016/j.stem.2017.06.013.
Hirche, C., Frenz, T., Haas, S. F., Döring, M., Borst, K., Tegtmeyer, P. K., Brizic, I., Jordan, S., Keyser, K., Chhatbar, C., Pronk, E., Lin, S., Messerle, M., Jonjic, S., Falk, C. S., Trumpp, A., Essers, M., & Kalinke, U. (2017). Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Reports, 19(11), 2345–2356. https://doi.org/10.1016/j.celrep.2017.05.063.
Matatall, K. A., Jeong, M., Chen, S., Sun, D., Chen, F., Mo, Q., Kimmel, M., & King, K. Y. (2016). Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Reports, 17(10), 2584–2595. https://doi.org/10.1016/j.celrep.2016.11.031.
Schulte-Schrepping, J., Reusch, N., Paclik, D., Baßler, K., Schlickeiser, S., Zhang, B., Krämer, B., Krammer, T., Brumhard, S., Bonaguro, L., De Domenico, E., Wendisch, D., Grasshoff, M., Kapellos, T. S., Beckstette, M., Pecht, T., Saglam, A., Dietrich, O., Mei, H. E., Schulz, A. R., et al. (2020). Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell, 182(6), 1419–1440.e23. Advance online publication. https://doi.org/10.1016/j.cell.2020.08.001.
Zhang, H., Rodriguez, S., Wang, L., Wang, S., Serezani, H., Kapur, R., Cardoso, A. A., & Carlesso, N. (2016). Sepsis induces hematopoietic stem cell exhaustion and myelosuppression through distinct contributions of TRIF and MYD88. Stem Cell Reports, 6(6), 940–956. https://doi.org/10.1016/j.stemcr.2016.05.002.
Josefsdottir, K. S., Baldridge, M. T., Kadmon, C. S., & King, K. Y. (2017). Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood, 129(6), 729–739. https://doi.org/10.1182/blood-2016-03-708594.
Balmer, M. L., Schürch, C. M., Saito, Y., Geuking, M. B., Li, H., Cuenca, M., Kovtonyuk, L. V., McCoy, K. D., Hapfelmeier, S., Ochsenbein, A. F., Manz, M. G., Slack, E., & Macpherson, A. J. (2014). Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. Journal of Immunology (Baltimore, Md. : 1950), 193(10), 5273–5283. https://doi.org/10.4049/jimmunol.1400762.
Meisel, M., Hinterleitner, R., Pacis, A., Chen, L., Earley, Z. M., Mayassi, T., Pierre, J. F., Ernest, J. D., Galipeau, H. J., Thuille, N., Bouziat, R., Buscarlet, M., Ringus, D. L., Wang, Y., Li, Y., Dinh, V., Kim, S. M., McDonald, B. D., Zurenski, M. A., Musch, M. W., et al. (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature, 557(7706), 580–584. https://doi.org/10.1038/s41586-018-0125-z.
Zhang, Q., & Casanova, J. L. (2020). Human TET2 bridges cancer and immunity. Blood, 136(9), 1018–1019. https://doi.org/10.1182/blood.2020006881.
Stremenova Spegarova, J., Lawless, D., Mohamad, S., Engelhardt, K. R., Doody, G., Shrimpton, J., Rensing-Ehl, A., Ehl, S., Rieux-Laucat, F., Cargo, C., Griffin, H., Mikulasova, A., Acres, M., Morgan, N. V., Poulter, J. A., Sheridan, E. G., Chetcuti, P., O'Riordan, S., Anwar, R., Carter, C. R., et al. (2020). Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood, 136(9), 1055–1066. https://doi.org/10.1182/blood.2020005844.
Zeng, H., He, H., Guo, L., Li, J., Lee, M., Han, W., Guzman, A. G., Zang, S., Zhou, Y., Zhang, X., Goodell, M. A., King, K. Y., Sun, D., & Huang, Y. (2019). Antibiotic treatment ameliorates Ten-eleven translocation 2 (TET2) loss-of-function associated hematological malignancies. Cancer Letters, 467, 1–8. https://doi.org/10.1016/j.canlet.2019.09.013.
Cai, Z., Kotzin, J. J., Ramdas, B., Chen, S., Nelanuthala, S., Palam, L. R., Pandey, R., Mali, R. S., Liu, Y., Kelley, M. R., Sandusky, G., Mohseni, M., Williams, A., Henao-Mejia, J., & Kapur, R. (2018). Inhibition of inflammatory signaling in tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell, 23(6), 833–849.e5. https://doi.org/10.1016/j.stem.2018.10.013.
Reynaud, D., Pietras, E., Barry-Holson, K., Mir, A., Binnewies, M., Jeanne, M., Sala-Torra, O., Radich, J. P., & Passegué, E. (2011). IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell, 20(5), 661–673. https://doi.org/10.1016/j.ccr.2011.10.012.
Mager, L. F., Riether, C., Schürch, C. M., Banz, Y., Wasmer, M. H., Stuber, R., Theocharides, A. P., Li, X., Xia, Y., Saito, H., Nakae, S., Baerlocher, G. M., Manz, M. G., McCoy, K. D., Macpherson, A. J., Ochsenbein, A. F., Beutler, B., & Krebs, P. (2015). IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. The Journal of Clinical Investigation, 125(7), 2579–2591. https://doi.org/10.1172/JCI77347.
Zambetti, N. A., Ping, Z., Chen, S., Kenswil, K., Mylona, M. A., Sanders, M. A., Hoogenboezem, R. M., Bindels, E., Adisty, M. N., Van Strien, P., van der Leije, C. S., Westers, T. M., Cremers, E., Milanese, C., Mastroberardino, P. G., van Leeuwen, J., van der Eerden, B., Touw, I. P., Kuijpers, T. W., Kanaar, R., et al. (2016). Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell, 19(5), 613–627. https://doi.org/10.1016/j.stem.2016.08.021.
Hasselbalch, H. C. (2013). Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leukemia Researsch, 37(2), 214–220. https://doi.org/10.1016/j.leukres.2012.10.020.
Pietras, E. M., Mirantes-Barbeito, C., Fong, S., Loeffler, D., Kovtonyuk, L. V., Zhang, S., Lakshminarasimhan, R., Chin, C. P., Techner, J. M., Will, B., Nerlov, C., Steidl, U., Manz, M. G., Schroeder, T., & Passegué, E. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nature Cell Biology, 18(6), 607–618. https://doi.org/10.1038/ncb3346.
Muto, T., Walker, C. S., Choi, K., Hueneman, K., Smith, M. A., Gul, Z., Garcia-Manero, G., Ma, A., Zheng, Y., & Starczynowski, D. T. (2020). Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nature Immunology, 21(5), 535–545. https://doi.org/10.1038/s41590-020-0663-z.
Mann, M., Mehta, A., de Boer, C. G., Kowalczyk, M. S., Lee, K., Haldeman, P., Rogel, N., Knecht, A. R., Farouq, D., Regev, A., & Baltimore, D. (2018). Heterogeneous responses of hematopoietic stem cells to inflammatory stimuli are altered with age. Cell Reports, 25(11), 2992–3005.e5. https://doi.org/10.1016/j.celrep.2018.11.056.
Kovtonyuk, L. V., Fritsch, K., Feng, X., Manz, M. G., & Takizawa, H. (2016). Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Frontiers in Immunology, 7, 502. https://doi.org/10.3389/fimmu.2016.00502.
Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., Lindsley, R. C., Mermel, C. H., Burtt, N., Chavez, A., Higgins, J. M., Moltchanov, V., Kuo, F. C., Kluk, M. J., Henderson, B., Kinnunen, L., Koistinen, H. A., Ladenvall, C., Getz, G., Correa, A., et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. The New England Journal of Medicine, 371(26), 2488–2498. https://doi.org/10.1056/NEJMoa1408617.
Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., McConkey, M., Gupta, N., Gabriel, S., Ardissino, D., Baber, U., Mehran, R., Fuster, V., Danesh, J., Frossard, P., Saleheen, D., Melander, O., Sukhova, G. K., Neuberg, D., Libby, P., et al. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. The New England Journal of Medicine, 377(2), 111–121. https://doi.org/10.1056/NEJMoa1701719.
Abegunde, S. O., Buckstein, R., Wells, R. A., & Rauh, M. J. (2018). An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Experimental Hematology, 59, 60–65. https://doi.org/10.1016/j.exphem.2017.11.002.
Yu, V., Yusuf, R. Z., Oki, T., Wu, J., Saez, B., Wang, X., Cook, C., Baryawno, N., Ziller, M. J., Lee, E., Gu, H., Meissner, A., Lin, C. P., Kharchenko, P. V., & Scadden, D. T. (2016). Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell, 167(5), 1310–1322.e17. https://doi.org/10.1016/j.cell.2016.10.045.
Ramdas, B., Mali, R. S., Palam, L. R., Pandey, R., Cai, Z., Pasupuleti, S. K., Burns, S. S., & Kapur, R. (2020). Driver mutations in leukemia promote disease pathogenesis through a combination of cell-autonomous and niche modulation. Stem Cell Reports, 15(1), 95–109. https://doi.org/10.1016/j.stemcr.2020.05.002.
Chavakis, T., Mitroulis, I., & Hajishengallis, G. (2019). Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nature Immunology, 20(7), 802–811. https://doi.org/10.1038/s41590-019-0402-5.
Gnani, D., Crippa, S., Della Volpe, L., Rossella, V., Conti, A., Lettera, E., Rivis, S., Ometti, M., Fraschini, G., Bernardo, M. E., & Di Micco, R. (2019). An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell, 18(3), e12933. https://doi.org/10.1111/acel.12933.
Batsivari, A., Haltalli, M., Passaro, D., Pospori, C., Lo Celso, C., & Bonnet, D. (2020). Dynamic responses of the haematopoietic stem cell niche to diverse stresses. Nature Cell Biology, 22(1), 7–17. https://doi.org/10.1038/s41556-019-0444-9.
Dragoljevic, D., Westerterp, M., Veiga, C. B., Nagareddy, P., & Murphy, A. J. (2018). Disordered haematopoiesis and cardiovascular disease: a focus on myelopoiesis. Clinical Science (London, England : 1979), 132(17), 1889–1899. https://doi.org/10.1042/CS20180111.
Zhang, C., Nix, D., Gregory, M., Ciorba, M. A., Ostrander, E. L., Newberry, R. D., Spencer, D. H., & Challen, G. A. (2019). Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Experimental Hematology, 80, 36–41.e3. https://doi.org/10.1016/j.exphem.2019.11.008.
Ricard, L., Hirsch, P., Largeaud, L., Deswarte, C., Jachiet, V., Mohty, M., Rivière, S., Malard, F., Tenon, M., de Vassoigne, F., Fain, O., Gaugler, B., Rossignol, J., Delhommeau, F., & Mekinian, A. (2020). Clonal haematopoiesis is increased in early onset in systemic sclerosis. Rheumatology (Oxford, England), keaa282. https://doi.org/10.1093/rheumatology/keaa282. Advance online publication.
Burns, S. S., & Kapur, R. (2020). Clonal hematopoiesis of indeterminate potential as a novel risk factor for donor-derived leukemia. Stem Cell Reports, 15(2), 279–291. https://doi.org/10.1016/j.stemcr.2020.07.008.
Bogeska, R., Kaschutnig, P., Fawaz, M., Mikecin, A-M., Büchler-Schäff, M., Paffenholz, S., Asada, N., Frauhammer, F., Buettner, F., Ball, M., Knoch, J., Stäble, S., Walter, D., Petri, A., Carreño-Gonzalez, M.J., Wagner, V., Brors, B., Haas, S., Lipka, D.B., Essers, M.A.G., Holland-Letz, T., Mallm, J-P., Rippe, K., Frenette, P.S., Rieger, M.A., & Milsom, M.D. (2020). Hematopoietic stem cells fail to regenerate following inflammatory challenge. bioRxiv 2020.08.01.230433. https://doi.org/10.1101/2020.08.01.230433.
Rossi, D. J., Jamieson, C. H., & Weissman, I. L. (2008). Stems cells and the pathways to aging and cancer. Cell, 132(4), 681–696. https://doi.org/10.1016/j.cell.2008.01.036.
Moehrle, B. M., & Geiger, H. (2016). Aging of hematopoietic stem cells: DNA damage and mutations? Experimental Hematology, 44(10), 895–901. https://doi.org/10.1016/j.exphem.2016.06.253.
Lombard, D. B., Chua, K. F., Mostoslavsky, R., Franco, S., Gostissa, M., & Alt, F. W. (2005). DNA repair, genome stability, and aging. Cell, 120(4), 497–512. https://doi.org/10.1016/j.cell.2005.01.028.
Ou, H. L., & Schumacher, B. (2018). DNA damage responses and p53 in the aging process. Blood, 131(5), 488–495. https://doi.org/10.1182/blood-2017-07-746396.
Sen, P., Shah, P. P., Nativio, R., & Berger, S. L. (2016). Epigenetic mechanisms of longevity and aging. Cell, 166(4), 822–839. https://doi.org/10.1016/j.cell.2016.07.050.
Rossi, D. J., Bryder, D., Seita, J., Nussenzweig, A., Hoeijmakers, J., & Weissman, I. L. (2007). Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature, 447(7145), 725–729. https://doi.org/10.1038/nature05862.
Florian, M. C., Nattamai, K. J., Dörr, K., Marka, G., Uberle, B., Vas, V., Eckl, C., Andrä, I., Schiemann, M., Oostendorp, R. A., Scharffetter-Kochanek, K., Kestler, H. A., Zheng, Y., & Geiger, H. (2013). A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature, 503(7476), 392–396. https://doi.org/10.1038/nature12631.
Flach, J., Bakker, S. T., Mohrin, M., Conroy, P. C., Pietras, E. M., Reynaud, D., Alvarez, S., Diolaiti, M. E., Ugarte, F., Forsberg, E. C., Le Beau, M. M., Stohr, B. A., Méndez, J., Morrison, C. G., & Passegué, E. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature, 512(7513), 198–202. https://doi.org/10.1038/nature13619.
Adams, P. D., Jasper, H., & Rudolph, K. L. (2015). Aging-induced stem cell mutations as drivers for disease and cancer. Cell Stem Cell, 16(6), 601–612. https://doi.org/10.1016/j.stem.2015.05.002.
Beerman, I., Bock, C., Garrison, B. S., Smith, Z. D., Gu, H., Meissner, A., & Rossi, D. J. (2013). Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell, 12(4), 413–425. https://doi.org/10.1016/j.stem.2013.01.017.
Beerman, I., & Rossi, D. J. (2015). Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell, 16(6), 613–625. https://doi.org/10.1016/j.stem.2015.05.009.
Sun, D., Luo, M., Jeong, M., Rodriguez, B., Xia, Z., Hannah, R., Wang, H., Le, T., Faull, K. F., Chen, R., Gu, H., Bock, C., Meissner, A., Göttgens, B., Darlington, G. J., Li, W., & Goodell, M. A. (2014). Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell, 14(5), 673–688. https://doi.org/10.1016/j.stem.2014.03.002.
Hennrich, M. L., Romanov, N., Horn, P., Jaeger, S., Eckstein, V., Steeples, V., Ye, F., Ding, X., Poisa-Beiro, L., Lai, M. C., Lang, B., Boultwood, J., Luft, T., Zaugg, J. B., Pellagatti, A., Bork, P., Aloy, P., Gavin, A. C., & Ho, A. D. (2018). Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nature Communications, 9(1), 4004. https://doi.org/10.1038/s41467-018-06353-4.
Grover, A., Sanjuan-Pla, A., Thongjuea, S., Carrelha, J., Giustacchini, A., Gambardella, A., Macaulay, I., Mancini, E., Luis, T. C., Mead, A., Jacobsen, S. E., & Nerlov, C. (2016). Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nature Communications, 7, 11075. https://doi.org/10.1038/ncomms11075.
Singh, S. K., Singh, S., Gadomski, S., Sun, L., Pfannenstein, A., Magidson, V., Chen, X., Kozlov, S., Tessarollo, L., Klarmann, K. D., & Keller, J. R. (2018). Id1 ablation protects hematopoietic stem cells from stress-induced exhaustion and aging. Cell Stem Cell, 23(2), 252–265.e8. https://doi.org/10.1016/j.stem.2018.06.001.
Chandel, N. S., Jasper, H., Ho, T. T., & Passegué, E. (2016). Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nature Cell Biology, 18(8), 823–832. https://doi.org/10.1038/ncb3385.
Balaban, R. S., Nemoto, S., & Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell, 120(4), 483–495. https://doi.org/10.1016/j.cell.2005.02.001.
Wiley, C. D., & Campisi, J. (2016). From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metabolism, 23(6), 1013–1021. https://doi.org/10.1016/j.cmet.2016.05.010.
Snoeck, H. W. (2015). Can metabolic mechanisms of stem cell maintenance explain aging and the immortal germline? Cell Stem Cell, 16(6), 582–584. https://doi.org/10.1016/j.stem.2015.04.021.
Ahlqvist, K. J., Suomalainen, A., & Hämäläinen, R. H. (2015). Stem cells, mitochondria and aging. Biochimica et Biophysica Acta, 1847(11), 1380–1386. https://doi.org/10.1016/j.bbabio.2015.05.014.
Mohrin, M., & Chen, D. (2016). The mitochondrial metabolic checkpoint and aging of hematopoietic stem cells. Current Opinion in Hematology, 23(4), 318–324. https://doi.org/10.1097/MOH.0000000000000244.
Norddahl, G. L., Pronk, C. J., Wahlestedt, M., Sten, G., Nygren, J. M., Ugale, A., Sigvardsson, M., & Bryder, D. (2011). Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell stem cell, 8(5), 499–510. https://doi.org/10.1016/j.stem.2011.03.009.
Shao, L., Li, H., Pazhanisamy, S. K., Meng, A., Wang, Y., & Zhou, D. (2011). Reactive oxygen species and hematopoietic stem cell senescence. International Journal of Hematology, 94(1), 24–32. https://doi.org/10.1007/s12185-011-0872-1.
Khatri, R., Krishnan, S., Roy, S., Chattopadhyay, S., Kumar, V., & Mukhopadhyay, A. (2016). Reactive oxygen species limit the ability of bone marrow stromal cells to support hematopoietic reconstitution in aging mice. Stem Cells and Development, 25(12), 948–958. https://doi.org/10.1089/scd.2015.0391.
Yang, S. R., Park, J. R., & Kang, K. S. (2015). Reactive oxygen species in mesenchymal stem cell aging: implication to lung diseases. Oxidative Medicine and Cellular Longevity, 2015, 486263. https://doi.org/10.1155/2015/486263.
Mantel, C., Messina-Graham, S., Moh, A., Cooper, S., Hangoc, G., Fu, X. Y., & Broxmeyer, H. E. (2012). Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood, 120(13), 2589–2599. https://doi.org/10.1182/blood-2012-01-404004.
Katajisto, P., Döhla, J., Chaffer, C. L., Pentinmikko, N., Marjanovic, N., Iqbal, S., Zoncu, R., Chen, W., Weinberg, R. A., & Sabatini, D. M. (2015). Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science (New York, N.Y.), 348(6232), 340–343. https://doi.org/10.1126/science.1260384.
van Galen, P., Kreso, A., Mbong, N., Kent, D. G., Fitzmaurice, T., Chambers, J. E., Xie, S., Laurenti, E., Hermans, K., Eppert, K., Marciniak, S. J., Goodall, J. C., Green, A. R., Wouters, B. G., Wienholds, E., & Dick, J. E. (2014). The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature, 510(7504), 268–272. https://doi.org/10.1038/nature13228.
Mohrin, M., Shin, J., Liu, Y., Brown, K., Luo, H., Xi, Y., Haynes, C. M., & Chen, D. (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science (New York, N.Y.), 347(6228), 1374–1377. https://doi.org/10.1126/science.aaa2361.
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly-Y, M., Gidlöf, S., Oldfors, A., Wibom, R., Törnell, J., Jacobs, H. T., & Larsson, N. G. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature, 429(6990), 417–423. https://doi.org/10.1038/nature02517.
Jeong, M., Piao, Z. H., Kim, M. S., Lee, S. H., Yun, S., Sun, H. N., Yoon, S. R., Chung, J. W., Kim, T. D., Jeon, J. H., Lee, J., Kim, H. N., Choi, J. Y., & Choi, I. (2009). Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. Journal of Immunology (Baltimore, Md. : 1950), 183(4), 2495–2505. https://doi.org/10.4049/jimmunol.0804221.
Jung, H., Kim, M. J., Kim, D. O., Kim, W. S., Yoon, S. J., Park, Y. J., Yoon, S. R., Kim, T. D., Suh, H. W., Yun, S., Min, J. K., Lee, H. G., Lee, Y. H., Na, H. J., Lee, D. C., Kim, H. C., & Choi, I. (2013). TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metabolism, 18(1), 75–85. https://doi.org/10.1016/j.cmet.2013.06.002.
Jung, H., & Choi, I. (2014). Thioredoxin-interacting protein, hematopoietic stem cells, and hematopoiesis. Current Opinion in Hematology, 21(4), 265–270. https://doi.org/10.1097/MOH.0000000000000037.
Florian, M. C., Dörr, K., Niebel, A., Daria, D., Schrezenmeier, H., Rojewski, M., Filippi, M. D., Hasenberg, A., Gunzer, M., Scharffetter-Kochanek, K., Zheng, Y., & Geiger, H. (2012). Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell, 10(5), 520–530. https://doi.org/10.1016/j.stem.2012.04.007.
Borodkina, A., Shatrova, A., Abushik, P., Nikolsky, N., & Burova, E. (2014). Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging, 6(6), 481–495. https://doi.org/10.18632/aging.100673.
Li-Harms, X., Milasta, S., Lynch, J., Wright, C., Joshi, A., Iyengar, R., Neale, G., Wang, X., Wang, Y. D., Prolla, T. A., Thompson, J. E., Opferman, J. T., Green, D. R., Schuetz, J., & Kundu, M. (2015). Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood, 125(1), 162–174. https://doi.org/10.1182/blood-2014-07-586396.
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., Adachi, H., Adams, C. M., Adams, P. D., Adeli, K., Adhihetty, P. J., Adler, S. G., Agam, G., Agarwal, R., Aghi, M. K., Agnello, M., Agostinis, P., Aguilar, P. V., Aguirre-Ghiso, J., Airoldi, E. M., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12(1), 1–222. https://doi.org/10.1080/15548627.2015.1100356.
Rubinsztein, D. C., Mariño, G., & Kroemer, G. (2011). Autophagy and aging. Cell, 146(5), 682–695. https://doi.org/10.1016/j.cell.2011.07.030.
García-Prat, L., Martínez-Vicente, M., Perdiguero, E., Ortet, L., Rodríguez-Ubreva, J., Rebollo, E., Ruiz-Bonilla, V., Gutarra, S., Ballestar, E., Serrano, A. L., Sandri, M., & Muñoz-Cánoves, P. (2016). Autophagy maintains stemness by preventing senescence. Nature, 529(7584), 37–42. https://doi.org/10.1038/nature16187.
Leveque, L., Le Texier, L., Lineburg, K. E., Hill, G. R., & MacDonald, K. P. (2015). Autophagy and haematopoietic stem cell transplantation. Immunology and Cell Biology, 93(1), 43–50. https://doi.org/10.1038/icb.2014.95.
Lin, W., Yuan, N., Wang, Z., Cao, Y., Fang, Y., Li, X., Xu, F., Song, L., Wang, J., Zhang, H., Yan, L., Xu, L., Zhang, X., Zhang, S., & Wang, J. (2015). Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Scientific Reports, 5, 12362. https://doi.org/10.1038/srep12362.
Ho, T. T., Warr, M. R., Adelman, E. R., Lansinger, O. M., Flach, J., Verovskaya, E. V., Figueroa, M. E., & Passegué, E. (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature, 543(7644), 205–210. https://doi.org/10.1038/nature21388.
Säwén, P., Lang, S., Mandal, P., Rossi, D. J., Soneji, S., & Bryder, D. (2016). Mitotic history reveals distinct stem cell populations and their contributions to hematopoiesis. Cell Reports, 14(12), 2809–2818. https://doi.org/10.1016/j.celrep.2016.02.073.
Baar, M. P., Brandt, R., Putavet, D. A., Klein, J., Derks, K., Bourgeois, B., Stryeck, S., Rijksen, Y., van Willigenburg, H., Feijtel, D. A., van der Pluijm, I., Essers, J., van Cappellen, W. A., van IJcken, W. F., Houtsmuller, A. B., Pothof, J., de Bruin, R., Madl, T., Hoeijmakers, J., Campisi, J., et al. (2017). Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell, 169(1), 132–147.e16. https://doi.org/10.1016/j.cell.2017.02.031.
Killackey, S. A., Philpott, D. J., & Girardin, S. E. (2020). Mitophagy pathways in health and disease. The Journal of Cell Biology, 219(11), e202004029. https://doi.org/10.1083/jcb.202004029.
Ikeda, F. (2020). Mitophagy is induced by short ubiquitin chains on mitochondria. The Journal of Cell Biology, 219(9), e202008031. https://doi.org/10.1083/jcb.202008031.
Bharath, L. P., Agrawal, M., McCambridge, G., Nicholas, D. A., Hasturk, H., Liu, J., Jiang, K., Liu, R., Guo, Z., Deeney, J., Apovian, C. M., Snyder-Cappione, J., Hawk, G. S., Fleeman, R. M., Pihl, R., Thompson, K., Belkina, A. C., Cui, L., Proctor, E. A., Kern, P. A., et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metabolism, 32(1), 44–55.e6. https://doi.org/10.1016/j.cmet.2020.04.015.
Coleman, C. N., Blakely, W. F., Fike, J. R., MacVittie, T. J., Metting, N. F., Mitchell, J. B., Moulder, J. E., Preston, R. J., Seed, T. M., Stone, H. B., Tofilon, P. J., & Wong, R. S. (2003). Molecular and cellular biology of moderate-dose (1-10 Gy) radiation and potential mechanisms of radiation protection: report of a workshop at Bethesda, Maryland, December 17-18, 2001. Radiation Research, 159(6), 812–834. https://doi.org/10.1667/rr3021.
Dainiak, N., Waselenko, J. K., Armitage, J. O., MacVittie, T. J., & Farese, A. M. (2003). The hematologist and radiation casualties. Hematology. American Society of Hematology. Education Program, 473–496. https://doi.org/10.1182/asheducation-2003.1.473.
Chua, H. L., Plett, P. A., Sampson, C. H., Katz, B. P., Carnathan, G. W., MacVittie, T. J., Lenden, K., & Orschell, C. M. (2014). Survival efficacy of the PEGylated G-CSFs Maxy-G34 and neulasta in a mouse model of lethal H-ARS, and residual bone marrow damage in treated survivors. Health Physics, 106(1), 21–38. https://doi.org/10.1097/HP.0b013e3182a4df10.
Chua, H. L., Plett, P. A., Sampson, C. H., Joshi, M., Tabbey, R., Katz, B. P., MacVittie, T. J., & Orschell, C. M. (2012). Long-term hematopoietic stem cell damage in a murine model of the hematopoietic syndrome of the acute radiation syndrome. Health Physics, 103(4), 356–366. https://doi.org/10.1097/HP.0b013e3182666d6f.
Chua, H. L., Plett, P. A., Fisher, A., Sampson, C. H., Vemula, S., Feng, H., Sellamuthu, R., Wu, T., MacVittie, T. J., & Orschell, C. M. (2019). Lifelong residual bone marrow damage in murine survivors of the hematopoietic acute radiation syndrome (H-ARS): a compilation of studies comprising the Indiana University Experience. Health Physics, 116(4), 546–557. https://doi.org/10.1097/HP.0000000000000950.
Botnick, L. E., Hannon, E. C., & Hellman, S. (1979). A long lasting proliferative defect in the hematopoietic stem cell compartment following cytotoxic agents. International Journal of Radiation Oncology, Biology, Physics, 5(9), 1621–1625. https://doi.org/10.1016/0360-3016(79)90785-5.
Mauch, P., Rosenblatt, M., & Hellman, S. (1988). Permanent loss in stem cell self renewal capacity following stress to the marrow. Blood, 72(4), 1193–1196.
Wu, T., Plett, P.A., Chua, H.L., Jacobsen, M., Sandusky, G.E., MacVittie, T.J. , & Orschell, C.M. (2020). Immune reconstitution and thymic involution in the acute and delayed hematopoietic radiation syndromes. Health Physics. 2020 (in press).
Patterson, A. M., Plett, P. A., Chua, H. L., Sampson, C. H., Fisher, A., Feng, H., Unthank, J. L., Miller, S. J., Katz, B. P., MacVittie, T. J., & Orschell, C. M. (2020). Development of a model of the acute and delayed effects of high dose radiation exposure in Jackson diversity outbred mice; comparison to inbred C57BL/6 Mice. Health Physics. https://doi.org/10.1097/HP.0000000000001344. Advance online publication.
Hellman, S., & Botnick, L. E. (1977). Stem cell depletion: an explanation of the late effects of cytotoxins. International Journal of Radiation Oncology, Biology, Physics, 2(1-2), 181–184. https://doi.org/10.1016/0360-3016(77)90028-1.
Wang, Y., Schulte, B. A., LaRue, A. C., Ogawa, M., & Zhou, D. (2006). Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood, 107(1), 358–366. https://doi.org/10.1182/blood-2005-04-1418.
Meng, A., Wang, Y., Van Zant, G., & Zhou, D. (2003). Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Research, 63(17), 5414–5419.
Fish, B. L., Gao, F., Narayanan, J., Bergom, C., Jacobs, E. R., Cohen, E. P., Moulder, J. E., Orschell, C. M., & Medhora, M. (2016). Combined hydration and antibiotics with lisinopril to mitigate acute and delayed high-dose radiation injuries to multiple organs. Health Physics, 111(5), 410–419. https://doi.org/10.1097/HP.0000000000000554.
Traycoff, C., Yoder, M., Hiatt, K., & Srour, E. (1996). Cell cycle stage-specific expression of adhesion molecules may augment engraftment potential of quiescent but not mitotically active hematopoietic progenitor cells. Blood, 88(10), 475.
Abramson, S., Miller, R. G., & Phillips, R. A. (1977). The identification in adult bone marrow of pluripotent and restricted stem cells of the myeloid and lymphoid systems. The Journal of Experimental Medicine, 145(6), 1567–1579. https://doi.org/10.1084/jem.145.6.1567.
Jones, R. J., Celano, P., Sharkis, S. J., & Sensenbrenner, L. L. (1989). Two phases of engraftment established by serial bone marrow transplantation in mice. Blood, 73(2), 397–401.
Jones, R. J., Wagner, J. E., Celano, P., Zicha, M. S., & Sharkis, S. J. (1990). Separation of pluripotent haematopoietic stem cells from spleen colony-forming cells. Nature, 347(6289), 188–189. https://doi.org/10.1038/347188a0.
Keller, G., & Snodgrass, R. (1990). Life span of multipotential hematopoietic stem cells in vivo. The Journal of Experimental Medicine, 171(5), 1407–1418. https://doi.org/10.1084/jem.171.5.1407.
Visser, J. W. M., Bauman, J. G. J., Mulder, A. H., Eliason, J. F., & DeLeeuw, A. M. (1984). Isolation of murine pluripotent hematopoietic stem cells. The Journal of Experimental Medicine, 159, 1576.
Hall, E. (2000). Acute effects of total-body irradiation. In E. Hall (Ed.), Radiobiology for the radiologist (pp. 124–135). Philadelphia: Lippincott Williams & Wilkins.
Till, J. E., & Mcculloch, E. A. (1961). A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiation Research, 14, 213–222.
Yahata, T., Takanashi, T., Muguruma, Y., Ibrahim, A. A., Matsuzawa, H., Uno, T., Sheng, Y., Onizuka, M., Ito, M., Kato, S., & Ando, K. (2011). Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood, 118(11), 2941–2950. https://doi.org/10.1182/blood-2011-01-330050.
Simonnet, A. J., Nehmé, J., Vaigot, P., Barroca, V., Leboulch, P., & Tronik-Le Roux, D. (2009). Phenotypic and functional changes induced in hematopoietic stem/progenitor cells after gamma-ray radiation exposure. Stem Cells (Dayton, Ohio), 27(6), 1400–1409. https://doi.org/10.1002/stem.66.
Zhao, W., Diz, D. I., & Robbins, M. E. (2007). Oxidative damage pathways in relation to normal tissue injury. The British Journal of Radiology, 80 Spec No 1, S23–S31. https://doi.org/10.1259/bjr/18237646.
Delanian, S., & Lefaix, J. L. (2007). Current management for late normal tissue injury: radiation-induced fibrosis and necrosis. Seminars in Radiation Oncology, 17(2), 99–107. https://doi.org/10.1016/j.semradonc.2006.11.006.
Yarnold, J., & Brotons, M. C. (2010). Pathogenetic mechanisms in radiation fibrosis. Radiotherapy and Oncology : Journal of the European Society for Therapeutic Radiology and Oncology, 97(1), 149–161. https://doi.org/10.1016/j.radonc.2010.09.002.
Zhao, W., & Robbins, M. E. (2009). Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Current Medicinal Chemistry, 16(2), 130–143. https://doi.org/10.2174/092986709787002790.
Stone, H. B., Coleman, C. N., Anscher, M. S., & McBride, W. H. (2003). Effects of radiation on normal tissue: consequences and mechanisms. The Lancet. Oncology, 4(9), 529–536. https://doi.org/10.1016/s1470-2045(03)01191-4.
Yan, X., Sasi, S. P., Gee, H., Lee, J., Yang, Y., Mehrzad, R., Onufrak, J., Song, J., Enderling, H., Agarwal, A., Rahimi, L., Morgan, J., Wilson, P. F., Carrozza, J., Walsh, K., Kishore, R., & Goukassian, D. A. (2014). Cardiovascular risks associated with low dose ionizing particle radiation. PLoS ONE, 9(10), e110269. https://doi.org/10.1371/journal.pone.0110269.
Sasi, S. P., Yan, X., Lee, J., Sisakyan, H., Carrozza, J., & Goukassian, D. A. (2014). Radiation-associated degenerative cardiovascular risks during normal aging and after adverse CV event 10 months post-initial exposure. Journal of Radiation Research, 55(Suppl 1), i111–i1i2.
Di Maggio, F. M., Minafra, L., Forte, G. I., Cammarata, F. P., Lio, D., Messa, C., Gilardi, M. C., & Bravatà, V. (2015). Portrait of inflammatory response to ionizing radiation treatment. Journal of Inflammation (London, England), 12, 14. https://doi.org/10.1186/s12950-015-0058-3.
Fliedner, T. M., Nothdurft, W., & Calvo, W. (1986). The development of radiation late effects to the bone marrow after single and chronic exposure. International Journal of radiation biology and Related Studies in Physics, Chemistry, and Medicine, 49(1), 35–46. https://doi.org/10.1080/09553008514552211.
Soucy, K. G., Lim, H. K., Attarzadeh, D. O., Santhanam, L., Kim, J. H., Bhunia, A. K., Sevinc, B., Ryoo, S., Vazquez, M. E., Nyhan, D., Shoukas, A. A., & Berkowitz, D. E. (2010). Dietary inhibition of xanthine oxidase attenuates radiation-induced endothelial dysfunction in rat aorta. Journal of Applied Physiology (Bethesda, Md. : 1985), 108(5), 1250–1258. https://doi.org/10.1152/japplphysiol.00946.2009.
Kajimura, J., Kyoizumi, S., Kubo, Y., Misumi, M., Yoshida, K., Hayashi, T., Imai, K., Ohishi, W., Nakachi, K., Weng, N. P., Young, L. F., Shieh, J. H., Moore, M. A., van den Brink, M. R., & Kusunoki, Y. (2016). Relationship between spontaneous γH2AX foci formation and progenitor functions in circulating hematopoietic stem and progenitor cells among atomic-bomb survivors. Mutation research. Genetic toxicology and environmental mutagenesis, 802, 59–65. https://doi.org/10.1016/j.mrgentox.2016.04.006.
Kusunoki, Y., & Hayashi, T. (2008). Long-lasting alterations of the immune system by ionizing radiation exposure: implications for disease development among atomic bomb survivors. International Journal of Radiation Biology, 84(1), 1–14. https://doi.org/10.1080/09553000701616106.
Soucy, K. G., Lim, H. K., Kim, J. H., Oh, Y., Attarzadeh, D. O., Sevinc, B., Kuo, M. M., Shoukas, A. A., Vazquez, M. E., & Berkowitz, D. E. (2011). HZE 56Fe-ion irradiation induces endothelial dysfunction in rat aorta: role of xanthine oxidase. Radiation Research, 176(4), 474–485. https://doi.org/10.1667/rr2598.1.
Datta, K., Suman, S., Kallakury, B. V., & Fornace Jr., A. J. (2012). Exposure to heavy ion radiation induces persistent oxidative stress in mouse intestine. PLoS ONE, 7(8), e42224. https://doi.org/10.1371/journal.pone.0042224.
Collins-Underwood, J. R., Zhao, W., Sharpe, J. G., & Robbins, M. E. (2008). NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radical Biology & Medicine, 45(6), 929–938. https://doi.org/10.1016/j.freeradbiomed.2008.06.024.
Raju, U., Gumin, G. J., & Tofilon, P. J. (2000). Radiation-induced transcription factor activation in the rat cerebral cortex. International Journal of Radiation Biology, 76(8), 1045–1053. https://doi.org/10.1080/09553000050111514.
Tak, P. P., & Firestein, G. S. (2001). NF-kappaB: a key role in inflammatory diseases. The Journal of Clinical Investigation, 107(1), 7–11. https://doi.org/10.1172/JCI11830.
Donato, A. J., Pierce, G. L., Lesniewski, L. A., & Seals, D. R. (2009). Role of NFkappaB in age-related vascular endothelial dysfunction in humans. Aging, 1(8), 678–680. https://doi.org/10.18632/aging.100080.
Pan, J., Li, D., Xu, Y., Zhang, J., Wang, Y., Chen, M., Lin, S., Huang, L., Chung, E. J., Citrin, D. E., Wang, Y., Hauer-Jensen, M., Zhou, D., & Meng, A. (2017). Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. International Journal of Radiation Oncology, Biology, Physics, 99(2), 353–361. https://doi.org/10.1016/j.ijrobp.2017.02.216.
He, Y., Thummuri, D., Zheng, G., Okunieff, P., Citrin, D. E., Vujaskovic, Z., & Zhou, D. (2019). Cellular senescence and radiation-induced pulmonary fibrosis. Translational Research: the Journal of Laboratory and Clinical Medicine, 209, 14–21. https://doi.org/10.1016/j.trsl.2019.03.006.
Chang, J., Wang, Y., Shao, L., Laberge, R. M., Demaria, M., Campisi, J., Janakiraman, K., Sharpless, N. E., Ding, S., Feng, W., Luo, Y., Wang, X., Aykin-Burns, N., Krager, K., Ponnappan, U., Hauer-Jensen, M., Meng, A., & Zhou, D. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature Medicine, 22(1), 78–83. https://doi.org/10.1038/nm.4010.
Yuan, R., Tsaih, S. W., Petkova, S. B., Marin de Evsikova, C., Xing, S., Marion, M. A., Bogue, M. A., Mills, K. D., Peters, L. L., Bult, C. J., Rosen, C. J., Sundberg, J. P., Harrison, D. E., Churchill, G. A., & Paigen, B. (2009). Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell, 8(3), 277–287. https://doi.org/10.1111/j.1474-9726.2009.00478.x.
Turturro, A., Witt, W. W., Lewis, S., Hass, B. S., Lipman, R. D., & Hart, R. W. (1999). Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. The journals of gerontology. Series A, Biological Sciences and Medical Sciences, 54(11), B492–B501. https://doi.org/10.1093/gerona/54.11.b492.
Garrett, J., Sampson, C. H., Plett, P. A., Crisler, R., Parker, J., Venezia, R., Chua, H. L., Hickman, D. L., Booth, C., MacVittie, T., Orschell, C. M., & Dynlacht, J. R. (2019). Characterization and etiology of swollen muzzles in irradiated mice. Radiation Research, 191(1), 31–42. https://doi.org/10.1667/RR14724.1.
Ericsson, A. C., Davis, J. W., Spollen, W., Bivens, N., Givan, S., Hagan, C. E., McIntosh, M., & Franklin, C. L. (2015). Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS ONE, 10(2), e0116704. https://doi.org/10.1371/journal.pone.0116704.
Mantel, C., & Broxmeyer, H. E. (2008). Sirtuin 1, stem cells, aging, and stem cell aging. Current Opinion in Hematology, 15(4), 326–331. https://doi.org/10.1097/MOH.0b013e3283043819.
Ou, X., Chae, H. D., Wang, R. H., Shelley, W. C., Cooper, S., Taylor, T., Kim, Y. J., Deng, C. X., Yoder, M. C., & Broxmeyer, H. E. (2011). SIRT1 deficiency compromises mouse embryonic stem cell hematopoietic differentiation, and embryonic and adult hematopoiesis in the mouse. Blood, 117(2), 440–450. https://doi.org/10.1182/blood-2010-03-273011.
Rimmelé, P., Bigarella, C. L., Liang, R., Izac, B., Dieguez-Gonzalez, R., Barbet, G., Donovan, M., Brugnara, C., Blander, J. M., Sinclair, D. A., & Ghaffari, S. (2014). Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Reports, 3(1), 44–59. https://doi.org/10.1016/j.stemcr.2014.04.015.
Guarente, L., & Picard, F. (2005). Calorie restriction--the SIR2 connection. Cell, 120(4), 473–482. https://doi.org/10.1016/j.cell.2005.01.029.
Ocampo, A., & Izpisua Belmonte, J. C. (2015). Stem cells. Holding your breath for longevity. Science (New York, N.Y.), 347(6228), 1319–1320. https://doi.org/10.1126/science.aaa9608.
Baur, J. A., Ungvari, Z., Minor, R. K., Le Couteur, D. G., & de Cabo, R. (2012). Are sirtuins viable targets for improving healthspan and lifespan? Nature Reviews Drug Discovery, 11(6), 443–461. https://doi.org/10.1038/nrd3738.
Mitchell, S. J., Madrigal-Matute, J., Scheibye-Knudsen, M., Fang, E., Aon, M., González-Reyes, J. A., Cortassa, S., Kaushik, S., Gonzalez-Freire, M., Patel, B., Wahl, D., Ali, A., Calvo-Rubio, M., Burón, M. I., Guiterrez, V., Ward, T. M., Palacios, H. H., Cai, H., Frederick, D. W., Hine, C., et al. (2016). Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metabolism, 23(6), 1093–1112. https://doi.org/10.1016/j.cmet.2016.05.027.
Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J., Liu, Y., Zhang, D., Scadden, D. T., & Chen, D. (2013). SIRT3 reverses aging-associated degeneration. Cell Reports, 3(2), 319–327. https://doi.org/10.1016/j.celrep.2013.01.005.
Wrighton, K. H. (2015). Stem cells: SIRT7, the UPR and HSC ageing. Nature Reviews Molecular Cell Biology, 16(5), 266–267. https://doi.org/10.1038/nrm3981.
Yan, H., Baldridge, M. T., & King, K. Y. (2018). Hematopoiesis and the bacterial microbiome. Blood, 132(6), 559–564. https://doi.org/10.1182/blood-2018-02-832519.
Manzo, V. E., & Bhatt, A. S. (2015). The human microbiome in hematopoiesis and hematologic disorders. Blood, 126(3), 311–318. https://doi.org/10.1182/blood-2015-04-574392.
Broxmeyer, H. E., Cooper, S., Cacalano, G., Hague, N. L., Bailish, E., & Moore, M. W. (1996). Involvement of Interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. The Journal of Experimental Medicine, 184(5), 1825–1832. https://doi.org/10.1084/jem.184.5.1825.
Khosravi, A., Yáñez, A., Price, J. G., Chow, A., Merad, M., Goodridge, H. S., & Mazmanian, S. K. (2014). Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host & Microbe, 15(3), 374–381. https://doi.org/10.1016/j.chom.2014.02.006.
Staffas, A., Burgos da Silva, M., Slingerland, A. E., Lazrak, A., Bare, C. J., Holman, C. D., Docampo, M. D., Shono, Y., Durham, B., Pickard, A. J., Cross, J. R., Stein-Thoeringer, C., Velardi, E., Tsai, J. J., Jahn, L., Jay, H., Lieberman, S., Smith, O. M., Pamer, E. G., Peled, J. U., et al. (2018). Nutritional support from the intestinal microbiota improves hematopoietic reconstitution after bone marrow transplantation in mice. Cell Host & Microbe, 23(4), 447–457.e4. https://doi.org/10.1016/j.chom.2018.03.002.
Theilgaard-Mönch, K. (2017). Gut microbiota sustains hematopoiesis. Blood, 129(6), 662–663. https://doi.org/10.1182/blood-2016-12-754481.
Luo, Y., Chen, G. L., Hannemann, N., Ipseiz, N., Krönke, G., Bäuerle, T., Munos, L., Wirtz, S., Schett, G., & Bozec, A. (2015). Microbiota from obese mice regulate hematopoietic stem cell differentiation by altering the bone niche. Cell Metabolism, 22(5), 886–894. https://doi.org/10.1016/j.cmet.2015.08.020.
Lee, S., Kim, H., You, G., Kim, Y. M., Lee, S., Le, V. H., Kwon, O., Im, S. H., Kim, Y. M., Kim, K. S., Sung, Y. C., Kim, K. H., Surh, C. D., Park, Y., & Lee, S. W. (2019). Bone marrow CX3CR1+ mononuclear cells relay a systemic microbiota signal to control hematopoietic progenitors in mice. Blood, 134(16), 1312–1322. https://doi.org/10.1182/blood.2019000495.
Velders, G. A., van Os, R., Hagoort, H., Verzaal, P., Guiot, H. F., Lindley, I. J., Willemze, R., Opdenakker, G., & Fibbe, W. E. (2004). Reduced stem cell mobilization in mice receiving antibiotic modulation of the intestinal flora: involvement of endotoxins as cofactors in mobilization. Blood, 103(1), 340–346. https://doi.org/10.1182/blood-2002-07-2270.
Severyn, C. J., Brewster, R., & Andermann, T. M. (2019). Microbiota modification in hematology: still at the bench or ready for the bedside? Hematology. American Society of Hematology. Education Program, 2019(1), 303–314. https://doi.org/10.1182/hematology.2019000365.
Khosravi, A., Yáñez, A., Price, J. G., Chow, A., Merad, M., Goodridge, H. S., & Mazmanian, S. K. (2014). Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host & Microbe, 15(3), 374–381. https://doi.org/10.1016/j.chom.2014.02.006.
Wallis, C. (2020). New player in cancer’s spread. A commonplace mouth bacterium now is tied to metastasis of some tumors. Scientific American, 323(4), 28.
Thaiss, C. A., Levy, M., Korem, T., Dohnalová, L., Shapiro, H., Jaitin, D. A., David, E., Winter, D. R., Gury-BenAri, M., Tatirovsky, E., Tuganbaev, T., Federici, S., Zmora, N., Zeevi, D., Dori-Bachash, M., Pevsner-Fischer, M., Kartvelishvily, E., Brandis, A., Harmelin, A., Shibolet, O., et al. (2016). Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell, 167(6), 1495–1510.e12. https://doi.org/10.1016/j.cell.2016.11.003.
Kostic, A. D., Chun, E., Robertson, L., Glickman, J. N., Gallini, C. A., Michaud, M., Clancy, T. E., Chung, D. C., Lochhead, P., Hold, G. L., El-Omar, E. M., Brenner, D., Fuchs, C. S., Meyerson, M., & Garrett, W. S. (2013). Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host & Microbe, 14(2), 207–215. https://doi.org/10.1016/j.chom.2013.07.007.
Sears, C. L., & Garrett, W. S. (2014). Microbes, microbiota, and colon cancer. Cell Host & Microbe, 15(3), 317–328. https://doi.org/10.1016/j.chom.2014.02.007.
Abed, J., Emgård, J. E., Zamir, G., Faroja, M., Almogy, G., Grenov, A., Sol, A., Naor, R., Pikarsky, E., Atlan, K. A., Mellul, A., Chaushu, S., Manson, A. L., Earl, A. M., Ou, N., Brennan, C. A., Garrett, W. S., & Bachrach, G. (2016). Fap2 mediates fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc. Cell Host & Microbe, 20(2), 215–225. https://doi.org/10.1016/j.chom.2016.07.006.
Yan, X., Liu, L., Li, H., Qin, H., & Sun, Z. (2017). Clinical significance of Fusobacterium nucleatum, epithelial-mesenchymal transition, and cancer stem cell markers in stage III/IV colorectal cancer patients. OncoTargets and Therapy, 10, 5031–5046. https://doi.org/10.2147/OTT.S145949.
Shang, F. M., & Liu, H. L. (2018). Fusobacterium nucleatum and colorectal cancer: a review. World Journal of Gastrointestinal Oncology, 10(3), 71–81. https://doi.org/10.4251/wjgo.v10.i3.71.
Sun, C. H., Li, B. B., Wang, B., Zhao, J., Zhang, X. Y., Li, T. T., Li, W. B., Tang, D., Qiu, M. J., Wang, X. C., Zhu, C. M., & Qian, Z. R. (2019). The role of Fusobacterium nucleatum in colorectal cancer: from carcinogenesis to clinical management. Chronic Diseases and Translational Medicine, 5(3), 178–187. https://doi.org/10.1016/j.cdtm.2019.09.001.
Kang, W., Ji, X., Zhang, X., Tang, D., & Feng, Q. (2019). Persistent exposure to fusobacterium nucleatum triggers chemokine/cytokine release and inhibits the proliferation and osteogenic differentiation capabilities of human gingiva-derived mesenchymal stem cells. Frontiers in Cellular and Infection Microbiology, 9, 429. https://doi.org/10.3389/fcimb.2019.00429.
Chen, Y., Chen, Y., Zhang, J., Cao, P., Su, W., Deng, Y., Zhan, N., Fu, X., Huang, Y., & Dong, W. (2020). Fusobacterium nucleatum promotes metastasis in colorectal cancer by activating autophagy signaling via the upregulation of CARD3 expression. Theranostics, 10(1), 323–339. https://doi.org/10.7150/thno.38870.
Furtek, K. J., Kubiak, D. W., Barra, M., Varughese, C. A., Ashbaugh, C. D., & Koo, S. (2016). High incidence of neutropenia in patients with prolonged ceftaroline exposure. The Journal of Antimicrobial Chemotherapy, 71(7), 2010–2013. https://doi.org/10.1093/jac/dkw062.
Vinh, D. C., & Rubinstein, E. (2009). Linezolid: a review of safety and tolerability. The Journal of Infection, 59(Suppl 1), S59–S74. https://doi.org/10.1016/S0163-4453(09)60009-8.
Iwamura, C., Bouladoux, N., Belkaid, Y., Sher, A., & Jankovic, D. (2017). Sensing of the microbiota by NOD1 in mesenchymal stromal cells regulates murine hematopoiesis. Blood, 129(2), 171–176. https://doi.org/10.1182/blood-2016-06-723742.
Shintouo, C. M., Mets, T., Beckwee, D., Bautmans, I., Ghogomu, S. M., Souopgui, J., Leemans, L., Meriki, H. D., & Njemini, R. (2020). Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Experimental Gerontology, 141, 111079. Advance online publication. https://doi.org/10.1016/j.exger.2020.111079.
Chen, F., Liu, Y., Wong, N. K., Xiao, J., & So, K. F. (2017). Oxidative stress in stem cell aging. Cell Transplantation, 26(9), 1483–1495. https://doi.org/10.1177/0963689717735407.
Broxmeyer, H. E., Capitano, M. L., Cooper, S., Potchanant, E. S., & Clapp, D. W. (2020). Numbers of long-term hematopoietic stem cells from bone marrow of fanca and fancc knockout mice can be greatly enhanced by their collection and processing in physioxia conditions. Blood Cells, Molecules & Diseases, 86, 102492. Advance online publication. https://doi.org/10.1016/j.bcmd.2020.102492.
Broxmeyer, H. E., Cooper, S., & Capitano, M. L. (2020). Enhanced collection of phenotypic and engrafting human cord blood hematopoietic stem cells at 4°C. Stem Cells (Dayton, Ohio), 38(10), 1326–1331. https://doi.org/10.1002/stem.3243.
Säwén, P., Lang, S., Mandal, P., Rossi, D. J., Soneji, S., & Bryder, D. (2016). Mitotic history reveals distinct stem cell populations and their contributions to hematopoiesis. Cell Reports, 14(12), 2809–2818. https://doi.org/10.1016/j.celrep.2016.02.073.
Calderwood, S. K., Murshid, A., & Prince, T. (2009). The shock of aging: molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology, 55(5), 550–558. https://doi.org/10.1159/000225957.
Pandya, J. D., Valdez, M., Royland, J. E., MacPhail, R. C., Sullivan, P. G., & Kodavanti, P. (2020). Age- and organ-specific differences in mitochondrial bioenergetics in brown norway rats. Journal of Aging Research, 2020, 7232614. https://doi.org/10.1155/2020/7232614.
Valente, L. J., Tarangelo, A., Li, A. M., Naciri, M., Raj, N., Boutelle, A. M., Li, Y., Mello, S. S., Bieging-Rolett, K., DeBerardinis, R. J., Ye, J., Dixon, S. J., & Attardi, L. D. (2020). p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. The Journal of Cell Biology, 219(11), e201908212. https://doi.org/10.1083/jcb.201908212.
Nombela-Arrieta, C., Pivarnik, G., Winkel, B., Canty, K. J., Harley, B., Mahoney, J. E., Park, S. Y., Lu, J., Protopopov, A., & Silberstein, L. E. (2013). Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nature Cell Biology, 15(5), 533–543. https://doi.org/10.1038/ncb2730.
Conboy, I. M., Conboy, M. J., Wagers, A. J., Girma, E. R., Weissman, I. L., & Rando, T. A. (2005). Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature, 433(7027), 760–764. https://doi.org/10.1038/nature03260.
Maryanovich, M., Zahalka, A. H., Pierce, H., Pinho, S., Nakahara, F., Asada, N., Wei, Q., Wang, X., Ciero, P., Xu, J., Leftin, A., & Frenette, P. S. (2018). Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nature Medicine, 24(6), 782–791. https://doi.org/10.1038/s41591-018-0030-x.
Aguilar-Navarro, A. G., Meza-León, B., Gratzinger, D., Juárez-Aguilar, F. G., Chang, Q., Ornatsky, O., Tsui, H., Esquivel-Gómez, R., Hernández-Ramírez, A., Xie, S. Z., Dick, J. E., & Flores-Figueroa, E. (2020). Human aging alters the spatial organization between CD34+ hematopoietic cells and adipocytes in bone marrow. Stem Cell Reports, 15(2), 317–325. https://doi.org/10.1016/j.stemcr.2020.06.011.
Singh, P., Kacena, M. A., Orschell, C. M., & Pelus, L. M. (2020). Aging-related reduced expression of CXCR4 on bone marrow mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell defects. Stem Cell Reviews and Reports, 16(4), 684–692. https://doi.org/10.1007/s12015-020-09974-9.
Wang, Y. P., & Lei, Q. Y. (2018). Metabolite sensing and signaling in cell metabolism. Signal Transduction and Targeted Therapy, 3, 30. https://doi.org/10.1038/s41392-018-0024-7.
Shapira, S. N., & Christofk, H. R. (2020). Metabolic regulation of tissue stem cells. Trends in cell Biology, 30(7), 566–576. https://doi.org/10.1016/j.tcb.2020.04.004.
Xia, P., Wang, S., Du, Y., Huang, G., Satoh, T., Akira, S., & Fan, Z. (2015). Insulin-InsR signaling drives multipotent progenitor differentiation toward lymphoid lineages. The Journal of Experimental Medicine, 212(13), 2305–2321. https://doi.org/10.1084/jem.20150618.
Zhang, Y., Xue, Y., Cao, C., Huang, J., Hong, Q., Hai, T., Jia, Q., Wang, X., Qin, G., Yao, J., Wang, X., Zheng, Q., Zhang, R., Li, Y., Luo, A., Zhang, N., Shi, G., Wang, Y., Ying, H., Liu, Z., et al. (2017). Thyroid hormone regulates hematopoiesis via the TR-KLF9 axis. Blood, 130(20), 2161–2170. https://doi.org/10.1182/blood-2017-05-783043.
Stewart, M. H., Gutierrez-Martinez, P., Beerman, I., Garrison, B., Gallagher, E. J., LeRoith, D., & Rossi, D. J. (2014). Growth hormone receptor signaling is dispensable for HSC function and aging. Blood, 124(20), 3076–3080. https://doi.org/10.1182/blood-2014-05-575308.
Friedman, J. M., & Halaas, J. L. (1998). Leptin and the regulation of body weight in mammals. Nature, 395(6704), 763–770. https://doi.org/10.1038/27376.
La Cava, A., & Matarese, G. (2004). The weight of leptin in immunity. Nature Reviews Immunology, 4(5), 371–379. https://doi.org/10.1038/nri1350.
Ongrádi, J., & Kövesdi, V. (2010). Factors that may impact on immunosenescence: an appraisal. Immunity & Ageing : I & A, 7, 7. https://doi.org/10.1186/1742-4933-7-7.
Frasca, D., Diaz, A., Romero, M., & Blomberg, B. B. (2020). Leptin induces immunosenescence in human B cells. Cellular Immunology, 348, 103994. https://doi.org/10.1016/j.cellimm.2019.103994.
Frasca, D., & Blomberg, B. B. (2017). Adipose tissue inflammation induces B cell inflammation and decreases B cell function in aging. Frontiers in Immunology, 8, 1003. https://doi.org/10.3389/fimmu.2017.01003.
Gupta, S., Agrawal, S., & Gollapudi, S. (2013). Increased activation and cytokine secretion in B cells stimulated with leptin in aged humans. Immunity & Ageing : I & A, 10(1), 3. https://doi.org/10.1186/1742-4933-10-3.
Koenig, S., Luheshi, G. N., Wenz, T., Gerstberger, R., Roth, J., & Rummel, C. (2014). Leptin is involved in age-dependent changes in response to systemic inflammation in the rat. Brain, Behavior, and Immunity, 36, 128–138. https://doi.org/10.1016/j.bbi.2013.10.019.
Gabriely, I., Ma, X. H., Yang, X. M., Rossetti, L., & Barzilai, N. (2002). Leptin resistance during aging is independent of fat mass. Diabetes, 51(4), 1016–1021. https://doi.org/10.2337/diabetes.51.4.1016.
Filippi, B. M., & Lam, T. K. (2014). Leptin and aging. Aging, 6(2), 82–83. https://doi.org/10.18632/aging.100637.
Zhou, B. O., Yu, H., Yue, R., Zhao, Z., Rios, J. J., Naveiras, O., & Morrison, S. J. (2017). Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nature Cell Biology, 19(8), 891–903. https://doi.org/10.1038/ncb3570.
Himburg, H. A., Termini, C. M., Schlussel, L., Kan, J., Li, M., Zhao, L., Fang, T., Sasine, J. P., Chang, V. Y., & Chute, J. P. (2018). Distinct bone marrow sources of pleiotrophin control hematopoietic stem cell maintenance and regeneration. Cell stem cell, 23(3), 370–381.e5. https://doi.org/10.1016/j.stem.2018.07.003.
Comazzetto, S., Murphy, M. M., Berto, S., Jeffery, E., Zhao, Z., & Morrison, S. J. (2019). Restricted hematopoietic progenitors and erythropoiesis require SCF from leptin receptor+ niche cells in the bone marrow. Cell stem cell, 24(3), 477–486.e6. https://doi.org/10.1016/j.stem.2018.11.022.
Ding, L., Saunders, T. L., Enikolopov, G., & Morrison, S. J. (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature, 481(7382), 457–462. https://doi.org/10.1038/nature10783.
Krings, A., Rahman, S., Huang, S., Lu, Y., Czernik, P. J., & Lecka-Czernik, B. (2012). Bone marrow fat has brown adipose tissue characteristics, which are attenuated with aging and diabetes. Bone, 50(2), 546–552. https://doi.org/10.1016/j.bone.2011.06.016.
Ambrosi, T. H., Scialdone, A., Graja, A., Gohlke, S., Jank, A. M., Bocian, C., Woelk, L., Fan, H., Logan, D. W., Schürmann, A., Saraiva, L. R., & Schulz, T. J. (2017). Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell, 20(6), 771–784.e6. https://doi.org/10.1016/j.stem.2017.02.009.
Trinh, T., Ropa, J., Aljoufi, A., Cooper, S., Sinn, A., Srour, E.F., & Broxmeyer, H.E. (2020). Leptin receptor as a marker for long-term functional hematopoietic stem cells. Leukemia. In Press.
Nakao, T., Hino, M., Yamane, T., Nishizawa, Y., Morii, H., & Tatsumi, N. (1998). Expression of the leptin receptor in human leukaemic blast cells. British journal of haematology, 102(3), 740–745. https://doi.org/10.1046/j.1365-2141.1998.00843.x.
Lu, Z., Xie, J., Wu, G., Shen, J., Collins, R., Chen, W., Kang, X., Luo, M., Zou, Y., Huang, L. J., Amatruda, J. F., Slone, T., Winick, N., Scherer, P. E., & Zhang, C. C. (2017). Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nature Medicine, 23(1), 79–90. https://doi.org/10.1038/nm.4252.
Wex, H., Ponelis, E., Wex, T., Dressendörfer, R., Mittler, U., & Vorwerk, P. (2002). Plasma leptin and leptin receptor expression in childhood acute lymphoblastic leukemia. International Journal of Hematology, 76(5), 446–452. https://doi.org/10.1007/BF02982810.
Ozturk, K., Avcu, F., & Ural, A. U. (2012). Aberrant expressions of leptin and adiponectin receptor isoforms in chronic myeloid leukemia patients. Cytokine, 57(1), 61–67. https://doi.org/10.1016/j.cyto.2011.10.004.
He, H., Xu, P., Zhang, X., Liao, M., Dong, Q., Cong, T., Tang, B., Yang, X., Ye, M., Chang, Y., Liu, W., Wang, X., Ju, Z., & Wang, J. (2020). Aging-induced IL27Ra signaling impairs hematopoietic stem cells. Blood, 136(2), 183–198. https://doi.org/10.1182/blood.2019003910.
Kucia, M., Masternak, M., Liu, R., Shin, D. M., Ratajczak, J., Mierzejewska, K., Spong, A., Kopchick, J. J., Bartke, A., & Ratajczak, M. Z. (2013). The negative effect of prolonged somatotrophic/insulin signaling on an adult bone marrow-residing population of pluripotent very small embryonic-like stem cells (VSELs). Age (Dordrecht, Netherlands), 35(2), 315–330. https://doi.org/10.1007/s11357-011-9364-8.
Ratajczak, M. Z., Bartke, A., & Darzynkiewicz, Z. (2017). Prolonged growth hormone/insulin/insulin-like growth factor nutrient response signaling pathway as a silent killer of stem cells and a culprit in aging. Stem Cell Reviews and Reports, 13(4), 443–453. https://doi.org/10.1007/s12015-017-9728-2.
Boyiadzis, M., & Whiteside, T. L. (2017). The emerging roles of tumor-derived exosomes in hematological malignancies. Leukemia, 31(6), 1259–1268. https://doi.org/10.1038/leu.2017.91.
Shah, R., Patel, T., & Freedman, J. E. (2018). Circulating extracellular vesicles in human disease. The New England Journal of Medicine, 379(10), 958–966. https://doi.org/10.1056/NEJMra1704286.
Kowal, J., Tkach, M., & Théry, C. (2014). Biogenesis and secretion of exosomes. Current Opinion in Cell Biology, 29, 116–125. https://doi.org/10.1016/j.ceb.2014.05.004.
Dreyer, F., & Baur, A. (2016). Biogenesis and functions of exosomes and extracellular vesicles. Methods in Molecular Biology (Clifton, N.J.), 1448, 201–216. https://doi.org/10.1007/978-1-4939-3753-0_15.
Kalluri, R. (2016). The biology and function of exosomes in cancer. The Journal of Clinical Investigation, 126(4), 1208–1215. https://doi.org/10.1172/JCI81135.
Guo, W., Li, Y., Pang, W., & Shen, H. (2020). Exosomes: a potential therapeutic tool targeting communications between tumor cells and macrophages. Molecular Therapy : the Journal of the American Society of Gene Therapy, 28(9), 1953–1964. https://doi.org/10.1016/j.ymthe.2020.06.003.
Namburi, S., Broxmeyer, H. E., Hong, C. S., Whiteside, T. L., & Boyiadzis, M. (2020). DPP4+ exosomes in aml patient’s plasma suppress proliferation of hematopoietic progenitor cells. Leukemia. In Press.
Gliech, C. R., & Holland, A. J. (2020). Keeping track of time: the fundamentals of cellular clocks. The Journal of Cell Biology, 219(11), e202005136. https://doi.org/10.1083/jcb.202005136.
Kuintzle, R. C., Chow, E. S., Westby, T. N., Gvakharia, B. O., Giebultowicz, J. M., & Hendrix, D. A. (2017). Circadian deep sequencing reveals stress-response genes that adopt robust rhythmic expression during aging. Nature Communications, 8, 14529. https://doi.org/10.1038/ncomms14529.
Golan, K., Kumari, A., Kollet, O., Khatib-Massalha, E., Subramaniam, M. D., Ferreira, Z. S., Avemaria, F., Rzeszotek, S., García-García, A., Xie, S., Flores-Figueroa, E., Gur-Cohen, S., Itkin, T., Ludin-Tal, A., Massalha, H., Bernshtein, B., Ciechanowicz, A. K., Brandis, A., Mehlman, T., Bhattacharya, S., et al. (2018). Daily onset of light and darkness differentially controls hematopoietic stem cell differentiation and maintenance. Cell Stem Cell, 23(4), 572–585.e7. https://doi.org/10.1016/j.stem.2018.08.002.
Butler, T. D., & Gibbs, J. E. (2020). Circadian host-microbiome interactions in immunity. Frontiers in Immunology, 11, 1783. https://doi.org/10.3389/fimmu.2020.01783.
Anderson, S. T., & FitzGerald, G. A. (2020). Sexual dimorphism in body clocks. Science (New York, N.Y.), 369(6508), 1164–1165. https://doi.org/10.1126/science.abd4964.
Liesveld, J. L., Sharma, N., & Aljitawi, O. S. (2020). Stem cell homing: from physiology to therapeutics. Stem cells (Dayton, Ohio). https://doi.org/10.1002/stem.3242. Advance online publication.
Huang, X., & Broxmeyer, H. E. (2019). Progress towards improving homing and engraftment of hematopoietic stem cells for clinical transplantation. Current Opinion in Hematology, 26(4), 266–272. https://doi.org/10.1097/MOH.0000000000000510.
Guo, B., Huang, X., Cooper, S., & Broxmeyer, H. E. (2017). Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nature Medicine, 23(4), 424–428. https://doi.org/10.1038/nm.4298.
Huang, X., Guo, B., Liu, S., Wan, J., & Broxmeyer, H. E. (2018). Neutralizing negative epigenetic regulation by HDAC5 enhances human haematopoietic stem cell homing and engraftment. Nature Communications, 9(1), 2741. https://doi.org/10.1038/s41467-018-05178-5.
Xu, D., Yang, M., Capitano, M., Guo, B., Liu, S., Wan, J., Broxmeyer, H. E., & Huang, X. (2020). Pharmacological activation of nitric oxide signaling promotes human hematopoietic stem cell homing and engraftment. Leukemia. https://doi.org/10.1038/s41375-020-0787-z. Advance online publication.
Capitano, M. L., Chitteti, B. R., Cooper, S., Srour, E. F., Bartke, A., & Broxmeyer, H. E. (2015). Ames hypopituitary dwarf mice demonstrate imbalanced myelopoiesis between bone marrow and spleen. Blood Cells, Molecules & Diseases, 55(1), 15–20. https://doi.org/10.1016/j.bcmd.2015.03.004.
Dong, E., Du, H., & Gardner, L. (2020). An interactive web-based dashboard to track COVID-19 in real time. The Lancet. Infectious Diseases, 20(5), 533–534. https://doi.org/10.1016/S1473-3099(20)30120-1.
Banerjee, A., Nasir, J. A., Budylowski, P., Yip, L., Aftanas, P., Christie, N., Ghalami, A., Baid, K., Raphenya, A. R., Hirota, J. A., Miller, M. S., McGeer, A. J., Ostrowski, M., Kozak, R. A., McArthur, A. G., Mossman, K., & Mubareka, S. (2020). Isolation, sequence, infectivity, and replication kinetics of severe acute respiratory syndrome coronavirus 2. Emerging Infectious Diseases, 26(9), 2054–2063. https://doi.org/10.3201/eid2609.201495.
Korber, B., Fischer, W. M., Gnanakaran, S., Yoon, H., Theiler, J., Abfalterer, W., Hengartner, N., Giorgi, E. E., Bhattacharya, T., Foley, B., Hastie, K. M., Parker, M. D., Partridge, D. G., Evans, C. M., Freeman, T. M., de Silva, T. I., Sheffield COVID-19 Genomics Group, McDanal, C., Perez, L. G., Tang, H., et al. (2020). Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell, 182(4), 812–827.e19. https://doi.org/10.1016/j.cell.2020.06.043.
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., Schiergens, T. S., Herrler, G., Wu, N. H., Nitsche, A., Müller, M. A., Drosten, C., & Pöhlmann, S. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181(2), 271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Chan, J. F., Yuan, S., Kok, K. H., To, K. K., Chu, H., Yang, J., Xing, F., Liu, J., Yip, C. C., Poon, R. W., Tsoi, H. W., Lo, S. K., Chan, K. H., Poon, V. K., Chan, W. M., Ip, J. D., Cai, J. P., Cheng, V. C., Chen, H., Hui, C. K., et al. (2020). A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet (London, England), 395(10223), 514–523. https://doi.org/10.1016/S0140-6736(20)30154-9.
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., Cheng, Z., Yu, T., Xia, J., Wei, Y., Wu, W., Xie, X., Yin, W., Li, H., Liu, M., Xiao, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, England), 395(10223), 497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Pence, B. D. (2020). Severe COVID-19 and aging: are monocytes the key? GeroScience, 42(4), 1051–1061. https://doi.org/10.1007/s11357-020-00213-0.
COVID-19 Hospitalization and Death by Age | CDC [Online]. Available at: https://www.cdc.gov/coronavirus/2019-ncov/covid-data/investigations-discovery/hospitalization-death-by-age.html. Accessed 25 Sept 2020.
Heald-Sargent, T., Muller, W. J., Zheng, X., Rippe, J., Patel, A. B., & Kociolek, L. K. (2020). Age-related differences in nasopharyngeal severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) levels in patients with mild to moderate coronavirus disease 2019 (COVID-19). JAMA Pediatrics, 174(9), 902–903. Advance online publication. https://doi.org/10.1001/jamapediatrics.2020.3651.
Zhang, Y., Geng, X., Tan, Y., Li, Q., Xu, C., Xu, J., Hao, L., Zeng, Z., Luo, X., Liu, F., & Wang, H. (2020). New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie, 127, 110195. https://doi.org/10.1016/j.biopha.2020.110195.
Wen, W., Su, W., Tang, H., Le, W., Zhang, X., Zheng, Y., Liu, X., Xie, L., Li, J., Ye, J., Dong, L., Cui, X., Miao, Y., Wang, D., Dong, J., Xiao, C., Chen, W., & Wang, H. (2020). Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discovery, 6, 31. https://doi.org/10.1038/s41421-020-0168-9.
Terpos, E., Ntanasis-Stathopoulos, I., Elalamy, I., Kastritis, E., Sergentanis, T. N., Politou, M., Psaltopoulou, T., Gerotziafas, G., & Dimopoulos, M. A. (2020). Hematological findings and complications of COVID-19. American Journal of Hematology, 95(7), 834–847. https://doi.org/10.1002/ajh.25829.
Ratajczak, M. Z., Bujko, K., Ciechanowicz, A., Sielatycka, K., Cymer, M., Marlicz, W., & Kucia, M. (2020). SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45- precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Reviews and Reports, 1–12. https://doi.org/10.1007/s12015-020-10010-z. Advance online publication.
Ropa, J., Cooper, S., Capitano, M.L., Van't Hof, W., & Broxmeyer, H.E. (2020). Human hematopoietic stem, progenitor, and immune cells respond ex vivo to SARS-CoV-2 spike protein. Stem Cell Reviews and Reports. In Press.
Mehta, P., McAuley, D. F., Brown, M., Sanchez, E., Tattersall, R. S., Manson, J. J., & HLH Across Speciality Collaboration, UK. (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (London, England), 395(10229), 1033–1034. https://doi.org/10.1016/S0140-6736(20)30628-0.
Yang, Y., Shen, C., Li, J., Yuan, J., Wei, J., Huang, F., Wang, F., Li, G., Li, Y., Xing, L., Peng, L., Yang, M., Cao, M., Zheng, H., Wu, W., Zou, R., Li, D., Xu, Z., Wang, H., Zhang, M., et al. (2020). Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. The Journal of Allergy and Clinical Immunology, 146(1), 119–127.e4. https://doi.org/10.1016/j.jaci.2020.04.027.
Jiang, F., Yang, J., Zhang, Y., Dong, M., Wang, S., Zhang, Q., Liu, F. F., Zhang, K., & Zhang, C. (2014). Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nature Reviews Cardiology, 11(7), 413–426. https://doi.org/10.1038/nrcardio.2014.59.
Hadley, E. C., Lakatta, E. G., Morrison-Bogorad, M., Warner, H. R., & Hodes, R. J. (2005). The future of aging therapies. Cell, 120(4), 557–567. https://doi.org/10.1016/j.cell.2005.01.030.
Montecino-Rodriguez, E., Berent-Maoz, B., & Dorshkind, K. (2013). Causes, consequences, and reversal of immune system aging. The Journal of Clinical Investigation, 123(3), 958–965. https://doi.org/10.1172/JCI64096.
McHugh, D., & Gil, J. (2018). Senescence and aging: causes, consequences, and therapeutic avenues. The Journal of Cell Biology, 217(1), 65–77. https://doi.org/10.1083/jcb.201708092.
Campisi, J., Kapahi, P., Lithgow, G. J., Melov, S., Newman, J. C., & Verdin, E. (2019). From discoveries in ageing research to therapeutics for healthy ageing. Nature, 571(7764), 183–192. https://doi.org/10.1038/s41586-019-1365-2.
Spehar, K., Pan, A., & Beerman, I. (2020). Restoring aged stem cell functionality: current progress and future directions. Stem Cells (Dayton, Ohio). https://doi.org/10.1002/stem.3234. Advance online publication.
Serebryannyy, L., & Misteli, T. (2018). Protein sequestration at the nuclear periphery as a potential regulatory mechanism in premature aging. The Journal of Cell Biology, 217(1), 21–37. https://doi.org/10.1083/jcb.201706061.
Klaips, C. L., Jayaraj, G. G., & Hartl, F. U. (2018). Pathways of cellular proteostasis in aging and disease. The Journal of Cell Biology, 217(1), 51–63. https://doi.org/10.1083/jcb.201709072.
Hu, J. L., Todhunter, M. E., LaBarge, M. A., & Gartner, Z. J. (2018). Opportunities for organoids as new models of aging. The Journal of Cell Biology, 217(1), 39–50. https://doi.org/10.1083/jcb.201709054.
Wang, H., & Zhang, X. H. (2020). Molecules in the blood of older people promote cancer spread. Nature, 585(7824), 187–188. https://doi.org/10.1038/d41586-020-02381-7.
Gomes, A. P., Ilter, D., Low, V., Endress, J. E., Fernández-García, J., Rosenzweig, A., Schild, T., Broekaert, D., Ahmed, A., Planque, M., Elia, I., Han, J., Kinzig, C., Mullarky, E., Mutvei, A. P., Asara, J., de Cabo, R., Cantley, L. C., Dephoure, N., Fendt, S. M., et al. (2020). Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature, 585(7824), 283–287. https://doi.org/10.1038/s41586-020-2630-0.
Sinclair, D., LaPlante, M. D., & Delphia, C. (2019). Lifespan: why we age--and why we don’t have to. Publisher: New York: Atria Books, 2019.
Silwal, P., Kim, J. K., Kim, Y. J., & Jo, E. K. (2020). Mitochondrial reactive oxygen species: double-edged weapon in host defense and pathological inflammation during infection. Frontiers in Immunology, 11, 1649. https://doi.org/10.3389/fimmu.2020.01649.
Hormaechea-Agulla, D., Le, D. T., & King, K. Y. (2020). Common sources of inflammation and their impact on hematopoietic stem cell biology. Current Stem Cell Reports, 1–12. https://doi.org/10.1007/s40778-020-00177-z. Advance online publication.
Haas, S. (2020). Hematopoietic stem cells in health and disease—insights from single-cell multi-omic approaches. Current Stem Cell Reports, 6, 67–76. https://doi.org/10.1007/s40778-020-00174-2.
Horton, P. D., Dumbali, S., & Wenzel, P. L. (2020). Mechanoregulation in hematopoiesis and hematologic disorders. Current Stem Cell Reports, 6, 86–95. https://doi.org/10.1007/s40778-020-00172-4.
Golan, K., Singh, A. K., Kollet, O., Bertagna, M., Althoff, M., Khatib-Massalha, E., Petrovich-Kopitman, E., Wellendorf, A., Massalha, H., Levin-Zaidman, S., Dadosh, T., Bohan, B., Gawali, M. V., Dasgupta, B., Lapidot, T., & Cancelas, J. A. (2020). Bone marrow regeneration requires mitochondrial transfer from donor Cx43-expressing hematopoietic progenitors to stroma. Blood, 2020005399. https://doi.org/10.1182/blood.2020005399. Advance online publication.
Challen, G. A., & Goodell, M. A. (2020). Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood, 136(14), 1590–1598. https://doi.org/10.1182/blood.2020006510.
Warren, J. T., & Link, D. C. (2020). Clonal hematopoiesis and risk for hematologic malignancy. Blood, 136(14), 1599–1605. https://doi.org/10.1182/blood.2019000991.
Jaiswal, S. (2020). Clonal hematopoiesis and nonhematologic disorders. Blood, 136(14), 1606–1614. https://doi.org/10.1182/blood.2019000989.
Shaheen, M., & Broxmeyer, H. E. (2013). Principles of cytokine signaling. In R. Hoffman, E. J. Benz, L. E. Silberstein Jr., H. Heslop, J. I. Weitz, & J. Anastasi (Eds.), Hematology: Basic principles and practice (6th Edition, Chapter 14 ed., pp. 136–146). Philadelphia: Elsevier Saunders.
Shaheen, M., & Broxmeyer, H. E. (2018). Cytokine/Receptor families and signal transduction. In R. Hoffman, E. Benz, L. Silberstein, H. Heslop, J. I. Weitz, J. Anastasi, M. E. Salama, & S. A. Abutalib (Eds.), Hematology: Basic principles and practice (7th Edition, Chapter 16 ed., pp. 163–175).
Shao, L., Elujoba-Bridenstine, A., Zink, K. E., Sanchez, L. M., Cox, B. J., Pollok, K. E., Sinn, A., Bailey, B. J., Sims, E., Cooper, S., Broxmeyer, H. E., Pajcini, K. V., & Tamplin, O. J. (2020). The neurotransmitter receptor Gabbr1 regulates proliferation and function of hematopoietic stem and progenitor cells. Blood, 2019004415. https://doi.org/10.1182/blood.2019004415. Advance online publication.
Wang, X. and Broxmeyer, H.E. (2020). DUSP16 is a regulator of hematopoietic stem and progenitor cells and promotes their expansion ex-vivo. Leukemia. In Press.
Christopherson 2nd, K. W., Hangoc, G., Mantel, C. R., & Broxmeyer, H. E. (2004). Modulation of hematopoietic stem cell homing and engraftment by CD26. Science (New York, N.Y.), 305(5686), 1000–1003. https://doi.org/10.1126/science.1097071.
Broxmeyer, H. E., Hoggatt, J., O'Leary, H. A., Mantel, C., Chitteti, B. R., Cooper, S., Messina-Graham, S., Hangoc, G., Farag, S., Rohrabaugh, S. L., Ou, X., Speth, J., Pelus, L. M., Srour, E. F., & Campbell, T. B. (2012). Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nature Medicine, 18(12), 1786–1796. https://doi.org/10.1038/nm.2991.
Farag, S. S., Srivastava, S., Messina-Graham, S., Schwartz, J., Robertson, M. J., Abonour, R., Cornetta, K., Wood, L., Secrest, A., Strother, R. M., Jones, D. R., & Broxmeyer, H. E. (2013). In vivo DPP-4 inhibition to enhance engraftment of single-unit cord blood transplants in adults with hematological malignancies. Stem Cells and Development, 22(7), 1007–1015. https://doi.org/10.1089/scd.2012.0636.
Vélez de Mendizábal, N., Strother, R. M., Farag, S. S., Broxmeyer, H. E., Messina-Graham, S., Chitnis, S. D., & Bies, R. R. (2014). Modelling the sitagliptin effect on dipeptidyl peptidase-4 activity in adults with haematological malignancies after umbilical cord blood haematopoietic cell transplantation. Clinical Pharmacokinetics, 53(3), 247–259. https://doi.org/10.1007/s40262-013-0109-y.
Farag, S. S., Nelson, R., Cairo, M. S., O'Leary, H. A., Zhang, S., Huntley, C., Delgado, D., Schwartz, J., Zaid, M. A., Abonour, R., Robertson, M., & Broxmeyer, H. (2017). High-dose sitagliptin for systemic inhibition of dipeptidylpeptidase-4 to enhance engraftment of single cord umbilical cord blood transplantation. Oncotarget, 8(66), 110350–110357. https://doi.org/10.18632/oncotarget.22739.
Broxmeyer, H. E., Farag, S. S., & Rocha, V. (2016). Cord blood hematopoietic cell transplantation. In S. J. Forman, R. S. Negrin, J. H. Antin, & F. R. Appelbaum (Eds.), Thomas Hematopoietic Cell Transplantation (5th Edition, Chapter 39 ed., pp. 437–455). Oxford, England: John Wiley & Sons, Ltd.
Farag, S., Zaid, M. A., Nelson, R. P., Schwartz, J. E., Thakran, T. C., Blakley, A. J., Broxmeyer, H. E., & Zhang, S. (2020). Dipeptidyl peptidase-4 inhibition for the prevention of acute graft versus host disease following myeloablative allogeneic peripheral blood stem cell transplantation. New England Journal of Medicine. In Press.
Ou, X., O'Leary, H. A., & Broxmeyer, H. E. (2013). Implications of DPP4 modification of proteins that regulate stem/progenitor and more mature cell types. Blood, 122(2), 161–169. https://doi.org/10.1182/blood-2013-02-487470.
Ropa, J., & Broxmeyer, H. E. (2020). An expanded role for dipeptidylpeptidase4 (DPP4) in cell regulation. Current Opinion in Hematology, 27(4), 215–224. https://doi.org/10.1097/MOH.0000000000000590.
Broxmeyer, H. E., Capitano, M., Campbell, T. B., Hangoc, G., & Cooper, S. (2016). Modulation of hematopoietic chemokine effects in vitro and in vivo by DPP-4/CD26. Stem Cells and Development, 25(8), 575–585. https://doi.org/10.1089/scd.2016.0026.
Liggett, L. A., & Sankaran, V. G. (2020). Unraveling hematopoiesis through the lens of genomics. Cell, 182(6), 1384–1400. https://doi.org/10.1016/j.cell.2020.08.030.
Morrison, S. J., & Scadden, D. T. (2014). The bone marrow niche for haematopoietic stem cells. Nature, 505(7483), 327–334. https://doi.org/10.1038/nature12984.
Acknowledgements
Some of the studies reported in this review, and the writing of this review were supported by the following U.S. Public Health Grants from the National Institutes of Health to H.E.B.: R35 HL139599 (Outstanding Investigator Award), R01 DK109188, U54 DK106846; A.A., J.P.R., and T.T. were supported by T32 DK007519 to H.E.B. R01 HL150624, R56 DK119524, R56 AG052501, DoD W81XWH-13-1-0187, DoD W81XWH-18-1-0265 and DoD W81XWH-19-1-0575, the Leukemia and Lymphoma Society Translational Research Program award 6581-20 and the St. Baldrick’s Foundation Scholar Award to Y.L. MLC was supported by R01 DK109188. CO was supported by National Institute of Allergy and Infectious Diseases (NIAID) contracts HHSN266200500043C and HHSN272201000046C and grants 1U01AI107340-01 and 2R44 AI088288-03A1, National Institute on Aging (NIA) grant R01AG046246-01, and Department of Defense grants PR140896, PR141527, and PR140433P1.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Broxmeyer, H.E., Liu, Y., Kapur, R. et al. Fate of Hematopoiesis During Aging. What Do We Really Know, and What are its Implications?. Stem Cell Rev and Rep 16, 1020–1048 (2020). https://doi.org/10.1007/s12015-020-10065-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-020-10065-y