
Abstract
The gdaA gene encoding S12 family glycine–d-alanine aminopeptidase (GdaA) was found in the industrial fungus Aspergillus oryzae. GdaA shares 43% amino acid sequence identity with the d-aminopeptidase of the Gram-negative bacterium Ochrobactrum anthropi. GdaA purified from an A. oryzae gdaA-overexpressing strain exhibited high d-stereospecificity and efficiently released N-terminal glycine and d-alanine of substrates in a highly specific manner. The optimum pH and temperature were 8 to 9 and 40°C, respectively. This enzyme was stable under alkaline conditions at pH 8 to 11 and relatively resistant to acidic conditions until pH 5.0. The chelating reagent EDTA, serine protease inhibitors such as AEBSF, benzamidine, TPCK, and TLCK, and the thiol enzyme inhibitor PCMB inhibited the enzyme. The aminopeptidase inhibitor bestatin did not affect the activity. GdaA was largely responsible for intracellular glycine and d-alanine aminopeptidase activities in A. oryzae during stationary-phase growth in liquid media. In addition, the activity increased in response to the depletion of nitrogen or carbon sources in the growth media, although the GdaA-independent glycine aminopeptidase activity highly increased simultaneously. Aminopeptidases of A. oryzae attract attention because the enzymatic release of a variety of amino acids and peptides is important for the enhancement of the palatability of fermented foods. GdaA activity was found in extracts of a solid-state rice culture of A. oryzae (rice koji), which is widely used as a starter culture for Japanese traditional fermented foods, and was largely responsible for the glycine and d-alanine aminopeptidase activity detected at a pH range of 6 to 9.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The filamentous fungus Aspergillus oryzae produces a variety of enzymes such as amylases and proteases. This fungus has been domesticated and used for the production of Japanese traditional fermented foods and industrial enzymes. A distinct feature of the industrial use of A. oryzae is solid-state cultivation using rice, soybean, wheat, and wheat bran as the culture media (Machida et al. 2008). For example, the solid-sate rice culture, known as “rice koji” is widely used for the starter culture in rice wine (sake), sweet cooking rice wine (mirin), and soybean paste (miso) fermentation. Moreover, it is used as an ingredient of some pickled foods in Japan.
Since the genome sequence became available (Machida et al. 2005), genomics-based research on A. oryzae for academic and industrial purposes has greatly accelerated (Machida et al. 2008). Genomic information has also been utilized for the improvement of heterologous protein production of A. oryzae by disrupting endogenous protease genes (Yoon et al. 2011). In addition to the applications in the food and enzyme industry, a genomics-based approach using A. oryzae has been applied to construct a reporter system for exploring antifungal compounds and target genes (Marui et al. 2010).
A. oryzae proteolytic enzymes are thought to play important roles in the digestion of proteins to a variety of peptides and amino acids that enhance the palatability of fermented foods. The availability of an annotated genome sequence for A. oryzae has enabled comprehensive and reverse genetic study of the proteolytic enzymes. In the A. oryzae genome, 134 peptidase genes consisting of 69 exopeptidases and 65 endopeptidases were predicted. This number is strikingly larger than those for other fungi, and the genes represent roughly 1% of the total genes in the A. oryzae genome (Kobayashi et al. 2007), although only a limited number of peptidases have been characterized in detail. Exopeptidases such as aminopeptidases and carboxypeptidase, which hydrolytically release amino acids from the amino and carboxyl termini of short peptides, respectively, should have a direct role in liberating free amino acids during the fermentation process. These enzymes have been extensively studied in A. oryzae, particularly since the genome sequence became available. In the past 3 years, three aminopeptidases (Kusumoto et al. 2008; Matsushita-Morita et al. 2010, 2011; Watanabe et al. 2007), one cysteinyl dipeptidase with leucine aminopeptidase activity (Hattori et al. 2011), and five carboxypeptidases (Morita et al. 2009, 2010, 2011) of the fungus were characterized at the genetic level.
Aminopeptidases are widely distributed in bacteria, fungi, plants, and animals (Gonzales and Robert-Baudouy 1996; Taylor 1993). These enzymes physiologically function in the degradation of intracellular or extracellular peptides to obtain free amino acids as nutrients or for new protein syntheses (Gonzales and Robert-Baudouy 1996). In addition, many aminopeptidases are involved in protein maturation and stability and degradation of peptide hormones (Taylor 1993). In the A. oryzae genome, 19 aminopeptidase genes were predicted (Kobayashi et al. 2007), while 4 extracellular leucine aminopeptidases have been purified (Nakadai et al. 1973a, b, c; Nakadai and Nasuno 1977). To date, two intracellular aminopeptidase genes encoding the M18 family aspartic aminopeptidase and the S33 family prolyl aminopeptidase that specifically release N terminus acidic amino acids and proline, respectively (Kusumoto et al. 2008; Matsushita-Morita et al. 2010; Watanabe et al. 2007), and two extracellular aminopeptidase genes encoding the family 28A non-specific aminopeptidase (Blinkovsky et al. 2000) and the family 28E leucine aminopeptidase (Matsushita-Morita et al. 2011) have been cloned from A. oryzae; the encoded enzymes were characterized biochemically. Involvement of these aminopeptidases in fermented food production has not been clearly demonstrated at the molecular level.
The hydrolytic activity of aminopeptidases has been studied mainly on release of amino acids in the l-form, which are the predominant constituents of proteins and polypeptides. However, there exists an aminopeptidase that is specific toward the N-terminal d-amino acids. The d-stereospecific aminopeptidase (DAP) has been purified, and the dap gene has been isolated from the Gram-negative bacterium Ochrobactrum anthropi (Asano et al. 1989a, b). DAP releases N-terminal d-alanine from substrates in a highly specific manner (Asano et al. 1989b). In addition, it was highly active toward methyl-esterified or amidated glycine (Asano et al. 1989b), which is unique among the proteinogenic amino acids in that it does not contain an asymmetric carbon.
To date, little is known about the d-stereospecific aminopeptidase activity in eukaryotic organisms. In the present study, we identified the dap ortholog from the A. oryzae genome and designated the gene as glycine d-alanine aminopeptidase A (gdaA) based on the strict substrate specificity of the purified GdaA. To understand the physiological role, time-dependent changes in endogenous intracellular GdaA activity were specifically monitored using a gdaA disruptant and control strain. The aminopeptidase activity that liberates glycine as well as d-alanine has attracted attention from the standpoint of Japanese food bioindustries because glycine is known to enhance preferable tastes such as sweetness and umami, which is the fifth taste sensation (Kawai et al. 2002; Wada et al. 2001). In this study, we demonstrated GdaA activity existed in solid-state rice cultures of A. oryzae.
Materials and methods
Strains, media, and culture conditions
The A. oryzae ΔligD ΔpyrG strain, which is derived from the A. oryzae RIB40 strain, was used as the host for generating the gdaA disruptant (ΔgdaA), overexpressing (OE-gdaA), and control strains. The RIB40 strain was the DNA donor for genome sequencing analysis (Machida et al. 2005). Genomic DNA extracted from A. oryzae RIB40 and Aspergillus nidulans A89 were used as PCR templates. Minimal (0.3% NaNO3, 0.2% KCl, 0.1% KH2PO4, 0.05% MgSO4, 2% glucose, 1.0 × 10−4% FeSO4 • 7H2O, 1.0 × 10−5% Na2B4O7 • 10H2O, 4.0 × 10−5% CuSO4 • 5H2O, 1.5 × 10−5% MnSO4 • 4H2O, 5.0 × 10−6% (NH4)6Mo7O24 • 4H2O, 8.8 × 10−4% ZnSO4 • 7H2O, pH 6.5) and complete media (2% malt extract, 1% peptone, 2% glucose) were used for fungal growth. Uridine (5 mM) and uracil (10 mM) were added if required, and 1.5% agar was added to solidify the media for plate culture. The solidified minimal medium was used for conidiation. The conidia for the liquid and plate cultures were suspended in a solution consisting of 0.01% Tween 80 and 0.09% NaCl. The conidial suspension for solid-state rice culture was prepared with sterile water. For the liquid culture, 108 conidia were inoculated into 100 ml of liquid minimal medium prepared in a 300-ml Erlenmeyer flask without a baffle, followed by rotation culture at 30°C. For the plate culture, 105 conidia in a 5-μl spot were inoculated onto the solidified media, followed by incubation at 30°C. For solid-state rice culture, 2 ml of conidial suspension (2 × 106/ml) was mixed with 4 g of autoclaved α-preprocessed rice in a 100-ml Erlenmeyer flask, followed by incubation at 30°C and 90% humidity. Escherichia coli DH5α competent cells (Toyobo, Osaka, Japan) were used for the construction of the gdaA-overexpressing plasmid. LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl) was used to grow the bacterium. Bacillus stearothermophilus (NBRC100862) was used as a test bacterium for the in vitro penicillin plate assay. Bacto tryptic soy broth (Wako, Osaka, Japan) was used for the assay.
Disruption of the gdaA gene
To create the A. oryzae gdaA gene disruptant (ΔgdaA), the entire open reading frame of the gdaA gene in A. oryzae ΔligD ΔpyrG was replaced by the A. nidulans pyrG gene by homologous recombination. The gene disruption construct was created by PCR using the PrimeSTAR HS DNA polymerase (Takara, Shiga, Japan). Sequences of PCR primers are presented in the Supplementary Table S1. The A. nidulans pyrG gene was PCR amplified using a pair of primers, An_pryG-cF and An_pyrG-cR. Approximately 1 kb of the 5′ and 3′ flanking regions of the gdaA gene were PCR amplified from A. oryzae RIB40 genomic DNA using 0375-5F/0375-5R and 0375-3F/0375-3R primer pairs, respectively. A mixture of these PCR products was then used as a template for PCR using a primer pair, 0375-5F and 0375-3R, to fuse the 5′ and 3′ regions of the gdaA gene at each end of the A. nidulans pyrG gene. The resulting PCR product was introduced into the host strain by fungal transformation, as described previously (Matsushita-Morita et al. 2011). Disruption of the gdaA gene in the uridine prototrophic transformants was confirmed by PCR with 0375-5Rc/0375-3Fc and 0375-iF/0375-iR primer pairs using genomic DNA extracted from the transformants as templates. The resulting transformant was used as the A. oryzae gdaA disruptant (ΔgdaA) in this study. The A. oryzae pyrG gene was PCR amplified with the Ao_pyrG-F and Ao_pyrG-R primer pair and was introduced into the host strain to generate the control strain.
Overexpression of the gdaA gene
The gdaA gene was cloned into the gene overexpression plasmid pPTF5QC2 for overexpression in A. oryzae under the control of the A. oryzae TEF1 gene promoter, which has strong gene expression activity (Kitamoto et al. 1998). The plasmid was constructed as follows. The A. nidulans pyrG gene, PCR amplified with the primer pair An_pyrG-F and An_pyrG-R, was cloned into the ClaI–EcoRI site of the pBSIIKS(+), and the TEF1 gene promoter, PCR amplified with the primer pair tef1p-F and tef1p-R, was cloned into the EcoRI–PstI site to generate the plasmid pPTF5. The BamHI site originally located in the pyrG gene cloned in the pPTF5 was deleted using a QuikChange Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA) with a pair of primers, A2118C-s and A2118C-as. The resulting plasmid was designated as the gene-overexpressing plasmid pPTF5QC2. The gdaA gene, PCR amplified with the primer pair tef1p-0375-if-F and 0375-if-BamHI-R was cloned into the SmaI–BamHI site of the pPTF5QC2 plasmid using an In-Fusion Advantage PCR Cloning Kit with Cloning Enhancer (Takara). The resulting plasmid pPTF5QC2-0375 was introduced into the host strain by fungal transformation. Integration of the TEF1 gene promoter::gdaA fusion gene in the uridine prototrophic transformant was confirmed by PCR with a pair of primers, tef1p-F and 0375-if-BamH I-R.
Purification of GdaA
GdaA was purified 20-fold and obtained a yield of 3.9% from a cell extract of the OE-gdaA strain grown in the liquid complete medium at 30°C for 24 h. The mycelia harvested by filtration were washed with sterile purified water and frozen rapidly in liquid nitrogen. Approximately 20 g of frozen mycelia was rapidly ground to fine powder using Auto-Mill (Tokken. Inc., Chiba, Japan) and suspended in 200 ml of 20 mM Tris–HCl pH 7.0, which was used throughout the purification. The suspension was placed on ice for 30 min, followed by centrifugation at 20,000×g for 15 min at 4°C. The supernatant was used as the cell extract for purification of the GdaA. The purification step is summarized in Supplementary Table S2. AKTA prime (GE Healthcare, Buckinghamshire, UK) was used for column chromatography. All procedures were performed at temperatures lower than 4°C. The enzyme activity of GdaA was traced by measuring the hydrolysis of d-Ala-p-nitroanilide (pNA) in 100 mM potassium phosphate buffer at pH 8.0. The cell extract was mixed with ammonium sulfate to 30% saturation, followed by centrifugation at 20,000×g for 15 min. The supernatant, filtered using a DISMIC-25CS membrane (0.45 μm; ADVANTEC, Tokyo, Japan), was applied to a HiPrep Butyl FF 16/10 column (GE Healthcare) equilibrated with buffer containing ammonium sulfate to 30% saturation and eluted with a linear gradient of ammonium sulfate (30–0% saturation). The active fractions were combined and dialyzed against a buffer containing ammonium sulfate to 30% saturation. The enzyme solution was applied to a HiTrap Butyl HP column (GE Healthcare) equilibrated with the buffer containing ammonium sulfate to 30% saturation and eluted with a linear gradient of ammonium sulfate (30–0% saturation). The active fractions were combined and dialyzed against a buffer without ammonium sulfate. The enzyme solution was applied to a Mono Q 5/50 column (GE Healthcare) equilibrated with the same buffer and eluted with a gradient of 0–0.4 M NaCl. The active fractions were combined and dialyzed against a buffer without NaCl. The enzyme solution was applied to Superdex 200 10/300 GL (GE Healthcare) and eluted with same buffer. The active fractions eluted in a single peak were collected and used for analyses in the present study. The molecular mass of the purified enzyme was determined using Superdex 200 10/300 GL (GE Healthcare) calibrated using a Gel Filtration Calibration Kit (GE Healthcare) with a 50-mM sodium phosphate buffer at pH 7.5.
Quantitative RT-PCR
Fungal RNAs were extracted using RNAiso (Takara). The extracted RNAs were treated with DNase using an RNeasy Plant Mini Kit with RNase-free DNase (Qiagen, Hilden, Germany). The total RNAs (200 ng) were used as templates for cDNA synthesis using a high capacity cDNA reverse transcription kit (Life Technologies, Foster City, CA). The 10-fold diluted reaction mixtures (5 μl) were applied to quantitative PCR analysis using an Mx300P Real-Time QPCR System (Agilent Technologies Inc., Santa Clara, CA, USA) with Brilliant III Ultra-fast SYBR Green QPCR Master Mix (Agilent Technologies Inc.). The primer pair qrt0375-F4 and qrt0375-4 was used to amplify the gdaA genes. As an internal control, the primer pair qrt0495-F and qrt0495-R was used to amplify a part of the histone H3 gene (AO090012000495). Primer sequences are presented in Supplementary Table S1.
Aminopeptidase assay
For the aminopeptidase assay, amino acids coupled with p-nitroanilide (pNA), i.e., l-Leu-, l-Ala-, l-Lys-, and l-Glu-pNA (Peptide Institute, Osaka, Japan), l-Pro-, l-Met-, l-Gly-, l-Arg-, and l-Val-pNA (Sigma-Aldrich Japan, Tokyo, Japan), l-Asp-, l-Ile-, l-His-, d-Ala-, d-Leu-, d-Phe-, and β-Ala-pNA (Bachem, Bubendorf, Switzerland) or synthetic peptides, i.e., Gly-l-Ala, l-Ala-Gly, l-Cys-Gly, Gly-l-Cys-Gly, Gly-Gly-Gly, and l-Cys-Gly-Gly (Bachem), Gly-Gly (Peptide Institute), d-Ala-d-Ala, d-Ala-l-Ala, l-Ala-d-Ala, d-Ala-Gly, and d-Ala-Gly-Gly (Bachem) were used as substrates.
In the standard enzyme assay, a mixture containing 1 mM substrate in the reaction buffer was preincubated at 30°C for 10 min, followed by enzyme reaction at the same temperature. The reaction was terminated by adding a quarter volume of 40% (v/v) acetic acid. For the quantification of liberated d-Ala from peptide substrates, the hydrolytic reactions using GdaA were terminated by heat inactivation at 95°C for 5 min.
Hydrolysis of the amino acid–pNA substrate was quantified by measuring the absorbance at 415 nm, as described previously (Matsushita-Morita et al. 2011). The enzyme activity unit (U) was defined as the amount of enzyme that liberated 1 μmol of pNA per minute. Glycine liberated from peptide substrates by purified GdaA (0.01 mg/ml in 20 mM Tris–HCl pH 9.0) was measured using an Agilent 1100 HPLC system (Agilent Technology Inc.). Liberated d-alanine from peptide substrates was measured by the colorimetric method (Nagata et al. 1985) using d-amino acid oxidase (Sigma).
Biochemical characterization of GdaA
Amino acid–pNA substrates were used in the standard enzyme assay to determine the substrate specificity of the aminopeptidase activity of purified GdaA. The K m and K cat values of the enzyme reacted for 10 min with d-Ala-, Gly-, or l-Ala-pNA under the standard assay condition using 20 mM Tris–HCl (pH 9.0) were calculated from Lineweaver–Burk plots.
The pH–activity profile of the purified GdaA was analyzed using 10 mM Britton–Robinson buffer (10 mM H3BO3, 10 mM KH2PO4, 10 mM CH3COOH) at pH values ranging from 3.5 to 12 and 20 mM Tris–HCl at pH values ranging from 7 to 9.5 with Gly-pNA. To test the pH stability, purified GdaA was mixed with 10 mM Britton–Robinson buffer and incubated on ice for 1 h. Aliquots of the mixtures were subjected to standard enzyme assay with Gly-pNA. The temperature–activity profile was analyzed with Gly-pNA in 20 mM Tris–HCl pH 9.0 at temperatures ranging from 0°C to 80°C. To test thermal stability, the purified enzyme was preincubated at temperatures ranging from 0°C to 80°C for 1 h, followed by the standard enzyme assay with Gly-pNA.
The effects of protease inhibitors (i.e., EDTA, AEBSF, benzamidine, TPCK, TLCK, PCBM, leupeptin, pepstatin A, bestatin, and DTT), chloride salts of divalent metal ions (i.e., ZnCl2, CdCl2, CuCl2, CuCl2, NiCl2, CoCl2, CaCl2, MnCl2, FeCl2), ampicillin anhydrous (Wako), ampicillin sodium salt (Wako), penicillin G sodium salt (Sigma-Aldrich Japan), and penicillin G potassium salt (Wako) were measured. The purified enzyme was preincubated with each compound in 20 mM Tris–HCl (pH 9.0) at 30°C for 30 min, followed by the standard enzyme assay with Gly-pNA.
Preparation of culture extracts for the time-course analyses of Gly- and d-Ala aminopeptidase activities
For time-course analysis of the intracellular glycine or d-alanine aminopeptidase activities of liquid-cultured mycelia, the A. oryzae ΔgdaA and control strains grown in liquid complete media at 30°C for 24, 48, or 72 h were harvested and frozen in liquid nitrogen. To observe the time-dependent changes of the enzyme activities of ΔgdaA and the control strain under nitrogen- or carbon-starved conditions, the two strains precultured in the liquid minimal medium for 24 h were harvested and washed with minimal medium without sodium nitrate or glucose. Subsequently, the mycelia were transferred to media that were similar in composition to those used for the washing. After additional growth for 3, 6, 9, 12, and 24 h, the mycelia were harvested and frozen in liquid nitrogen. Approximately 0.1 g of frozen mycelia samples was rapidly ground to fine powder using an Auto-Mill (Tokken Inc.) and suspended in 1 ml of 20 mM Tris–HCl (pH 7.5) on ice for 10 min. The suspensions were centrifuged at 20,000×g for 15 min at 4°C, and the supernatants were assayed for glycine and d-alanine aminopeptidase activities.
To prepare extracts of the solid-state rice cultures of A. oryzae strains, the cultures grown for 36 and 48 h were suspended in 12 ml of sterile purified water and stirred for 60 min at 4°C. The suspensions were centrifuged at 20,000×g for 15 min at 4°C, and the supernatants were assayed for the glycine and d-alanine aminopeptidase activities.
Computational sequence analysis
A homology search was performed using the BLASTp search of the NCBI database (http://www.ncbi.nlm.nih.gov/) and MEROPS proteolytic enzyme database (http://merops.sanger.ac.uk/index.shtml). The search for functional domains was performed using the sequence search of the Pfam database (http://pfam.sanger.ac.uk/). Searches for signal peptides and organellar targeting sequences were performed using Signal P 3.0 Server (http://www.cbs.dtu.dk/services/SignalP/) and PSORT II Prediction (http://psort.hgc.jp/form2.html), respectively.
In vitro penicillin plate assay
Penicillin G potassium salt (1 mM) and purified GdaA (0.01 or 0.1 mg/ml) were mixed in 20 mM Tris–HCl pH 9.0. After incubation at 30°C for 120 min, 5 μl of the 500-fold diluted reaction mixtures was spotted in 20 ml of solidified tryptic soy broth media containing Bacillus stearothsemophilus var. calidolactis (NBRC100862) as test bacteria at a final optical density of 0.1 in a 9-cm diameter petri dish, followed by incubation at 55°C for 16 h. Diameters of inhibition halos (mm) were measured to evaluate the inhibitory effect of GdaA to antibacterial activity of penicillin. Penicillinase from Bacillus cereus (Sigma-Aldrich Japan, Tokyo, Japan) was used (0.5 units) for the positive control of penicillin degradation (data not shown).
Other methods
Fungal DNA was extracted using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). 5′ RACE was performed as described previously (Matsushita-Morita et al. 2011) using a primer MK36RACE-R (Supplementary Table S1). Protein concentration was determined colorimetrically with a Protein assay kit (Bio-Rad Laboratory, Hercules, CA) using bovine serum albumin as the standard. SDS-PAGE was performed using MINI PROTEAN TGX gel Any kD (Bio-Rad Laboratory) following the manufacturer’s recommendation. N-terminal amino acid sequence analysis was performed by APRO Science Co. Ltd. (Tokushima, Japan).
Results
Features of the A. oryzae gdaA gene
The gene AO090010000375 annotated in the A. oryzae genome database (http://www.bio.nite.go.jp/dogan/project/view/AO) was identified as a homolog of the dap gene (genbank accession number M84523, MEROPS accession number MER000457) encoding the d-stereospecific aminopeptidase of the Gram-negative bacterium O. anthropi (Asano et al. 1989b). Based on the substrate specificity analysis as mentioned later, the AO090010000375 gene was referred to as glycine d-alanine aminopeptidase (gdaA). The open reading frame of the gdaA gene determined in the genome database was predicted to comprise 1,485 bp without an intron and to encode 494 amino acids. However, the amplicon obtained by 5′ RACE in the present study (genbank accession number AB641129) started 262 bp upstream from the deduced translation start site, and this 262-bp region contained a 58 bp intron, which interrupted most upstream in-frame ATG codon located 181 bp upstream of the in silico-predicted translation start site of the gdaA gene (Fig. 1a). Therefore, GdaA was concluded to contain an additional 41 amino acids at the N terminus of the deduced sequence in the genome database and comprise 535 amino acids with a molecular mass of 59.3 kDa. According to the family system of the MEROPS proteolytic enzyme database, GdaA belong to the S12 family. The corrected amino acid sequence (genbank accession number BR000934) shared a 43% identity with O. anthropi DAP with an E value of 3.0e−114. No other predicted proteins in the A. oryzae genome database showed significant homology to DAP. Bacterial homologs of GdaA (E value cut-off of 1.0e−50) that shared similar extents of sequence identities were found in other Gram-negative bacteria such as Brucella species, Rhodobacter sphaeroides, Gluconobacter oxydans, Pelagibaca bermudensis, and Citreicella sp. (data not shown). In eukaryotes, its homologs were found only in filamentous fungi such as Aspergillus flavus, Aspergillus niger, Nectria haematococca, Penicillium chrysogenum, and Gibberella zeae that shared 99%, 64%, 50%, 51%, and 47% amino acid sequence identities, respectively (data not shown).

Schematic representations of the gdaA gene and its encoded protein. a Structure of the gdaA gene. The exon and intron of the gdaA gene are represented as filled and open boxes, respectively. The transcription and translation start sites deduced in the present study are indicated by −81 and +1, respectively. The in silico-predicted translation start site in A. oryzae genome database is indicated at position +182 by an arrowhead. b Deduced functional domains of GdaA. The serine β-lactamase fold and the d-aminopeptidase domains B and C predicted by a sequence search of the Pfam database are indicated by dotted, hatched, and crosshatched boxes, respectively, referring to the N-terminal Met as position 1. The Ser-Xaa-Xaa-Lys motif at positions 58 to 61 in the serine β-lactamase fold is represented
High similarity between the primary structures of O. anthropi DAP and β-lactamases and the importance of the Ser-Xaa-Xaa-Lys motif in the serine β-lactamase fold for catalytic activity have been reported (Asano et al. 1992). Crystal structure analysis has shown that the two antiparallel eight-stranded β-barrels called d-aminopeptidase domains B and C (DAP_B and DAP_C) were connected to the serine β-lactamase fold in a row (Bompard-Gilles et al. 2000). In A. oryzae GdaA, the serine β-lactamase fold and the d-aminopeptidase domains B and C were predicted by sequence search of the Pfam database to be located at positions 3 to 341, 349 to 435, and 439 to 535 with bit scores of 119.1, 31.1, and 159.0 and E values of 1.6e−34, 1.1e−7, and 1.9e−47, respectively (Fig. 1b). The Ser-Xaa-Xaa-Lys motif was located at position 58 to 61 in the sequence (Fig. 1b). The secretion and organellar targeting signals were not identified by the PSORT II program. These facts suggested that GdaA was a cytosolic enzyme whose property resembled that of DAP.
Disruption and overexpression of the gdaA gene
The A. oryzae gdaA disruptant (ΔgdaA) and overexpressing strains (OE-gdaA) were created and used in this study to investigate the enzymatic properties and physiological role of GdaA. No significant differences were observed in the vegetative growth and conidiation on minimal and complete media in the ΔgdaA, OE-gdaA, and control strains (data not shown). We first selected d-Ala- and Gly-pNA as substrates for the aminopeptidase assay of those cell extracts because O. anthropi DAP exhibited highly selective reactivity toward d-alanine amide, d-alanine-containing peptides, and glycine methyl esters (Asano et al. 1989b). The endogenous glycine and d-alanine aminopeptidase activities in the control strain grown in the liquid complete medium were clearly detected at 24 h after inoculation and increased in a time-dependent manner (Fig. 2), and the activities at 72 h were 11.1 and 6.7 mU/mg protein, respectively. On the other hand, the activities in ΔgdaA remained low at all the time points examined (Fig. 2). These results strongly suggested that GdaA was expressed in A. oryzae in this culture condition and functioned as a major enzyme responsible for intracellular aminopeptidase activity that liberates d-alanine as well as glycine.

Time-dependent change of glycine and d-alanine aminopeptidase activities in the A. oryzae gdaA disruptant (ΔgdaA) and control strain grown in the liquid complete medium. The glycine and d-alanine aminopeptidase activities (mU/mg) in the cell extracts of the A. oryzae control strain (open bar) and the gdaA disruptant (ΔgdaA: filled bar) are presented; Gly- and d-Ala-pNA are used as substrates, respectively. The cell extracts prepared from the strains grown in the liquid complete medium for the time indicated were subjected to the aminopeptidase assay in 100 mM potassium phosphate at pH 8.0. The values are means of three replicates with standard deviations
The transcript level measured by qRT-PCR in the OE-gdaA strain grown in the liquid complete medium for 24 h at 30°C was approximately 5,000-fold higher than that of the control strain, while it was not detected in the ΔgdaA strain (data not shown). In accordance with the significant increase of the gdaA transcript level in the OE-gdaA strain, the glycine and d-alanine aminopeptidase activities assayed in 100 mM potassium phosphate at pH 8.0 increased to 7,445 and 5,490 mU/mg cell protein, respectively (data not shown). The level appeared to be sufficient for the purification of the enzyme.
Biochemical characterization of purified GdaA
For the characterization of the enzymatic properties, GdaA was purified from a cell extract of the OE-gdaA grown in the liquid complete medium at 30°C for 24 h. Purified GdaA was detected as a single peak by gel filtration chromatography, and the molecular mass was calculated to be 126.3 kDa (data not shown). Because the gdaA gene was deduced to encode a protein with a molecular mass of 59.3 kDa, GdaA was thought to exist as a homodimer in A. oryzae. SDS-PAGE of the active fraction of each purification step is presented in Fig. 3. Although purified GdaA was detected as a single peak by gel filtration chromatography, it was separated by SDS-PAGE into three bands. The sequences of the upper and middle bands started from the second amino acid residue from the deduced N-terminal initiator methionine, and the lower band started from amino acid position 347 of GdaA comprised of 535 amino acids (data not shown). These facts indicated that the purified GdaA was partially separated into two pieces comprised of 345 and 189 amino acids, and the molecular masses were 39 and 19 kDa, respectively, which were in reasonable agreement with the mobility observed in the SDS-PAGE (Fig. 3). These two bands did not appear in any purification steps other than the final one, and digestion did not appear to negatively affect the specific activity of GdaA (Supplementary Table S2). Hence, purified GdaA might be digested during the final step of purification without affecting the activity and dimerization under nondenaturing condition.

SDS-PAGE of the purified protein. The protein solution (5 μl) of each purification step was subjected to SDS-PAGE using MINI PROTEAN Any kD (Bio-Rad Laboratory) following the manufacturer’s recommendation. Lanes 1 cell extract, 2 ammonium sulfate, 3 HiPrep Butyl FF 16/10, 4 HiTrap Butyl HP, 5 Mono Q 5/50, 6 Superdex 200 10/300 GL
The substrate specificity of purified GdaA was analyzed using a number of amino acid–pNA substrates (Table 1). GdaA efficiently hydrolyzed Gly- and d-Ala-pNA and was weakly active to l-Ala-pNA. The kinetic parameters of GdaA with Gly-, d-Ala-, and l-Ala-pNA are presented in Table 2. The preference of GdaA to Gly-pNA was slightly higher than that to d-Ala-pNA. The K cat/K m value of the enzyme to d-Ala-pNA was 24.7-fold higher than that of l-Ala-pNA, clearly indicating the d-stereospecific aminopeptidase activity of GdaA. Based on these observations, we used Gly-pNA as the substrate for the enzymatic characterization of GdaA presented below.
GdaA was active at pH 5 to 11 and optimum at pH 8 to 9 (Fig. 4a). It was most stable at pH 8.5 and retained more than 80% and approximately 60% residual activity at pH 8 to 11 and 5 to 7, respectively (Fig. 4b). The enzyme was active between 0°C and 60°C and optimum at 40°C (Fig. 4c). It retained 80% residual activity up to 40°C (Fig. 4d).

Effects of pH and temperature on GdaA activity. a pH–activity profile. The specific activities (U/mg) of GdaA at each pH in 20 mM Tris–HCl (filled circle) and 10 mM Britton–Robinson buffer (open circle) are presented. b pH stability. The relative activities of GdaA at each pH are presented referring to the untreated control as 100%. c Temperature–activity profile. The specific activities (U/mg) of GdaA at each temperature are presented. d Thermal stability. The relative activities of GdaA treated at each temperature are presented referring to the untreated control as 100%. Gly-pNA was used as the substrate for the experiments. The values are means of three replicates with standard deviations
The enzyme activity was severely inhibited by the chelating reagent EDTA and the serine protease inhibitor AEBSF at concentrations of 10 mM (Fig. 5a). It was inhibited by 20–80% of the untreated control by 3 other serine protease inhibitors, benzamidine, TPCK, and TLCK, a serine–cysteine protease inhibitor, leupeptine, and a thiol enzyme inhibitor, PCMB (Fig. 5a). Among the possible protease inhibitors tested, the aspartic protease inhibitor pepstatin A, the reducing reagent DTT, and the aminopeptidase inhibitor bestatin did not inhibit the enzyme (Fig. 5a). The effects of divalent metal ions at 0.1 or 1 mM on GdaA activity are presented in Fig. 5b. The enzyme was severely inhibited by Zn2+ and Cd2+ at both concentrations. The activity was decreased to 5% and 20% of the untreated control by 1 mM Cu2+ and Ni2+, respectively, while the inhibitory effects of both cations at 0.1 mM remained at the 50% level. Moreover, 50% inhibition was exhibited by Co2+ at 0.1 and 1 mM. More than 80% of the residual activities were detected by treatment with Ca2+, Mn2+, Mg2+, and Fe2+ at 0.1 mM, although these cations inhibited enzyme activity by 20–80% of the untreated control at 1 mM.

Effects of protease inhibitors and divalent metal ions on GdaA activity. The relative activities of GdaA treated with a protease inhibitors and b divalent metal ions at the concentrations indicated are presented referring to the untreated control as 100%. Gly-pNA was used as the substrate for the experiments. The values are means of three replicates with standard deviations
Because the aminopeptidase activity of O. anthropi DAP was inhibited by β-lactam compounds (Asano et al. 1992), their effect on the aminopeptidase activity of GdaA was examined using ampicillin and penicillin G. We used sodium or potassium salts of the compounds as well as ampicillin anhydrous to examine the possible effect of the alkali metal ions associated with β-lactam compounds. GdaA was not inhibited by up to 10 mM ampicillin anhydrous (Fig. 6a), showing that ampicillin did not affect GdaA activity. However, the activity was decreased to approximately 30% and 20% of the untreated control by 10 mM of the sodium salts of ampicillin or penicillin G and sodium chloride, respectively (Fig. 6b). Although the activity was not affected by penicillin G potassium salt or potassium chloride at 1 mM, the activity was decreased to 80% of the untreated control by 10 mM of both compounds (Fig. 6c). These results strongly suggested that the GdaA activity was inhibited not by ampicillin and penicillin G but by the sodium or potassium ions associated with those β-lactam compounds.

Effects of ampicillin and penicillin G on GdaA activity. The relative activities of GdaA treated with a ampicillin anhydrous, b sodium salts of ampicillin (filled bar) and penicillin G (dotted bar) and sodium chloride (hatched bar), and c penicillin G potassium salt (dotted bar) and potassium chloride (open bar) at the concentrations indicated are presented referring to the untreated control as 100%. Gly-pNA was used as the substrate for the experiments. The values are means of three replicates with standard deviations
Liberation of N-terminal glycine and d-alanine by GdaA from the peptide substrate
In addition to synthetic substrates, the aminopeptidase activity of GdaA was tested using peptide substrates, of which glycine or d-alanine was positioned at the N-terminus (Tables 3 and 4). Peptide substrates (1 mM) were treated with purified GdaA (0.01 mg/ml) in 20 mM Tris–HCl (pH 9.0) at 30°C for 15 min. Hydrolytic release of N-terminal glycine and d-alanine was quantified with an amino acid analyzer and colorimetry using d-amino acid oxidase, respectively because d-alanine liberated from the peptide substrates could not be specifically detected by the amino acid analyzer under our experimental conditions. Liberated glycine was detected from Gly-l-Ala, Gly-l-Cys, and Gly-Gly dipeptides and Gly-l-Cys-Gly and Gly-Gly-Gly tripeptides but not from l-Ala-Gly and l-Cys-Gly dipeptides and l-Cys-Gly-Gly tripeptides (Table 3). This indicated that GdaA released only the N-terminal glycine of peptide substrates. The amount of N-terminal glycine released from the Gly-l-Cys-Gly tripeptide was 5.4- and 12.8-fold higher than those from Gly-l-Ala and Gly-l-Cys dipeptides, respectively (Table 3). Considering that the amount of liberated glycine from the Gly-Gly dipeptide is thought to double the volume of the digested substrate and the amount of liberated glycine from Gly-Gly-Gly is from the tripeptide and the resulting Gly-Gly dipeptides, the reactivity of GdaA to those two substrates appeared to be comparable or slightly higher than that to the Gly-Cys-Gly tripeptide (Table 3).
In addition, liberation of the N-terminal d-alanine from the peptide substrates was clearly detected (Table 4), and the amount tended to be higher than that of N-terminal glycine. The liberated d-alanine from l-Ala-d-Ala was under the detection limit in this experimental condition, indicating the strict d-stereospecific aminopeptidase activity of GdaA. The activities to d-Ala-Gly and d-Ala-Gly-Gly were comparable and markedly higher than those to the d-Ala-d-Ala and d-Ala-l-Ala dipeptides (Table 4).
Time-dependent change of intracellular GdaA activity under nutrient-limited conditions
The glycine aminopeptidase activity of the control and ΔgdaA strains precultured in minimal medium for 24 h was 11.6 and 6.4 mU/mg, respectively, while the d-alanine aminopeptidase activity of these strains were 3.1 and 0.3 mU/mg, respectively (Fig. 7). In the nitrogen-starved condition, the glycine aminopeptidase activity of the control and ΔgdaA strains increased significantly from 3 h after transfer, and reached 528.2 and 480.1 mU/mg, respectively, at 24 h (Fig. 7a, left panel). In the same condition, the d-alanine aminopeptidase activity in the control strain increased to the maximum level at 9 h (35.1 mU/mg) and slightly fluctuated until 24 h, while the activity in the ΔgdaA strain increased gradually to 12 mU/mg at 24 h (Fig. 7a, right panel).

Time-dependent change in intracellular GdaA activity in nutrient-limited conditions. The intracellular glycine and d-alanine aminopeptidase activities (mU/mg cell protein) of the A. oryzae control strain (open circle) and gdaA disruptant (ΔgdaA, filled circle) under a nitrogen- and b carbon-starved conditions are presented. Gly-pNA (left panel) and d-Ala-pNA (right panel) were used as substrates. The cell extract prepared from the strains grown under each starved condition for the time indicated was subjected to the aminopeptidase assay in 20 mM Tris–HCl at pH 7.5. The values are means of three replicates with standard deviations
Under the carbon-starved condition, the glycine aminopeptidase activities of both strains increased (Fig. 7b left panel), although the increasing rates were lower than those under the nitrogen-starved condition. The activities of the control and ΔgdaA strains at 24 h after transfer reached 53.2 and 29.7 mU/mg, respectively. An increase in the d-alanine aminopeptidase activity of the control strain was observed at 3 h after transfer (Fig. 7b, right panel). Although the increasing rate between 6 and 24 h was lower than that observed during the first 3 h, the activity increased over time to 20.0 mU/mg. On the other hand, the activity in ΔgdaA remained low throughout all the time points examined (Fig. 7b, right panel).
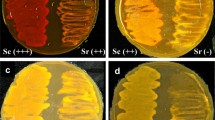
GdaA activity in solid-state rice culture (rice koji)
For the production of fermented food, solid-state rice culture of A. oryzae is mixed with fermentation feedstock, although the levels of agitation vary according to the type of fermented food. In this study, we examined whether GdaA activity could release from the solid-state rice culture by simple agitation. The glycine and d-alanine aminopeptidase activities of the solid-state rice cultures of the A. oryzae control and ΔgdaA strains were compared (Fig. 8). The incubation times of the cultures for the analysis were 36 and 48 h because preparation of the solid-state rice culture in the food industry generally takes no more than 2 days. The glycine and d-alanine aminopeptidase activities of the control strain were clearly detected at both time points under the acidic to alkaline pH conditions examined; the activities increased in a time-dependent manner and were higher under the alkaline condition than under the acidic condition, i.e., in good accordance with the pH–activity profile of GdaA. On the other hand, the d-Ala aminopeptidase activity of ΔgdaA was considerably low or undetectable under the same conditions. Furthermore, glycine aminopeptidase activities of ΔgdaA under each pH condition were significantly lower than those of the control strain, suggesting the importance of GdaA in the glycine aminopeptidase activity of the solid-state rice culture of A. oryzae (rice koji).

GdaA activity in solid-state rice culture (rice koji). The glycine (filled bar) and d-alanine (open bar)-aminopeptidase activities (mU/ml) of the extracts of the solid-state rice culture of the A. oryzae control strain (cont) and gdaA disruptant (Δ) at 36 and 48 h after inoculation are presented. Gly- and d-Ala-pNA were used as substrates in the reaction mixture prepared with 10 mM Britton–Robinson buffer at the pH conditions indicated. The values are means of three replicates with standard deviations
Discussion
In this study, the A. oryzae gdaA gene encoding glycine d-alanine aminopeptidase was identified through a homology search of the A. oryzae genome database. Homologous genes exist only in a limited number of species of Gram-negative bacteria and filamentous fungi. Using a reverse genetics approach, we first demonstrated that glycine and d-alanine aminopeptidase activities derived from GdaA existed in the industrial fungus A. oryzae, as is the case with Gram-negative bacterium O. anthropi, known as an opportunistic pathogen. Similar to O. anthropi DAP, GdaA showed strict substrate specificity and high reactivity for the N-terminal glycine and d-alanine of substrates. While the catalytic activities of GdaA toward the synthetic substrates Gly- and d-Ala-pNA were nearly identical, d-alanine at the N-terminal of peptide substrates was released more efficiently than glycine. Therefore, the catalytic activity of GdaA toward a peptide substrate might vary depending not only on the N-terminal amino acid but also on the variety and number of the subsequent amino acid(s).
It preferably functions under mesophilic conditions, although it hydrolyzes substrates even at low temperatures. The pH of the Aspergillus cytosol was reported to be 7.8 (Hesse et al. 2000). Purified GdaA is active and stable under alkaline conditions and relatively resistant to acidic conditions until pH 5.0; hence, GdaA could function in the A. oryzae cytosol. The chelating reagent EDTA inhibited the aminopeptidase activity of GdaA, indicating that the enzyme requires metal ion(s) to maintain activity, although purified GdaA tended to be negatively affected by the exogenous addition of divalent metal ions in the reaction mixture. Bestatin, which competitively inhibits a broad range of aminopeptidases (Suda et al. 1976; Umezawa et al. 1976), did not affect the aminopeptidase activity of GdaA, suggesting that the catalytic mechanism of GdaA differs from that of the bestatin-sensitive aminopeptidases. In good agreement with the prediction of a serine β-lactamase fold with a Ser-Xaa-Xaa-Lys motif containing the putative active Ser and Lys at positions 58 and 61, respectively, the aminopeptidase activity of GdaA was inhibited by serine protease inhibitors such as AEBSF, benzamidine, TPCK, and TLCK. These results suggest that GdaA possesses a serine type aminopeptidase activity. Moreover, the activity was inhibited by 10 mM PCMB, which is a thiol enzyme inhibitor, while O. anthropi Dap was inhibited by 0.0925 mM PCMB (Asano et al. 1992) The inhibition might be attributed to the mercaptide bond between PCMB and Cys at position 57 because in O. anthropi DAP, a change at Cys60, which is adjacent to the active Ser61 of the Ser-Xaa-Xaa-Lys motif, to Ser and Gly resulted in the production of enzyme that was less sensitive to PCMB (Asano et al. 1992). Unlike O. anthropi DAP, GdaA was not inhibited by the β-lactam compounds such as penicillin G and ampicillin. These differences in sensitivity to protease inhibitor and β-lactam compounds suggest that structures of these enzymes have been changed during the evolution without affecting the substrate specificity. It was previously reported that O. anthropi DAP did not possess β-lactamase activity (Asano et al. 1992). Inhibitory effect of the purified GdaA on antibacterial activity of penicillin was not detected by in vitro penicillin plate assay (Supplementary Table S3). In addition, the enzyme did not hydrolyze a chromogenic β-lactamase substrate such as nitrocefin (data not shown). These results suggested that GdaA did not possess β-lactamase activity.
d-stereospecific aminopeptidase activity is one of the distinct features of GdaA in A.oryzae. It efficiently released N-terminal d-alanine from the peptide substrates. The physiological role of the activity is of considerable interest. Furthermore, it questions whether peptide substrates containing d-alanine at the N terminus exist in nature. d-alanine is known to be an important constituent of the bacterial cell wall. The structural similarity of O. anthropi DAP to carboxypeptidase DD, which is involved in bacterial peptidoglycan biosynthesis, has been reported (Asano et al. 1992). Thus, d-alanine aminopeptidase might be involved in peptidoglycan metabolism. However, this does not appear to be the case for filamentous fungi because eukaryotic cell walls are not composed of peptidoglycan.
So far, virtually nothing is known about the existence of d-alanine or d-alanine-containing peptides in A. oryzae. However, in addition to bacterial peptidoglycan, d-amino acids have been found in both bacterial and eukaryotic peptides (Cava et al. 2011), although they occur less frequently in living organisms compared with proteinogenic l-amino acids in nature. In filamentous fungi, d-amino acids are often found in non-ribosomal peptides. For example, the cyclic peptides cyclosporine and HC-toxin of Tolypocladium niveum and Cochliobolus carbonum, respectively, contain d-alanine in the structure (Cheng and Walton 2000; Hoffmann et al. 1994). While d-amino acids in such non-ribosomal peptides tend to be generated during synthesis by an epimerization domain in the non-ribosomal peptide synthetase, alanine racemases involved in the cyclic non-ribosomal peptide synthesis exist in those fungi (Cheng and Walton 2000; Hoffmann et al. 1994), and the gene was isolated from C. carbonum (Cheng and Walton 2000). The putative homolog of the alanine racemase gene (AO090012000408), which shares 44% amino acid sequence identity with an E value of 1.0e−79, was found in the A. oryzae genome, although the alanine racemase activity of the encoded protein has not been investigated so far. These facts have led to the speculation that d-alanine-containing peptides could exist in A. oryzae. It is important to determine the content of d-alanine and d-alanine-containing peptides in cells.
It is also conceivable that the d-alanine aminopeptidase activity in A. oryzae is the remnant of an ancestral fungal–plant and fungal–bacterial interaction. In addition to bacterial peptidoglycan, d-alanine-containing peptides are known in plants. Indeed, d-Ala-Gly, which was the most preferred peptide substrate tested in the present study, exists in the Japonica rice plant (Manabe et al. 1981; Manabe 1990, 1992; Yamauchi et al. 1979). d-Ala-d-Ala and d-Ala-l-Ala that were hydrolyzed by GdaA were also found in other wild rice plants (Manabe 1990, 1992). These facts imply a close relationship between A. oryzae and rice plants, and it might be one of the reasons why the gdaA gene has been conserved in the fungus.
The gdaA gene does not appear to be essential for the growth of A. oryzae. Although the cDNA of gdaA was obtained by 5′ RACE in the present study, no expressed sequence tag (EST) clone was identified in the EST database of A. oryzae RIB40 in any culture conditions (http://nribf2.nrib.go.jp/EST2/) (Akao et al. 2007). Correspondingly, the relative transcriptional level of gdaA was considerably low in both liquid- and solid-state culture in a recent transcriptome analysis (Wang et al. 2010). However, we clearly detected the aminopeptidase activity of GdaA specifically by enzyme assay using ΔgdaA and a control strain. This should be attributed to the high specific activity of GdaA. The time-course experiments in complete medium indicated that GdaA is the major enzyme in A. oryzae responsible for the intracellular aminopeptidase activity that liberates glycine and d-alanine in the stationary phase growth, although unknown intracellular glycine aminopeptidase(s) simultaneously functioned in the fungus grown in minimal medium for 24 h. Time-dependent increases of intracellular glycine aminopeptidase activity under nitrogen- or carbon-starved conditions imply the importance of aminopeptidases for maintaining intracellular amino acid levels. Proteins and peptides in the cell should be rapidly digested to amino acids, particularly under nitrogen-starved conditions. From the results of the d-Ala-pNA hydrolysis, the specific activities of GdaA 24 h after transfer to the nitrogen- and carbon-starved conditions were calculated to increase to 9.2- and 6.9-fold, respectively. Furthermore, it is noteworthy that GdaA-independent glycine aminopeptidase activity was increased significantly in the nitrogen-starved condition in a time-dependent manner and constituted a majority of the intracellular glycine aminopeptidase activity. On the other hand, GdaA-independent d-alanine aminopeptidase activity was increased, albeit weakly, only in the nitrogen-starved condition. Overall, GdaA and the unknown enzyme(s) in A. oryzae may coordinately function in the digestion of peptides containing glycine in response to the nutritional condition, amino acid availability, and growth phase.
As observed in case of GdaA, aminopeptidases capable of liberating N-terminal glycine tend to possess strict substrate specificity, and the number was limited compared with those with broad substrate specificity (Schomburg and Schomburg 2002). In filamentous fungi, an aminopeptidase specific for glycine was purified from Actinomucor elegans, although the d-alanine aminopeptidase activity was not tested (Ito et al. 2003). Because genetic information is not available, the orthology between the enzyme and GdaA cannot be directly discussed. These enzymes exhibited similar temperature– and pH–activity profiles. However, the enzyme in A. elegans was insensitive to the serine protease inhibitor and highly sensitive to thiol enzyme inhibitors, suggesting that the catalytic mechanisms of the two enzymes are different. Aminopeptidase II of A. oryzae is known to be a non-specific extracellular aminopeptidase that exceptionally liberates various amino acids including glycine from the N terminus of peptides (Blinkovsky et al. 2000). Aminopeptidases exhibiting glycine aminopeptidase activity have attracted attention because the peptide bonds Gly-Ala, and particularly Gly-Gly, are thought to be exceptionally resistant to proteolysis probably due to the flexibility of the structure (Blinkovsky et al. 2000). Glycine is the smallest among the proteinogenic amino acids, and unlike the other amino acids, its side chain consists of a single hydrogen atom. Therefore, a glycine peptide bond is more flexible than those of the other amino acids. GdaA liberated N-terminal glycine of the peptide substrates, suggesting that A. oryzae utilizes the enzyme to overcome this so-called “bottleneck” of proteolysis and eventually leads to the complete degradation of proteins and peptides. This feature should also be valuable as industrial enzyme for efficient protein processing.
The solid-state culture of A. oryzae for fermented food production has been developed over a long period of time (Machida et al. 2008). The beneficial phenotypes responsible for safety and the amylolytic and proteolytic enzyme activities should have been conserved and refined over time. The enzyme activities of the culture have been skillfully controlled and empirically optimized for the production of fermented foods. These traditional techniques have been of scientific interest. While it is natural to assume that extracellular proteases function in the fermentation process, the involvement of intracellular enzymes such as GdaA without signal peptide has been unclear. We clearly demonstrated that GdaA activity existed in an extract of the solid-state rice culture of A. oryzae, suggesting that intracellular enzymes expressing under solid-state rice culture could be released probably by rupture of mycelia and function in the fermentation feedstock. It is also noteworthy that in the solid-state rice culture, GdaA was largely responsible for the aminopeptidase activity that liberated not only d-alanine but also glycine. Glycine is one of the major factors that determines the palatability of foods. In addition to the strong sweetness and taste-enhancing effect (Kawai et al. 2002; Nishimura and Kato 1988; Wada et al. 2001), glycine is used to mask the unpleasant taste of anticholesterolemic food, beverages, and pharmaceuticals containing saponin (Sohi et al. 2004), a glycoside that exists in plants including soybean and oriental soybean foods (Anderson and Wolf 1995). Furthermore, glycine moderates the saltiness of pickled vegetables (Shitomi and Ikeda 1974). These facts led to speculation that the enzymatic release of glycine by GdaA in fermentation feedstock might be important for the taste of high salt-containing seasonings such as soy sauce and soybean paste as well as salt-free fermented foods such as rice wine (sake) and sweet cooking rice wine (mirin). Although GdaA was inhibited by NaCl, the Gly-pNA hydrolytic activity of purified GdaA was still clearly detected (0.8 U/mg; data not shown) even in the reaction mixture using 20 mM Tris–HCl at pH 9.0 containing 3 M NaCl. It was presumably due to high specific activity of GdaA. The activity of the enzyme was approximately 400 U/mg in the same reaction buffer without NaCl (Fig. 4a). Therefore, GdaA expressed in solid-state rice culture probably plays a role in liberating glycine in mixtures of fermentation feedstocks not only under salt-free or low-salt conditions but also even under high-salt conditions, at least in the early phase of the fermentation process.
References
Akao T, Sano M, Yamada O, Akeno T, Fujii K, Goto K, Ohashi-Kunihiro S, Takase K, Yasukawa-Watanabe M, Yamaguchi K, Kurihara Y, Maruyama J, Juvvadi PR, Tanaka A, Hata Y, Koyama Y, Yamaguchi S, Kitamoto N, Gomi K, Abe K, Takeuchi M, Kobayashi T, Horiuchi H, Kitamoto K, Kashiwagi Y, Machida M, Akita O (2007) Analysis of expressed sequence tags from the fungus Aspergillus oryzae cultured under different conditions. DNA Res 14:47–57
Anderson RL, Wolf WJ (1995) Compositional changes in trypsin inhibitors, phytic acid, saponins and isoflavones related to soybean processing. J Nutrition 125:581S–588S
Asano Y, Mori T, Hanamoto S, Kato Y, Nakazawa A (1989a) A new d-stereospecific amino acid amidase from Ochrobactrum anthropi. Biochem Biophys Res Commun 162:470–474
Asano Y, Nakazawa A, Kato Y, Kondo K (1989b) Properties of a novel d-stereospecific aminopeptidase from Ochrobactrum anthropi. J Biol Chem 264:14233–14239
Asano Y, Kato Y, Yamada A, Kondo K (1992) Structural similarity of d-aminopeptidase to carboxypeptidase DD and β-lactamases. Biochemistry 31:2316–2328
Blinkovsky AM, Byun T, Brown KM, Golightly EJ, Klotz AV (2000) A non-specific aminopeptidase from Aspergillus. Biochim Biophys Acta 1480:171–181
Bompard-Gilles C, Remaut H, Villeret V, Prangé T, Fanuel L, Delmarcelle M, Joris B, Frère J, Van Beeumen J (2000) Crystal structure of a d-aminopeptidase from Ochrobactrum anthropi, a new member of the ‘penicillin-recognizing enzyme’ family. Structure 8:971–980
Cava F, Lam H, de Pedro MA, Waldor MK (2011) Emerging knowledge of regulatory roles of d-amino acids in bacteria. Cell Mol Life Sci 68:817–831
Cheng YQ, Walton JD (2000) A eukaryotic alanine racemase gene involved in cyclic peptide biosynthesis. J Biol Chem 275:4906–4911
Gonzales T, Robert-Baudouy J (1996) Bacterial aminopeptidases: properties and functions. FEMS Microbiol Rev 18:319–344
Hattori R, Matsushita-Morita M, Marui J, Tada S, Suzuki S, Furukawa I, Yamagata Y, Amano H, Ishida H, Takeuchi M, Kusumoto K (2011) Characterization of an Aspergillus oryzae cysteinyl dipeptidase expressed in Escherichia coli. Biosci Biotechnol Biochem 75:159–161
Hesse SJ, Ruijter GJ, Dijkema C, Visser J (2000) Measurement of intracellular (compartmental) pH by 31P NMR in Aspergillus niger. J Biotechnol 77:5–15
Hoffmann K, Schneider-Scherzer E, Kleinkauf H, Zocher R (1994) Purification and characterization of eucaryotic alanine racemase acting as key enzyme in cyclosporin biosynthesis. J Biol Chem 269:12710–12714
Ito K, Ma X, Azmi N, Huang HS, Fujii M, Yoshimoto T (2003) Novel aminopeptidase specific for glycine from Actinomucor elegans. Biosci Biotechnol Biochem 67:83–88
Kawai M, Okiyama A, Ueda Y (2002) Taste enhancements between various amino acids and IMP. Chem Senses 27:739–745
Kitamoto N, Matsui J, Kawai Y, Kato A, Yoshino S, Ohmiya K, Tsukagoshi N (1998) Utilization of the TEF1-alpha gene (TEF1) promoter for expression of polygalacturonase genes, pgaA and pgaB, in Aspergillus oryzae. Appl Microbiol Biotechnol 50:85–92
Kobayashi T, Abe K, Asai K, Gomi K, Juvvadi P, Kato M, Kitamoto K, Takeuchi M, Machida M (2007) Genomics of Aspergillus oryzae. Biosci Biotechnol Biochem 71:646–670
Kusumoto K, Matsushita-Morita M, Furukawa I, Suzuki S, Yamagata Y, Koide Y, Ishida H, Takeuchi M, Kashiwagi Y (2008) Efficient production and partial characterization of aspartyl aminopeptidase from Aspergillus oryzae. J Appl Microbiol 105:1711–1719
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K-I, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161
Machida M, Yamada O, Gomi K (2008) Genomics of Aspergillus oryzae: learning from the history of Koji mold and exploration of its future. DNA Res 15:173–183
Manabe H (1990) Distribution of dipeptides containing d-alanine in Oryza species. Phytochemistry 29:3143–3147
Manabe H (1992) Formations of dipeptides containing d-alanine in wild rice plants. Phytochemistry 31:527–529
Manabe H, Yamauchi M, Ohira K (1981) Studies on d-amino acids in rice plants: behaviors of d-alanylglycine in rice seedlings. Plant Cell Physiol 22:333–336
Marui J, Yoshimi A, Hagiwara D, Fujii-Watanabe Y, Oda K, Koike H, Tamano K, Ishii T, Sano M, Machida M, Abe K (2010) Use of the Aspergillus oryzae actin gene promoter in a novel reporter system for exploring antifungal compounds and their target genes. Appl Microbiol Biotechnol 87:1829–1840
Matsushita-Morita M, Furukawa I, Suzuki S, Yamagata Y, Koide Y, Ishida H, Takeuchi M, Kashiwagi Y, Kusumoto K (2010) Characterization of recombinant prolyl aminopeptidase from Aspergillus oryzae. J Appl Microbiol 109:156–165
Matsushita-Morita M, Tada S, Suzuki S, Hattori R, Marui J, Furukawa I, Yamagata Y, Amano H, Ishida H, Takeuchi M, Kashiwagi Y, Kusumoto K (2011) Overexpression and characterization of an extracellular leucine aminopeptidase from Aspergillus oryzae. Curr Microbiol 62:557–564
Morita H, Okamoto A, Yamagata Y, Kusumoto K, Koide Y, Ishida H, Takeuchi M (2009) Heterologous expression and characterization of CpI, OcpA, and novel serine-type carboxypeptidase OcpB from Aspergillus oryzae. Appl Microbiol Biotechnol 85:335–346
Morita H, Kuriyama K, Akiyama N, Okamoto A, Yamagata Y, Kusumoto K, Koide Y, Ishida H, Takeuchi M (2010) Molecular cloning of ocpO encoding carboxypeptidase O of Aspergillus oryzae IAM2640. Biosci Biotechnol Biochem 74:1000–1006
Morita H, Abo H, Okamoto A, Maeda H, Yamagata Y, Kusumoto K, Amano H, Ishida H, Takeuchi M (2011) Enzymatic properties of the recombinant serine-type carboxypeptidase OcpC, which is unique to Aspergillus oryzae. Biosci Biotechnol Biochem 75:662–668
Nagata Y, Akino T, Ohno K (1985) Microdetermination of serum d-amino acids. Anal Biochem 150:238–242
Nakadai T, Nasuno S (1977) Purification and properties of leucine aminopeptidase IV from Aspergillus oryzae. Agr Biol Chem 41:1657–1666
Nakadai T, Nasuno S, Iguchi N (1973a) Purification and properties of leucine aminopeptidase I from Aspergillus oryzae. Agr Biol Chem 37:757–765
Nakadai T, Nasuno S, Iguchi N (1973b) Purification and properties of leucine aminopeptidase II from Aspergillus oryzae. Agr Biol Chem 37:767–774
Nakadai T, Nasuno S, Iguchi N (1973c) Purification and properties of leucine aminopeptidase III from Aspergillus oryzae. Agr Biol Chem 37:775–782
Nishimura T, Kato H (1988) Taste of free amino acids and peptides. Food Rev Int 4:175–194
Schomburg I, Schomburg D (eds) (2002) Springer handbook of enzymes, vol. 6. Class 3.4 hydrolases I. Springer, Berlin
Shitomi H, Ikeda T (1974) A study of rice bran mash with glycine (in Japanese). Bulletin Seitoku Gakuen Junior College for Women 7:19–29
Sohi H, Sultana Y, Khar RK (2004) Taste masking technologies in oral pharmaceuticals: recent developments and approaches. Drug Dev Ind Pharm 30:429–448
Suda H, Aoyagi T, Takeuchi T, Umezawa H (1976) Inhibition of aminopeptidase B and leucine aminopeptidase by bestatin and its stereoisomer. Arch Biochem Biophys 177:196–200
Taylor A (1993) Aminopeptidases: structure and function. FASEB J 7:290–298
Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T (1976) Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J Antibiot (Tokyo) 29:97–99
Wada A, Isobe Y, Yamaguchi S, Yamaoka R, Ozaki M (2001) Taste-enhancing effects of glycine on the sweetness of glucose: a gustatory aspect of symbiosis between the ant, Camponotus japonicus, and the larvae of the lycaenid butterfly, Niphanda fusca. Chem Senses 26:983–992
Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L (2010) Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res 38:5075–5087
Watanabe J, Tanaka H, Akagawa T, Mogi Y, Yamazaki T (2007) Characterization of Aspergillus oryzae aspartyl aminopeptidase expressed in Escherichia coli. Biosci Biotechnol Biochem 71:2557–2560
Yamauchi M, Ohashi T, Ohira K (1979) Occurrence of d-alanylglycine in rice leaf blades. Plant Cell Physiol 20:671–673
Yoon J, Maruyama J, Kitamoto K (2011) Disruption of ten protease genes in the filamentous fungus Aspergillus oryzae highly improves production of heterologous proteins. Appl Microbiol Biotechnol 89:747–759
Acknowledgments
This study was partially supported by the Program for Promotion of Basic Research for Innovative Bioscience (PROBRAIN). We would like to thank Dr. Yoshio Tanaka at Amano Enzyme Inc. for the helpful discussions and support for the peptide hydrolysis analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00253-011-3690-8
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 53.2 kb)
Rights and permissions
About this article
Cite this article
Marui, J., Matsushita-Morita, M., Tada, S. et al. Enzymatic properties of the glycine d-alanine aminopeptidase of Aspergillus oryzae and its activity profiles in liquid-cultured mycelia and solid-state rice culture (rice koji). Appl Microbiol Biotechnol 93, 655–669 (2012). https://doi.org/10.1007/s00253-011-3610-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3610-y