
Abstract
A strain that exhibited intracellular proline-specific aminopeptidase (PAP) activity was isolated from soy sauce koji and identified as Aspergillus oryzae JN-412. The gene coding PAP was cloned and efficiently expressed in Escherichia coli BL21 in a biologically active form. The highest specific activity reached 52.28 U mg−1 at optimum cultivation conditions. The recombinant enzyme was purified 3.3-fold to homogeneity with a recovery of 36.7 % from cell-free extract using Ni-affinity column chromatography. It appeared as a single protein band on SDS-PAGE with molecular mass of approximately 50 kDa. The purified enzyme exhibited the highest activity at 60 °C and pH 7.5. The enzyme activity was inhibited by PMSF and ions like Zn2+ and Cu2+. DTT, β-mercaptoethanol, EDTA, and ions like Co2+, Mg2+, Mn2+, and Ca2+ had no influence on enzyme activity, whereas Ni2+ enhanced the enzyme activity. By using collagen as a substrate, the purified recombinant prolyl aminopeptidase contributed to the hydrolysis of collagen when used in combination with neutral protease, and free amino acids in collagen hydrolysates was significantly increased.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aminopeptidases (Aps; EC 3.4.11.) are exopeptidases that cleave N-terminal amino acids from proteins or peptides, sequently [1]. According to the mechanism of catalysis and the structure of their active site, aminopeptidases can be subdivided into three main families: metalloaminopeptidases, cysteine aminopeptidases, and serine aminopeptidases [2]. Prolyl aminopeptidases (PAPs; EC 3.4.11.5) belong to serine aminopeptidase and are classified into the S33 family in MEROPS [3], which contain a triad of serine, histidine, and aspartic acid in the active site [4, 5]. The serine active site appears within Gly-Xaa-Ser-Xbb-Gly, in which Xaa can be any amino acid residue and Xbb is most often a hydrophobic amino acid [3, 6]. PAPs can cleave a proline residue specifically from the N-terminus of substrates [7]. They are essential for the degradation of proline-rich peptides and proteins such as collagen and gelatin [8] because the unique structure of proline prevents its cleavage by many broad-specificity aminopeptidases [9]. PAPs also play an important role in treating wastewater in the food industry, especially that from meat industries, which contains plenty of collagen. Moreover, PAPs have the potential ability to reduce bitter peptides in daily products and enhanced the flavor of food [10, 11].
PAPs are widespread in various organisms. According to the similarity of an amino acid sequence of PAPs, PAPs can be further classified into two subfamilies: Neisseria gonorrhoeae PAP (S33.001) [12] and Aeromonas sobria (S33.008) [13]. PAPs in the S33.008 subfamily are found in eukaryotes (fungi and plants) and some bacteria [14–17], whereas aminopeptidases in the S33.001 subfamily only exist in bacteria [17, 18].
Aspergillus oryzae plays an important role in the production of traditional fermentation foods (soy sauce and soybean paste) and beverages (rice wine) in East Asia for a long history. Owing to its safety for human consumption, A. oryzae is generally regarded as safe by the US Food and Drug Administration [19]. Therefore, many enzymes produced by A. oryzae are widely used in food processing. Several aminopeptidases from A. oryzae, such as leucine aminopeptidase [20], aspartyl aminopeptidase [21, 22], proline aminopeptidase [23], lysine aminopeptidase [24], glycine d-alanine aminopeptidase [25], and d-stereoselective aminopeptidase [26], have been characterized, since the genome sequencing and analysis of A. oryzae had been completed in 2005 [27].
The E. coli expression system is widely used for the production of recombinant proteins to reveal their structures and functions. In this study, the gene coding PAP of A. oryzae JN-412 was cloned and overexpressed in E. coli BL21. The recombinant PAP was purified and characterized. The secondary structure was analyzed by circular dichroism (CD), and the 3D structure of the PAP monomer was predicted using the pGenTHREADER method. Furthermore, the ability of the recombinant PAP in collagen degradation was verified using an amino acid analyzer.
Materials and Methods
Chemicals and Reagents
All aminoacyl-p-nitroanilines were purchased from Bachem AG (Saffron Walden, Essex, UK). Collagen type Ι (bovine tendon) and protein inhibitors were obtained from Sigma (St. Louis, MO, USA). RNAiso Plus, complementary DNA (cDNA) Synthesis Kit, isopropyl-β-d-thiogalactoside (IPTG) and T-Vector pMD™19 (Simple) were all purchased from TaKaRa (Dalian, China). HisTrap HP (5 mL) was purchased from GE Healthcare (Buckinghamshire, UK). All other chemicals were standard reagent grade.
Strains and Vectors
A. oryzae JN-412 under the accession number of China General Microbiological Culture Collection (CGMCC) 8474 in the CGMCC was used in this paper. E. coli DH5α (Novagen, Darmstadt, Germany) was used for propagation of plasmids, and E. coli BL21 (DE3) (Novagen) was used as a host strain for pap expression. Plasmid pMD19-T was used as a cloning vector. Plasmid pET-28a(+) (Novagen) was used as the expression vector.
Enzyme Activity Assay and Protein Determination
Aminopeptidase activity was determined with artificial substrates, which were aminoacyl-p-nitroanilide [28]. The reaction mixture contained 2.5 mM l-pro-pNA in 50 mM Tris-HCl (pH 7.5) and an appropriate amount of enzyme and incubated at 50 °C for 10 min. The absorbance at 405 nm was measured using a spectrophotometer to determine the amount of liberated p-nitroanilide. One unit was defined as the amount of enzyme that released 1 μmol p-nitroanilide per minute.
The protein content of the samples was measured by the Bradford method with bovine serum albumin as a standard [29].
Cloning of Prolyl Aminopeptidase Gene and Sequence Analysis
Recombinant DNA techniques are as described by Sambrook and Russell [30]. For isolation of total RNA, A. oryzae JN-412 was grown at 30 °C in YPD liquid media for 3 days. Fungi mycelia were collected by centrifugation (8,000 × g, 15 min) and washed twice with water at 4 °C. The mycelia were frozen and ground to fine power in liquid nitrogen condition. The total RNA was isolated by an RNAiso Plus reagent. cDNA was obtained using cDNA Synthesis Kit. To clone the PAP gene, primers of papF1 and papR1 (Table 1) were designed based on the genome database of A. oryzae RIB40 (NBRC 100959) (database number AO090003000302) [23, 27]. Polymerase chain reaction (PCR) conditions were as follows: start at 98 °C for 5 min and 38 cycles of 98 °C for 10 s, 59 °C for 5 s, and 72 °C for 90 s, followed by 72 °C for 10 min. The amplified PCR product was purified, ligated to the pMD19-T vector and transferred into E. coli DH5α for sequencing and subjected to Basic Local Alignment Search Tool (BLAST) analysis.
Nucleotide and deduced amino-acid sequence were analyzed by BLAST. Search analysis of conserved domain and signature sequences was carried out by ScanProsite (http://www.expasy.ch/tools/ScanProsit). Signal peptide was analyzed by Signal P 3.0 service (http://www.cbs.dtu.dk/services/SigalP). N-Glycosylation site and O-glycosylation site prediction was based on NetNGlyc 1.0 service at http://www.cbs.dtu.dk/services/NetNGlyc/ and http://www.cbs.dtu.dk/services/NetOGlyc/, respectively. Disulfide bonds were predicted through the DiANNA 1.1 Web server at http://clavius.bc.edu/;clotelab/DiANNA/ and DISULFIND Web server at http://disulfind.dsi.unifi.it/. The amino-acid sequences were aligned by ClustalX Version 1.83.
Expression of the Gene in E. coli BL21
In order to express the PAP gene (pap) in E. coli BL21, papF2 and papR2 primers were designed (Table 1); BamH Ι and Not Ι sites were added to the forward primer and the reverse primer, respectively. PCR conditions were the same as above.
The amplified PCR product of the cDNA was purified and cloned into the pMD19-T vector and transformed into E. coli DH5α-competent cells. Finally, the positive clones were propagated. The recombinant plasmids which contained pap were extracted and digested with BamH Ι and Not Ι. Then, the pap gene was subcloned into the pET-28a(+) vector which was digested with the same restriction enzymes and transformed into E. coli BL21-competent cells. The positive clones were picked up, and then, the recombinant plasmids were extracted and tested by double digestion.
E. coli BL21 harboring pap in the pET-28a(+) vector was inoculated into 5-mL Luria-Bertani (LB) liquid media containing kanamycin (50 μg mL−1) on a rotary shaker (220 rpm) for 12 h at 37 °C which was used as seed. The prepared seed was inoculated into 50-mL (250-mL shaking flask) LB liquid media containing kanamycin (50 μg mL−1) on a rotary shaker (220 rpm) at 37 °C until the optical density at 600 nm reached 0.6–0.8. IPTG was added with a final concentration of 0.8 mM; the culture temperature was changed from 37 to 25 °C and the cultivation was continued for 8 h.
Purification of the Recombinant Prolyl Aminopeptidase
Fermentation broth was obtained according to the above-mentioned method. Cell pellet was collected by centrifugation (8,000 × g, 10 min) and washed with 50-mM Tris-HCl buffer (pH 7.5) twice. Finally, the cell pellet was resuspended in 40-mL binding buffer (40 mM imidazole and 0.5 M NaCl in 50 mM Tris-HCl (pH 7.5)), and the cell suspension was lysed by sonication followed by centrifugation at 10,000 × g for 15 min. Cell-free extract was collected for purification.
The crude enzyme solution was loaded onto a 5-mL Ni-NTA column, preequilibrated with binding buffer. After binding, the column was washed with six times the bed column of a binding buffer followed by eluting with an elution buffer (0.5 M imidazole and 0.5 M NaCl in 50 mM Tris-HCl (pH7.5)) for five times of the bed column. The active eluted fraction was pooled and dialyzed against 50-mM Tris-HCl buffer (pH 7.5) for 48 h.
SDS-PAGE Analysis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 12 % (w/v) acrylamide in gels [31] for analyzing the purified fractions. Protein was visualized by Coomassie brilliant blue R-250 staining.
The Secondary Structure Analysis and 3D Modeling of PAP
The secondary structure analysis was carried using MOS-450/AF circular dichroism (CD) spectrometer (Bio-Logic SAS, France) at room temperature. The light path was 0.1 nm, and the wavelength was varied from 190 to 250 nm. The scan rate was 1 nm per 2 s, and the bandwidth was 1 nm. The purified recombinant protein was diluted using 50-mM Tris-HCl (pH 7.5) buffer, and the final protein concentration was 0.4 mg mL−1. Tris-HCl (50 mM; pH 7.5) buffer was used as control. Selcon3 program was implemented for analyzing protein CD spectra and Origin 9.0 program was used for drawing the curve.
3D structure prediction was carried out using the pGenTHREADER method [32] (http://bioinf.cs.ucl.ac.uk). Analysis of the predicted structure was performed using PROCHECK (http://nihserver.mbi.ucla.edu/SAVS/), and the final model displayed good geometry with less than 2 % of residues in the disallowed region.
Properties of Purified Recombinant Prolyl Aminopeptidase
The optimum pH for purified recombinant PAP activity was determined using the following buffers (50 mM): citrate buffer pH 3.0–7.0, Tris-HCl buffer pH 7.0–9.0, and sodium glycine buffer pH 8.0–11.0. The pH stability was determined by preincubating purified recombinant PAP in buffer with varying pH for 30 min at 30 °C (citrate buffer pH 3.0–7.0, phosphate buffer pH 6.0–8.0, and sodium glycine buffer pH 8.0–11.0). The results were presented as the percentage of the activity measured compared with the activity at optimum pH.
The optimal temperature for purified recombinant PAP activity was determined over a range from 30 to 60 °C. The thermostability was determined by preincubating purified recombinant PAP at varying temperatures for 1 h. The enzyme activity was assayed under standard conditions. Enzyme incubated at 4 °C was used as control.
The effects of protease inhibitors and metal ions on purified enzyme were analyzed using Pro-pNA as a substrate, followed by preincubating purified recombinant PAP for 30 min at 30 °C with different chemicals (phenylmethanesulfonyl fluoride (PMSF) 1, 5, and 10 mM; dithiothreitol (DTT) 1 and 10 mM; β-mercaptoethanol 1 and 10 mM; and EDTA 1 and 10 mM) and metal ions (Co2+, Mg2+, Mn2+, Ca2+, Zn2+, and Ni2+) at the final concentration of 1 mM.
Substrate specificity of PAP was determined using ten different aminoacyl-pNAs (Pro-, Leu-, Arg-, Lys-, Met-, Ala-, Ile-, Val-, Phe-, and Glu-pNA). For determination of the Michaelis-Menten constants for Pro-pNA, an activity assay was performed with Pro-pNA at different concentrations, ranging from 0.1 to 1.0 mM, in Tris-HCl buffer (pH 7.5), at 50 °C for 10 min. K m and V max of PAP for Pro-pNA were calculated from Lineweaver-Burk plots.
Collagen Degradation and Hydrolysate Analysis
The reaction mixture of collagen degradation contains 50-mg collagen type Ι (bovine tendon), purified recombinant PAP (10 U mL−1), and/or neutral protease (50 mg, 100 U) in 50 mM Tris-HCl (pH 7.5), and the volume was 50 mL. The reaction system was agitated at 40 °C for 1 h, and the reaction was terminated by heating in a boiling water bath for 20 min. The supernatant was collected by centrifugation at 10,000 × g, 4 °C for 10 min. Amino acids in the supernatant were determined by an amino acid analyzer (L-8900, HITACHI, Japan).
Results and Discussion
Cloning and Characterization of pap Gene from A. oryzae JN-412
The nucleotide sequence of cDNA of the A. oryzae JN-412 PAP gene consisted of 1,344 bp and encodes a protein containing 447 amino acids. The BLAST search showed that the deduced amino acid sequence of PAP shares the highest similarities to proline aminopeptidase of A. oryzae RIB40 (98 %; GenBank accession No. BAE57503) [23] and proline iminopeptidase of Aspergillus niger CBS 513.88 (74 %; GenBank accession No. EHA19638) [33]. The theoretical molecular mass is 50,406.91 Da and pI is 5.53. Signal P analysis indicated that PAP does not possess a secretory signal peptide. Glycosylation analysis exhibited that PAP contains three possible N-glycosylation sites NYSR, NWSR, and NFSI (amino acid numbers 29–32, 316–319, and 328–331, respectively) and no possible O-glycosylation sites. Disulfide bonding state prediction indicated that there is no possible disulfide bond in protein.
Alignment of amino acid sequences of PAP with other known microbial proline aminopeptidases followed by conserved domain analysis of sequence revealed it to be an α/β-hydrolase fold serine peptidase, containing a Ser-Asp-His active triad (162, 396, and 424) and highly conserved motif G-X-S-X-G to serine aminopeptidase [6, 34]:GQSFG (160–165) (Fig. 1). This is conserved within multimeric PAP in most cases, but GGSWG, GQSWG, and GHSWG are also found in multimeric PAP [16, 17, 33]. Furthermore, multimeric PAPs are about 120 amino acids larger than monomeric PAPs.

Alignment of homologous prolyl aminopeptidases with prolyl aminopeptidase of the JN-412 strain. The sequences shown are S. marcescens BAA23336, A. sobria BAA06380, A. niger EHA19638, A. oryzae RIB40 BAE57503, and A. oryzae JN-412 in this paper. Conserved motif is underlined. Active sites are indicated with an asterisk
Expression and Purification of PAP in E. coli BL21
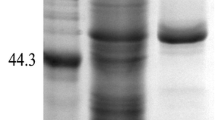
The pap gene from pMD19-pap was digested with BamH Ι and Not Ι and ligated into a pET-28a(+) expression vector to construct pET-28a-pap. The recombinant vector was transformed into E. coli BL21, and the pap gene was efficiently expressed in a biologically active form at an optimal condition. The highest specific activity was reached at 52.28 U mg−1 after an induction expression of 8 h at 25 °C in LB medium. This value was much higher than the original A. oryzae JN-412. Due to a 6 × His tag that exists in the N-terminus of a recombinant protein, the fusion protein was purified by Ni-affinity chromatography. This procedure represented a 3.3-fold purification with a final yield of 36.7 %. The specific activity toward Pro-pNA of purified PAP was 172.43 U mg−1 (Table 2). The purified PAP appeared as a single protein band on SDS-PAGE with a molecular mass of approximately 50 kDa (Fig. 2).
The Secondary Structure Analysis and 3D Modeling of PAP
The CD spectrum of purified recombinant protein exhibits one positive band and two negative bands in the far-UV region at 192, 208, and 222 nm which is the characteristic of the α-helix structure of protein (Fig. 3) [35]. The data of CD spectrum were analyzed by the Selcon3 program, and the result showed that the protein contains 39.8 % of α-helix, 12.9 % of β-sheet, and 47.3 % of other structures. Those values are basically in agreement with the prediction results by PSIPRED method (http://bioinf.cs.ucl.ac.uk/psipred/) which predicts that the content of α-helix, β-sheet, and other structures is of 37.58, 10.74, and 51.68 %, respectively.

SDS-PAGE of recombinant PAP purification steps. Lane M, low-molecular-weight standard protein markers; lane 1, cell-free extract; and lane 2, fraction of Ni-NTA (purified recombinant PAP). The arrowhead indicates the band of recombinant PAP protein
The 3D structure of A. oryzae JN-412 PAP was predicted using Xanthomonas campestris proline iminopeptidase crystal structure (PDB ID: 1azwa) [36] as a template through the pGenTHREADER method [32]. The predicted 3D structure of the PAP monomer has two continuous domains. One is the α/β-hydrolase fold, with a central seven-stranded β-sheet flanked by six helices on the other side. The other larger domain essentially consists of eight helices (Fig. 4a), which is different from monomeric PAP [37]. It may play a role in multimeric PAP multimerization. The active site is located at the bottom of a deep hydrophobic pocket at the interface between two domains (Fig. 4b).

Determination of the secondary structure of PAP by circular dichroism
Properties of Recombinant PAP
The effects of pH and temperature on recombinant PAP activity were examined. The pH curve displays a maximum activity of 52.28 U mg−1 at pH 7.5, which is similar to most reported PAPs (within 7.0–8.0) [10, 17, 33, 38, 39]. After incubation at 30 °C for 1 h, the loss in activity of this purified recombinant PAP was little, and the residual activity was still above 80 %. The purified recombinant PAP exhibits the highest activity at 60 °C. It is identical to proline aminopeptidase from Grifola frondosa [39] and much higher than the recombinant proline aminopeptidase from A. oryzae RIB40 (30 °C) [23], Debaryomyces hansenii (45 °C) [10], and Talaromyces emersonii (50 °C) [17]. Thermostability investigation showed that after 1-h preincubation in 50-mM Tris-HCl buffer (pH 7.5) in 30, 40, 50, and 60 °C; 94, 80.5, less than 10 %, and nondetectable activity remained, respectively. It differs from a thermophilic Aneurinibacillus sp. strain AM-1 [8] proline aminopeptidase, which completely loses its activity at 75 °C. However, it is similar to PAPs derived from Streptomyces aureofaciens TH-3 [38] and T. emersonii [17], which show poor thermostability over 50 °C.
The purified recombinant PAP was assayed for hydrolytic activity against ten different aminoacyl-pNA substrates (data not shown). PAP only showed catalytic activity toward Pro-pNA. This narrow specificity is the same as PAPs from Aeromonas [11], Hafnia [40], and Aspergillus [33]. However, proline aminopeptidase derived from Aneurinibacillus sp. strain AM-1 [8] showed alanine and leucine aminopeptidase activity, and PAP from G. frondosa [39] showed alanine aminopeptidase and glycine aminopeptidase. Hydrophobic interaction and a space suitable for proline in the hydrophobic pocket might cause the differences in substrate specificity. The Michaelis-Menten constants were further determined using Pro-pNA as the substrate. The K m and V max values were 0.13 mM and 0.13 μM min−1, respectively.

SDS-PAGE of recombinant PAP purification steps. Lane M, low-molecular-weight standard protein markers; lane 1, cell-free extract; and lane 2, fraction of Ni-NTA (purified recombinant PAP). The arrowhead indicates the band of recombinant PAP protein
The effects of a range of divalent cations and chemical reagents on purified recombinant PAP were shown in Table 3. PMSF, a serine protease inhibitor, showed strong inhibition on enzyme activity. Among the divalent cations studied, Co2+, Mg2+, Mn2+, Ca2+, and Ni2+ showed no inhibitation, whereas Zn2+ and Cu2+ strongly inhibit enzyme activity. Inactivation of proline aminopeptidase by Zn2+ and Cu2+ had also been reported by other studies [9, 41]. Metal chelator (EDTA) showed no effect on enzyme activity which indicates that the enzyme is metal independent, whereas the influence of EDTA was observed for PAPs from A. oryzae RIB40 [23] and Aneurinibacillus sp. strain AM-1 [8]. No inhibition of the enzyme activity was observed in the presence of reducing reagents, β-mercaptoethanol and DTT. These results suggest that the enzyme is likely to be an aminopeptidase without important disulfide bonds.
Collagen Hydrolysate Analysis
On account of collagen and fragments of degraded collagen is industrially attractive biomaterial; much more attention is being paid to generate collagen and gelatin fragments using proline aminopeptidase, coupled with other proteases. Accordingly, four experiments are designed as in Table 4 to evaluate the hydrolytic activity of A. oryzae PAP for collagen. Results are shown in Table 5. The recombinant PAP showed limited ability of collagen degradation when the collagen was in its native state. However, when combined with neutral protease, the free amino acids in collagen hydrolysates were significantly increased, which was noticeable in group 2; the reason is that neutral protease is related to the initial degradation of collagen. Therefore, the recombinant PAP is indeed contributed to the hydrolysis of collagen when used in combination with neutral protease. Moreover, essential amino acids Pro, Phe, Arg, Leu, Ile, Thr, Tyr, and His were observed in hydrolysates and all showed a high value (Table 5), which indicated that the collagen hydrolysates were rich in nutrients, and the amino acid composition would fulfill human requirements.
Conclusion
In this study, the PAP gene from A. oryzae JN-412 was cloned and successfully expressed in E. coli BL21 in a biologically active form. Besides the characterizations of purified recombinant PAP, the determination of secondary structure and 3D modeling were also performed. Moreover, the enzyme is applicable for the degradation of hard digestive proteins, e.g., collagen and gelatin fragments. Though, in combination with other enzymes, the enzyme possesses a high potential to hydrolyze collagen.
References
Taylor, A. (1993). Aminopeptidases: structure and function. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 7, 290–298.
Rawlings, N. D., & Barrett, A. J. (1993). Evolutionary families of peptidases. Biochemical Journal, 290, 205–218.
Rawlings, N. D., Morton, F. R., & Barrett, A. J. (2006). MEROPS: the peptidase database. Nucleic Acids Research, 34, D270–D272.
Morel, F., Gilbert, C., Geourjon, C., Frot-Coutaz, J., Portalier, R., & Atlan, D. (1999). The prolyl aminopeptidase from Lactobacillus delbrueckii subsp. bulgaricus belongs to the alpha/beta hydrolase fold family. Biochimica Et Biophysica Acta, 1429, 501–505.
Kitazono, A., Ito, K., & Yoshimoto, T. (1994). Prolyl aminopeptidase is not a sulfhydryl enzyme: identification of the active serine residue by site-directed mutagenesis. Journal of Biochemistry, 116, 943–945.
Polgar, L. (2002). The prolyl oligopeptidase family. Cellular and Molecular Life Sciences CMLS, 59, 349–362.
Arima, J., Uesugi, Y., Uraji, M., Iwabuchi, M., & Hatanaka, T. (2006). Dipeptide synthesis by an aminopeptidase from Streptomyces septatus TH-2 and its application to synthesis of biologically active peptides. Applied and Environmental Microbiology, 72, 4225–4231.
Murai, A., Tsujimoto, Y., Matsui, H., & Watanabe, K. (2004). An Aneurinibacillus sp. strain AM-1 produces a proline-specific aminopeptidase useful for collagen degradation. Journal of Applied Microbiology, 96, 810–818.
Nandan, A., Pandey, A., & Nampoothiri, K. M. (2011). Proline-specific extracellular aminopeptidase purified from Streptomyces lavendulae. Applied Biochemistry and Biotechnology, 163, 994–1001.
Bolumar, T., Sanz, Y., Aristoy, M. C., & Toldra, F. (2003). Purification and characterization of a prolyl aminopeptidase from Debaryomyces hansenii. Applied and Environmental Microbiology, 69, 227–232.
Izawa, N., Ishikawa, S., Tanokura, T., Ohta, K., & Hayashi, K. (1997). Purification and characterization of Aeromonas caviae aminopeptidase possessing debittering activity. Journal of Agricultural and Food Chemistry, 45, 4897–4902.
Chen, K. C., & Buchanan, T. M. (1980). Hydrolases from Neisseria gonorrhoeae. The study of gonocosin, an aminopeptidase-P, a proline iminopeptidase, and an asparaginase. The Journal of Biological Chemistry, 255, 1704–1710.
Kitazono, A., Kitano, A., Tsuru, D., & Yoshimoto, T. (1994). Isolation and characterization of the prolyl aminopeptidase gene (pap) from Aeromonas sobria: comparison with the Bacillus coagulans enzyme. Journal of Biochemistry, 116, 818–825.
Stepaniak, L. (2000). Isolation and characterization of proline iminopeptidase from Propionibacterium freudenreichii ATCC 9614. Die Nahrung, 44, 102–106.
Leenhouts, K., Bolhuis, A., Boot, J., Deutz, I., Toonen, M., Venema, G., et al. (1998). Cloning, expression, and chromosomal stabilization of the Propionibacterium shermanii proline iminopeptidase gene (pip) for food-grade application in Lactococcus lactis. Applied and Environmental Microbiology, 64, 4736–4742.
Szawlowska, U., Grabowska, A., Zdunek-Zastocka, E., & Bielawski, W. (2012). TsPAP1 encodes a novel plant prolyl aminopeptidase whose expression is induced in response to suboptimal growth conditions. Biochemical and Biophysical Research Communications, 419, 104–109.
Mahon, C. S., O'Donoghue, A. J., Goetz, D. H., Murray, P. G., Craik, C. S., & Tuohy, M. G. (2009). Characterization of a multimeric, eukaryotic prolyl aminopeptidase: an inducible and highly specific intracellular peptidase from the non-pathogenic fungus Talaromyces emersonii. Microbiology (Reading, England), 155, 3673–3682.
Rawlings, N. D., Waller, M., Barrett, A. J., & Bateman, A. (2013). MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Research, 42(D1), D503–D509.
Machida, M. (2002). Progress of Aspergillus oryzae genomics. Advances in Applied Microbiology, 51, 81–106.
Matsushita-Morita, M., Tada, S., Suzuki, S., Hattori, R., Marui, J., Furukawa, I., et al. (2011). Overexpression and characterization of an extracellular leucine aminopeptidase from Aspergillus oryzae. Current Microbiology, 62, 557–564.
Watanabe, J., Tanaka, H., Akagawa, T., Mogi, Y., & Yamazaki, T. (2007). Characterization of Aspergillus oryzae Aspartyl Aminopeptidase Expressed in Escherichia coli. Bioscience, Biotechnology, and Biochemistry, 71, 2557–2560.
Kusumoto, K. I., Matsushita-Morita, M., Furukawa, I., Suzuki, S., Yamagata, Y., Koide, Y., et al. (2008). Efficient production and partial characterization of aspartyl aminopeptidase from Aspergillus oryzae. Journal of Applied Microbiology, 105, 1711–1719.
Matsushita-Morita, M., Furukawa, I., Suzuki, S., Yamagata, Y., Koide, Y., Ishida, H., et al. (2010). Characterization of recombinant prolyl aminopeptidase from Aspergillus oryzae. Journal of Applied Microbiology, 109, 156–165.
Marui, J., Matsushita-Morita, M., Tada, S., Hattori, R., Suzuki, S., Amano, H., et al. (2012). Comparison of expression and enzymatic properties of Aspergillus oryzae lysine aminopeptidases ApsA and ApsB. World Journal of Microbiology and Biotechnology, 28, 2643–2650.
Marui, J., Matsushita-Morita, M., Tada, S., Hattori, R., Suzuki, S., Amano, H., et al. (2012). Enzymatic properties of the glycine d-alanine aminopeptidase of Aspergillus oryzae and its activity profiles in liquid-cultured mycelia and solid-state rice culture (rice koji). Applied Microbiology and Biotechnology, 93, 655–669.
Matsushita-Morita, M., Nakagawa, H., Tada, S., Marui, J., Hattori, R., Suzuki, S., et al. (2013). Characterization of a (D)-stereoselective aminopeptidase (DamA) exhibiting aminolytic activity and halophilicity from Aspergillus oryzae. Applied Biochemistry and Biotechnology, 171, 145–164.
Machida, M., Asai, K., Sano, M., Tanaka, T., Kumagai, T., Terai, G., et al. (2005). Genome sequencing and analysis of Aspergillus oryzae. Nature, 438, 1157–1161.
Atlan, D., Gilbert, C., Blanc, B., & Portalier, R. (1994). Cloning, sequencing and characterization of the pepIP gene encoding a proline iminopeptidase from Lactobacillus delbrueckii subsp. bulgaricus CNRZ 397. Microbiology (Reading, England), 140, 527–535.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Yoshimoto, T. (2007). Biochemistry and structural biology of microbial enzymes and their medical applications. Journal of the Pharmaceutical Society of Japan, 127, 1035–1045.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Lobley, A., Sadowski, M. I., & Jones, D. T. (2009). pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics (Oxford, England), 25, 1761–1767.
Basten, D. E., Moers, A. P., Ooyen, A. J., & Schaap, P. J. (2005). Characterisation of Aspergillus niger prolyl aminopeptidase. Molecular Genetics and Genomics : MGG, 272, 673–679.
Mitta, M., Miyagi, M., Kato, I., & Tsunasawa, S. (1998). Identification of the catalytic triad residues of porcine liver acylamino acid-releasing enzyme. Journal of Biochemistry, 123, 924–931.
Sreerama, N., Venyaminov, S. Y., & Woody, R. W. (1999). Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy. Protein Science, 8, 370–380.
Medrano, F., Alonso, J., Garcia, J., Romero, A., Bode, W., & Gomis-Ruth, F. (1998). Structure of proline iminopeptidase from Xanthomonas campestris pv. citri: a prototype for the prolyl oligopeptidase family. The EMBO Journal, 17, 1–9.
Yoshimoto, T., Kabashima, T., Uchikawa, K., Inoue, T., Tanaka, N., Nakamura, K. T., et al. (1999). Crystal structure of prolyl aminopeptidase from Serratia marcescens. Journal of Biochemistry, 126, 559–565.
Uraji, M., Arima, J., Uesugi, Y., Iwabuchi, M., & Hatanaka, T. (2007). Effect of salt on the activity of Streptomyces prolyl aminopeptidase. Biochimica et Biophysica Acta, 1774, 1462–1469.
Hiwatashi, K., Hori, K., Takahashi, K., Kagaya, A., Inoue, S., Sugiyama, T., et al. (2004). Purification and characterization of a novel prolyl aminopeptidase from Maitake (Grifola frondosa). Bioscience, Biotechnology, and Biochemistry, 68, 1395–1397.
Kitazono, A., Kitano, A., Kabashima, T., Ito, K., & Yoshimoto, T. (1996). Prolyl aminopeptidase is also present in Enterobacteriaceae: cloning and sequencing of the Hafnia alvei enzyme-gene and characterization of the expressed enzyme. Journal of Biochemistry, 119, 468–474.
Li, N., Wu, J. M., Zhang, L. F., Zhang, Y. Z., & Feng, H. (2010). Characterization of a unique proline iminopeptidase from white-rot basidiomycetes Phanerochaete chrysosporium. Biochimie, 92, 779–788.
Acknowledgments
This work was supported by the National High Technology Research and Development Program of China (863 Program, 2011AA100905) and the Program for New Century Excellent Talents in University (NCET-12-0878).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ding, GW., Zhou, ND. & Tian, YP. Over-Expression of a Proline Specific Aminopeptidase from Aspergillus oryzae JN-412 and Its Application in Collagen Degradation. Appl Biochem Biotechnol 173, 1765–1777 (2014). https://doi.org/10.1007/s12010-014-0963-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-0963-6