
Opinion statement
Dilated cardiomyopathy (DCM) is the third leading cause of heart failure in the USA. A major gene associated with DCM with cardiac conduction system disease is lamin A/C (LMNA) gene. Lamins are type V filaments that serve a variety of roles, including nuclear structure support, DNA repair, cell signaling pathway mediation, and chromatin organization. In 1999, LMNA was found responsible for Emery-Dreifuss muscular dystrophy (EDMD) and, since then, has been found in association with a wide spectrum of diseases termed laminopathies, including LMNA cardiomyopathy. Patients with LMNA mutations have a poor prognosis and a higher risk for sudden cardiac death, along with other cardiac effects like dysrhythmias, development of congestive heart failure, and potential need of a pacemaker or ICD. As of now, there is no specific treatment for laminopathies, including LMNA cardiomyopathy, because the mechanism of LMNA mutations in humans is still unclear. This review discusses LMNA mutations and how they relate to DCM, the necessity for further investigation to better understand LMNA mutations, and potential treatment options ranging from clinical and therapeutic to cellular and molecular techniques.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dilated cardiomyopathy (DCM), the most common type of cardiomyopathies, is characterized by ventricular dilation, systolic dysfunction, and fibrosis [1,2,3]. DCM can be acquired (idiopathic or environmental) or genetically inherited via genetic mutations, which accounts for approximately 40–50% of DCM [1,2,3,4]. About 30 such gene mutations have been identified and the number of genes responsible for DCM continues to increase [1, 4, 5]. Some DCM is associated with prominent cardiac conduction system disease, often referred to as conduction disease associated with DCM [1, 6]. One of the major genes accounting for approximately 6–8% of DCM with cardiac conduction system disease is the lamin A/C (LMNA) gene [5,8,, 7–9]. DCM patients carrying LMNA mutations have been reported to have a worse clinical prognosis than DCM patients carrying different pathologic DCM-associated gene mutations [10, 11, 12•, 13]. Therefore, further investigations are needed to understand how LMNA mutations alter signaling pathways, which will then guide us to find more specific treatments for this life-threatening disease [14,15,16,17]. To better understand the underpinnings of genetic mutations leading to disease manifestations and the potential therapeutic implications, a comprehensive review of recent insights into the structure and function of LMNA genes is warranted. In this review, we discuss the pathophysiology and clinical spectrum of LMNA mutations leading to DCM and the potential therapeutic strategies based on the recent cellular and molecular understandings of this disorder.
Structure and genetic determinants of lamin proteins
Lamins are type V intermediate filament proteins and are known to serve as major structural components of the nucleus [18, 19]. There are two major types of lamins, A type and B type [18, 20, 21]. A-type lamins include lamins A and C, which are alternative splice variants arising from a single gene LMNA (the gene of our interest), while B-type lamins include lamins B1 and B2, products of two separate genes, LMNB1 and LMNB2 [22,23,24,25,26]. The human LMNA gene comprises of 12 exons on chromosome 1q21.2–21.3 that encode A-type lamins: A, AD10, C, and C2, via alternative splicing [21, 27]. Type A lamins are widely expressed in all differentiated somatic cells, and their various mutations are known to cause laminopathies [20, 22, 28, 29]. Lamin A (664 amino acids) and lamin C (572 amino acids) proteins are structurally composed of a globular N-terminus head domain, a central coiled-coil rod domain implicated in dimerization of the proteins, and a C-terminal tail domain that includes an immunoglobulin-like domain where various posttranslational modifications occur [20, 22]. Most lamins undergo various posttranslational modifications such as phosphorylation, glycosylation, prenylation, sumoylation, nethylation, and malonylation, all affecting various downstream signaling pathways [30]. Posttranslational modification is especially important to produce mature lamin A from prelamin A [20, 30, 31].
The lamins A and C are identical in their first 566 amino acids which are encoded by exons 1–9. Exon 1 codes for the amino terminal head domain as well as the first part of the central rod domain; the rest of the central rod domain is coded by exons 2–6. It is in the C-terminal tail domain where the difference arises between the two proteins [21,32,, 22, 28, 31–33]. The C-terminal tail domain of lamin A is encoded by a part of exon 10 followed by full exons 11 and 12, while the tail domain of lamin C has a full exon 10 but lacks exons 11 and 12 [21, 31]. Exon 12 in lamin A contains a CAAX motif (C = cysteine, A = aliphatic, X = any residue) which serves as a site for sequential posttranslational modification processing. The process starts with cysteine undergoing isoprenylation, followed by the cleavage of the AAX motif. The final step is the carboxy-methylation of the farnesylated cysteine, which is followed by the cleavage of the last 15 amino acids (including the farnesylation) by the zinc metalloprotease Ste24 homologue (ZMPSTE24) protease, producing mature lamin A [34,35,36,37,38,39,40,41,42,43,44]. ZMPSTE24 is a membrane-associated enzyme that localizes to both the ER membrane and the inner nuclear membrane [12•, 13, 34, 37, 38]. It can farnesylate substrates such as prelamin A for proteolytic cleavage [45, 46]. Farnesylation and methylation lead to hydrophobicity, which facilitates the localization of lamin A to the nuclear envelope [14]. Conceivably, alteration in any step of this process may lead to functional impairment of translation at the nuclei, partially due to the abnormal/mutated prelamin A accumulation. Disruption in these cleavage events has been associated with various diseases such as premature aging syndrome, including Hutchinson-Gilford progeria syndrome, restrictive dermopathy, and lipodystrophy, including mandibuloacral dysplasia (MAD-B) [43,48,, 47–49]. In contrast, lamin C lacks the 98 amino acids at the C-terminus that are present in prelamin A (thereby lacking the CAAX motif). However, it contains a unique 6 amino acid C-terminus encoded by a part of exon 10. Nevertheless, it contains various sites for the lamin binding proteins that affect various downstream signaling cascades [21,32,, 22, 28, 31–33].
Although the human LMNA gene was first identified in 1986, it was not until 1999 that the LMNA mutation was found accountable for Emery-Dreifuss muscular dystrophy (EDMD), suggesting its role in human disease [32, 50]. EDMD is a type of muscular dystrophy that is characterized by slowly progressive muscle wasting and weakness, early tendon contractures, and dilated cardiomyopathy with conduction system disease [51]. Since 1999, approximately 400 new gene LMNA mutations have been reported (http://www.umd.be/LMNA/) and associated with a wide spectrum of human diseases termed laminopathies [19, 28, 32]. Currently, laminopathies comprise four distinct groups, depending on the affected tissue: (1) striated muscles (dystrophy, heart), (2) adipose tissue (lipodystrophy), (3) nervous system, and (4) accelerated aging syndrome. LMNA mutations can manifest as a multisystem disorder or tissue-specific disorder affecting the four tissues listed above [28,53,54,55,, 52–56]. Most laminopathies arise from missense mutations and disruption in posttranslational modifications that influence the function of lamins. These functions range from structural support of the nucleus to various cellular processes including gene regulations, protein-protein interactions, and DNA repair [19, 20, 22, 57, 58]. They can be autosomal dominant, autosomal recessive, or X-linked [23, 28, 34].
Functional role of lamin proteins
Lamins are major architectural proteins located within the nuclear matrix and provide a platform for the binding of proteins and chromatin [33]. They are attached to the internal face of the inner nuclear membrane, thereby conferring mechanical stability [20, 21, 33, 59]. Beyond their significant mechanical supporting roles for nuclear structures, lamins are now known to contribute to DNA repair/replication, transcription, mediating cellular signaling, chromatin organization, and cytoskeletal interactions [47,61,62,63,, 60–64]. Expression of A-type lamins was also proposed to be developmentally regulated in a tissue-specific manner and to be implicated in terminal tissue differentiation [65, 66]. Therefore, new findings of distinct LMNA mutations, including laminopathies with tissue-specific dysfunctions and distinctive phenotypes, suggest that more than just defective nuclear stability and deformation may play a role in the development of laminopathies from LMNA mutations [16,68,69,, 50, 57, 58, 67–70]. Indeed, diverse conditions of laminopathies have been characterized with features including not only misshapen nuclei or nucleus instability but also disorganization of heterochromatin, DNA repair dysfunction, impaired proliferation or survival, disruption of cellular signaling pathway, cell migration, senescence, stress response, and improper interaction with cytoskeleton [23, 31, 32, 63, 71].
Nuclear envelope support
As elements of the components that make up the nuclear envelope, lamins were primarily studied for their mechanical supporting roles in cells and during mitosis [72, 73]. Studies have demonstrated that lamin A/C defects induce changes in nuclear morphology in a subset of cells, such as misshapen nuclei, nuclear pore clustering, mislocalization of associated proteins, and aberrant intranuclear lamin foci [59, 61, 74]. Without lamins, assembled cell nuclei are small and fragile. Compared with those from LMNA +/+ mice, fibroblasts from LMNA −/− mice have more malleable and fragile nuclei that are less resistant to physical compression in an isotropic manner [74,75,76]. Lamin mutants known to have muscular phenotypes also demonstrate deformable nuclei with impaired stability and decreased nuclear stiffness [74]. It has been shown that cells from LMNA mutant patients have a range of different nuclear morphological phenotypes, suggesting lamins have a role as structural proteins [28].
Organizing chromatin
Lamins can organize and regulate chromatin position within the nuclear envelope [77, 78]. Studies with cardiomyocytes and MEFs derived from LMNA −/− mice showed a partial loss of peripheral heterochromatin, ectopic chromosome condensation, and mispositioned centromeric heterochromatin, a phenomenon also observed in cells with mutant lamin A proteins [59,80,81,82,, 79–83]. There are at least two chromatin-binding regions in lamins: one is located in the tail region and the other is within the rod domain [84, 85]. It is thought that the interactions between chromatin and lamins are mediated through histones and/or chromatin-associated proteins [85, 86]. Lamins associate with scaffold/matrix attachment regions that are involved in transcription, DNA replication, chromosome condensation, and chromatin organization [87]. The genome regions that preferentially associate with lamins are termed lamin-A-associated domains (LADs) [88]. The significance of their interaction and the exact molecular mechanisms leading to disease have yet to be determined, but there is growing evidence of altered interaction between lamin and chromatin in various laminopathies.
Participating in DNA repair
For studying the role of lamins in DNA repair, cells from Hutchinson-Gilford progeria syndrome (HGPS) patients were found to have a delayed recruitment of the repair factor p53-binding protein (53BP1) to damaged DNA sites. These cells showed increased levels of the double-stranded break marker γ-H2AX and were more sensitive to DNA damaging agents [89]. LMNA mutant cells causing muscular dystrophy or progeria are also shown to alter DNA damage regulators such as ATR and ATM signaling pathways [90]. Furthermore, it is now believed that LMNA mutations primarily affect not only myofibers but also the efficiency of satellite cells in muscle repair and regeneration [91]. LMNA mutations may therefore increase nuclear fragility by disrupting the mechanical coupling between the cytoskeleton and the nucleus and, consequently, lead to a greater susceptibility to physical stress, especially in tissues exposed to mechanical strain such as skeletal and cardiac muscle [61, 70, 79].
Transcription regulator
More and more evidence indicates that nuclear lamins can modulate gene expression either by directly interacting with chromatin or by sequestering transcriptional regulators at the nuclear periphery [60, 92, 93]. Several experimental results indicated that lamins mediate transcriptional regulation [92, 94, 95]. Lamin expression coincides with RNA polymerase II activity that changes according to the stages of development, and alteration of nuclear lamin organization can inhibit RNA polymerase II-dependent transcription [94]. A-type lamin also associates with numerous other transcriptional regulators, such as Rb, Gcl, Mok2, cFos, and Srebp1, affecting gene expression by sequestration of these factors or by influencing the assembly of core transcriptional complexes [93, 95, 96].
Mediating nucleo-cytoskeletal connections
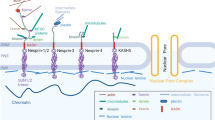
Lamins are considered an extended part of the LINC complex, which mechanically bridges nuclei with the cytoskeleton through lamin-interacting proteins that span the nuclear envelope [97,98,99,100]. The LINC complex is composed of two protein families—SUN (Sad1p/UNC-84) domain proteins at the inner nuclear membrane, where they in turn interact with a member of the nesprin family of proteins in the luminal space, and KASH domain proteins at the outer nuclear membrane [97, 100]. With two isoforms, Sun1 and Sun2, the SUN domains are conserved C-terminal protein regions about a few hundred amino acids long, followed by a transmembrane domain and a less conserved region of amino acids [101]. SUN proteins interact with the nuclear lamina, nuclear pore proteins, and other nuclear proteins at the nuclear interior and in the cytoplasm [102,103,104,105]. Nesprins span the outer nuclear membrane, where they associate with various cytoskeletal elements in the cytoplasm [105]. SUN domain proteins and nesprins together form the core of the LINC complex, which bridges the nucleus with the cytoskeleton to regulate proper function of transcription factors and gene expression [104, 105]. Any alteration in the LINC complex has been implicated for various nuclear functions including migration, positioning, morphology, and mechanics, and disturbed lamin function interferes with nuclear stability, gene regulation, and cytoskeletal functions [33, 61, 103]. Fibroblasts from mice with LMNA mutations linked to progeria demonstrated deformed nuclei and overaccumulation of protein Sun1, thereby negatively affecting lifespan and various tissues [106]. There is also evidence showing that proper expression and localization of nuclear lamin A/C and associated LINC complex are required for proper actin-cytoskeletal function during cell differentiation [107]. Studies also demonstrate that mutations in lamins A and C can disrupt LINC complex function and cause defects in skeletal and cardiac cells [108,109,110]. In addition to its role in muscle, proper nucleo-cytoskeletal coupling bridged by lamins is also essential during wound healing, inflammation, cell migration, cancer metastasis, and development [108,112,, 111–113]. Clinical outcomes and the molecular pathology in patients with laminopathies also indicated that the LINC complex was involved in LMNA mutations [76]. A-type lamin defects also affect different nuclear functions such as DNA replication, RNA transcription, and maturation by interacting with chromatin and many other transcription factors [98, 99].
Mediating cellular signaling pathway
Lamins also functionally interact with more than 30 direct and more than 100 indirect diverse proteins, indicating the function of lamins as a nuclear platform [33, 114, 115]. Moreover, the properties of variant proteins involved in these interactions may determine the tissue-specific roles of lamins [33, 116]. In addition, lamin A/C is also involved in a variety of signaling pathways affecting cell growth, survival, migration, and differentiation, including mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) pathways [60,118,, 117–119]. Through their interactions with the LINC complexes, cytoskeletal proteins, and chromatin, lamins regulate various signal transduction pathways that are important for diverse cellular functions [60, 120, 121]. Lamin and its associated nuclear proteins are shown to regulate the MAPK signaling pathway and its various downstream molecules as well as transforming growth factor-beta signaling pathways involving SMADs and other transcription factors [96, 122, 123].
LMNA mutations causing dilated cardiomyopathy
LMNA mutations are now known to account for 6–8% of dilated cardiomyopathy with conduction defect, portend a poor prognosis, and are associated with a high risk for sudden cardiac death [7,10,, 9–11, 12•, 13, 23, 124, 125]. Patients with LMNA mutations present with cardiac symptoms by mid-age (in their 20’s to 30’s) usually with mild low grade conduction system disease or atrial arrhythmia [7, 13, 126]. The conduction system disease gradually worsens and over 90% of patients develop dysrhythmia (bradyarrhythmias and tachyarrhythmias) with about half of patients needing either a pacemaker or ICD [7, 127]. There is a high incidence of sudden death (30%) and development of congestive heart failure (30%) [11, 13, 124, 125]. Moreover, almost half of the patients die suddenly before they reach the stage of overt heart failure [5, 11, 127]. A high cardiac disease penetrance and a high mortality were found in mutation carriers [7, 124, 125]. Male mutation carriers have a worse prognosis due to a higher prevalence of malignant ventricular arrhythmias and end-stage heart failure [125].
In addition to DCM and conduction disease disorder, LMNA mutations are also found in some patients with severe forms of arrhythmogenic right ventricular cardiomyopathy, a pathological pattern characterized by myocyte loss/fibro-fatty replacement, and reduction/absence of the intercalated discs of myocardium and LV noncompaction [128,129,130]. LMNA mutation carriers were also found to be associated with an increased risk of thromboembolic complications [131].
Cardiac effects of LMNA mutations have been demonstrated in a murine model as well [59, 79, 132, 133]. Targeted disruptions in mice led to development of cardiac and muscular dystrophy by interrupting nuclear envelope integrity [59, 79]. Homozygous (LMNA −/−) mice were also shown to exhibit significant growth retardation with premature death by 6–8 weeks of age with severe dilated cardiomyopathy with conduction disorder. They also demonstrated features similar to muscular dystrophy and suffered premature death by 6–8 weeks of age. These mice demonstrate nuclear deformation/instability as well as transport defects [79, 132]. LMNA knock-in mice carrying the H222P mutation, a missense mutation known to be responsible for EDMD in humans, developed dilated cardiomyopathy with conduction defects at adulthood in addition to muscular dystrophy. All of them died by 9 months of age with further histological analysis demonstrating extensive fibrosis and presumed altered gene expression from lamin mutation. Male mice had more prominent phenotypes and suffered earlier death compared to female mice [133].
Potential treatment options for LMNA cardiomyopathy
Clinical management
Currently, there is no specific treatment for laminopathies including LMNA cardiomyopathy. Current clinical management strategies for patients with LMNA cardiomyopathy are identical to those for patients with other cardiomyopathies or heart failure, which includes symptomatic and supportive treatment with pharmacologic and ventricular device therapies (neurohormonal antagonists, diuretics for congestion, vasodilators for hemodynamic unloading) [1, 3, 4]. Pacemakers can be considered in cases with the development of progressive conduction delays [1, 4, 5]. While sudden death from arrhythmias may be prevented by implantation of a defibrillator, progressive heart failure eventually becomes refractory to treatment, and heart transplantation is frequently necessary [1,2,3,4,5]. As patients with LMNA cardiomyopathy have shown to have a worse clinical course compared to non-LMNA cardiomyopathy, studies have tried to identify certain risk factors for sudden cardiac death among the LMNA mutation carriers for more aggressive therapy and earlier pacemaker/defibrillator placement [7, 124, 134]. Furthermore, in cardiomyopathy involving an additional system of laminopathies, medication for seizures and spasticity may be required for neuropathy, while physical therapy and/or corrective orthopedic surgery may be helpful for patients with muscular dystrophies [135,136,137].
Potential targeted pharmacologic therapies
Understanding how lamins control and alter signaling pathways holds great potential for therapeutic application in diverse laminopathies, including LMNA cardiomyopathy. Several mouse models have been used to study molecular pathways affected by LMNA mutations [138]. Identified signaling pathways deregulated by LMNA mutations include the MAPK pathway and mTOR pathway involving various downstream targets [118,144,145,, 139, 140•, 141, 142•, 143–146]. The MAPK/extracellular signal-regulated (ERK) pathway is activated by various stimuli that control signaling cascades that regulate cell proliferation, growth, differentiation, survival, migration, and apoptosis. It is expressed in all eukaryotic cells and disruptions in this pathway have been known to play a role in cancer and other numerous human diseases [117, 118, 147]. The MAPK pathway is also known to be involved in intracellular signaling of the ventricular myocytes. The MAPK pathway works as a multitiered pathway, involving various downstream target signaling molecules [148]. mTOR, the serine/threonine kinase, also plays an important role in regulating growth, proliferation, survival, and protein synthesis [149, 150]. It has been found that there is often cross-talk between mTOR and MAPK pathways [148].
LMNA knock-in mice carrying the H222P mutation, a missense mutation known to be responsible for Emery-Dreifuss muscular dystrophy and dilated cardiomyopathy in humans, are known to develop dilated cardiomyopathy with conduction defects that lead to eventual death by 9 months of age [117, 118, 142•, 143]. In the hearts of these mice, hyperactivation of the MAPK signaling pathway including ERK, JNK, elk, and c-jun, which are all downstream components of MAPK cascades, was observed [118, 141, 142•, 143]. Treatment of these mice with various inhibitors of MAPK signaling pathways (inhibitors of MAPK/ERK, JNK, or both) demonstrated a delay in LV dilation and improvement of LV systolic function in mice with dilated cardiomyopathy [141, 142•, 143].
Moreover, it has been shown that proper interaction of A-type lamin with activated ERK1/2 regulates activation of junction protein connexin43 (Cx43). Without normal A-type lamins, Cx43 activation increases due to inappropriate phosphorylation of ERK1/2, resulting in decreased gap junction function that may decrease cell communication and contribute to the arrhythmic pathology associated with laminopathies [151, 152].
These results provide genetic evidence that ERK1 and ERK2 contribute to the development of cardiomyopathy in laminopathies [146]. Hyperactivation of the MAPK/ERK signaling pathway was observed in explanted human hearts with LMNA cardiomyopathy, indicating that these inhibitors hold a great therapeutic potential for human subjects with LMNA cardiomyopathy [153, 154]. Various inhibitors of the MAPK or mTOR pathway are already in therapeutic use to treat other pathological diseases such as cancer, chronic pain, and inflammatory diseases in humans [139, 148, 150]. Recently, a phase II trial commenced using ARRY-797, a selective oral inhibitor of the p38 MAPK, in patients with LMNA cardiomyopathy. The company’s phase I trial demonstrated a favorable outcome for patients on ARRY-797, showing improved cardiac function on an echocardiogram. The final outcome from this trial has yet to be determined, but this trial exhibits a viable therapy for LMNA cardiomyopathy by attenuating left ventricular dilatation and deterioration (www.ClinicalTrials.gov, Identifier NCT02057341).
LMNA mutant mice also exhibited hyperactivation of the mTOR pathway in affected tissues such as cardiac and skeletal muscle [133, 155, 156]. LMNA mutant mice treated with mTOR pathway inhibitors (rapamycin or temsirolimus) showed improvement in their LV size and cardiac function [155].
Potential cellular and molecular therapies
Identifying the precise molecular mechanisms of LMNA mutations leading to laminopathies affecting striated muscles is critical for developing new therapeutic strategies to prevent cardiac dysfunction and sudden death. Lamin mutations are known to alter functions of various transcription factors [92]. Cells in mice with lamin mutations linked to muscular dystrophy and DCM were shown to have altered function of transcription factor megakaryoblastic leukemia 1 (MKL1) due to abnormal nuclear-cytoskeleton dynamics. This was rescued by expression of one of the nuclear proteins, emerin [157]. In addition, expression of cardiac-specific lamin A transgene in LMNA −/− mice demonstrated improvement in cardiac function with a preservation of a functional conductive system [132]. These findings indicate a novel mechanism that could provide insight into the disease etiology for the cardiac phenotype in laminopathies and also implies a potential therapeutic strategy for laminopathies.
Insights from noncardiomyopathy LMNA mutations
LMNA mutations are also known to cause accelerating syndromes such as HPGS [53, 158]. The most common mutation is due to the deletion of the C-terminal region required for posttransitional modification [63, 159]. This leads to an increase in farnesylated lamin A which is shown to cause mitochondrial dysfunction, abnormal chromatic interactions, DNA damage, and cell instability in both in vitro and the mice model [34, 63, 159, 160]. Several studies have shown that farnesyl transferase inhibitors showed some attenuation of progeria-like symptoms as well as restoration of nuclear morphology [161,162,163,164]. Currently, lonafarnib (FIT inhibitor) is undergoing a clinical trial in patients with HGPS with promising data showing improvement not only in weight gain but also in cardiovascular stiffness, bone strength, and hearing (neuropathy) [165].
Conclusions
Lamin A/C mutations are frequently reported as a cause of cardiomyopathy, often causing sudden death at a young age before patients even reach clinically overt heart failure. Mutations of LMNA associated with laminopathies are only beginning to be understood. Studies have demonstrated an intricate complexity of lamin function and how it affects a diverse spectrum of cellular and molecular changes responsible for laminopathies including LMNA cardiomyopathy. Further investigations are needed to examine how alternations in the lamin structure regulate various cellular and molecular processing such as transportation of trans-factors, signaling pathways, and/or processes of posttranslation. Such examinations can facilitate rational understanding of the pathology of laminopathies in order to design therapeutic strategies.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Luk A, Ahn E, Soor GS, Butany J. Dilated cardiomyopathy: a review. J Clin Pathol. 2009;62(3):219–25.
Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–16.
Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J. 1999;20(2):93–102.
Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57(16):1641–9.
Hershberger RE, Morales A, editors. LMNA-related dilated cardiomyopathy. Seattle: University of Washington; 2008.
Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52(15):1250–60.
Taylor MR, Fain PR, Sinagra G, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41(5):771–80.
Parks SB, Kushner JD, Nauman D, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156(1):161–9.
Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341(23):1715–24.
Perez-Serra A, Toro R, Campuzano O, et al. A novel mutation in lamin A/C causing familial dilated cardiomyopathy associated with sudden cardiac death. J Card Fail. 2015;21(3):217–25.
van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83(1):79–83.
• Arbustini E, Pilotto A, Repetto A, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. 2002;39(6):981–90. Important case series of LMNA cardiomyopathy.
Becane HM, Bonne G, Varnous S, et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin Electrophysiol. 2000;23(11 Pt 1):1661–6.
Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119(7):1825–36.
Zaremba-Czogalla M, Dubinska-Magiera M, Rzepecki R. Laminopathies: the molecular background of the disease and the prospects for its treatment. Cell Mol Biol Lett. 2011;16(1):114–48.
Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013;152(6):1365–75.
Mounkes L, Kozlov S, Burke B, Stewart CL. The laminopathies: nuclear structure meets disease. Curr Opin Genet Dev. 2003;13(3):223–30.
Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. 2015;84:131–64.
Szeverenyi I, Cassidy AJ, Chung CW, et al. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29(3):351–60.
Ho CY, Lammerding J. Lamins at a glance. J Cell Sci. 2012;125(Pt 9):2087–93.
Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268(22):16321–6.
Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol. 2013;14(1):13–24.
Mestroni L, Taylor MR. Lamin A/C gene and the heart: how genetics may impact clinical care. J Am Coll Cardiol. 2008;52(15):1261–2.
Sieprath T, Darwiche R, De Vos WH. Lamins as mediators of oxidative stress. Biochem Biophys Res Commun. 2012;421(4):635–9.
Machiels BM, Zorenc AH, Endert JM, et al. An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem. 1996;271(16):9249–53.
Hoger TH, Krohne G, Franke WW. Amino acid sequence and molecular characterization of murine lamin B as deduced from cDNA clones. Eur J Cell Biol. 1988;47(2):283–90.
Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A. 1986;83(17):6450–4.
Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313(10):2121–33.
Mounkes LC, Burke B, Stewart CL. The A-type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc Med. 2001;11(7):280–5.
Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15(3):163–77.
Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832–53.
Rankin J, Ellard S. The laminopathies: a clinical review. Clin Genet. 2006;70(4):261–74.
Dittmer TA, Misteli T. The lamin protein family. Genome Biol. 2011;12(5):222.
Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46(12):2531–58.
Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47(5):883–91.
Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet. 2009;10:153–74.
Barrowman J, Michaelis S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol Chem. 2009;390(8):761–73.
Bergo MO, Gavino B, Ross J, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99(20):13049–54.
Pendas AM, Zhou Z, Cadinanos J, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31(1):94–9.
Goldberg MW, Huttenlauch I, Hutchison CJ, Stick R. Filaments made from A- and B-type lamins differ in structure and organization. J Cell Sci. 2008;121(Pt 2):215–25.
Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol. 1990;110(5):1489–99.
Weber K, Plessmann U, Traub P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett. 1989;257(2):411–4.
Barrowman J, Hamblet C, Kane MS, Michaelis S. Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PLoS One. 2012;7:e32120.
Tam A, Schmidt WK, Michaelis S. The multispanning membrane protein Ste24p catalyzes CAAX proteolysis and NH2-terminal processing of the yeast a-factor precursor. J Biol Chem. 2001;276(50):46798–806.
Barrowman J, Hamblet C, George CM, Michaelis S. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell. 2008;19:5398–408.
Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc Natl Acad Sci U S A. 1998;95:11175–80.
Hutchison CJ. The role of DNA damage in laminopathy progeroid syndromes. Biochem Soc Trans. 2011;39(6):1715–8.
Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13(20):2493–503.
De Sandre-Giovannoli A, Levy N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog Mol Subcell Biol. 2006;44:199–232.
Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21(3):285–8.
Bonne G, Letcurcq F, Ben Yaou R. Emery-Dreifuss muscular dystrophy. In: GeneReviews. University of Washington, Seattle; 2004.
Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109–12.
Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86(3):967–1008.
Hegele RA, Cao H, Liu DM, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79(2):383–9.
Padiath QS, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38(10):1114–23.
Brown CA, Lanning RW, McKinney KQ, et al. Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am J Med Genet. 2001;102(4):359–67.
Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–6.
Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8.
Sullivan T, Escalante-Alcalde D, Bhatt H, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147(5):913–20.
Andres V, Gonzalez JM. Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol. 2009;187(7):945–57.
Lammerding J, Schulze PC, Takahashi T, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–8.
Gonzalez-Suarez I, Redwood AB, Perkins SM, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009;28(16):2414–27.
Dechat T, Shimi T, Adam SA, et al. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci U S A. 2007;104:4955–60.
Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17:626–38.
Röber RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development. 1989;105:365–78.
Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell. 1987;51:383–92.
Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9:1453–9.
De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055.
De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70(3):726–36.
Zwerger M, Jaalouk DE, Lombardi ML, et al. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet. 2013;22:2335–49.
Moriuchi T, Muraoka T, Mio K, Osumi T, Hirose F. Long-term expression of the lamin A mutant associated with dilated cardiomyopathy induces senescence. Genes Cells. 2014;19(12):901–18.
Moir RD, Yoon M, Khuon S, Goldman RD. Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J Cell Biol. 2000;151(6):1155–68.
Panorchan P, Schafer BW, Wirtz D, Tseng Y. Nuclear envelope breakdown requires overcoming the mechanical integrity of the nuclear lamina. J Biol Chem. 2004;279(42):43462–7.
Broers JL, Peeters EA, Kuijpers HJ, et al. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13:2567–80.
Newport JW, Wilson KL, Dunphy WG. A lamin-independent pathway for nuclear envelope assembly. J Cell Biol. 1990;111:2247–59.
Taranum S, Vaylann E, Meinke P, et al. LINC complex alterations in DMD and EDMD/CMT fibroblasts. Eur J Cell Biol. 2012;91:614–28.
Fawcett DW. On the occurrence of a fibrous lamina on the inner aspect of the nuclear envelope in certain cells of vertebrates. Am J Anat. 1966;119:129–45.
Belmont AS, Zhai Y, Thilenius A. Lamin B distribution and association with peripheral chromatin revealed by optical sectioning and electron microscopy tomography. J Cell Biol. 1993;123:1671–85.
Nikolova V, Leimena C, McMahon AC, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113(3):357–69.
Galiová G, Bártová E, Raska I, Krejcí J, Kozubek S. Chromatin changes induced by lamin A/C deficiency and the histone deacetylase inhibitor trichostatin A. Eur J Cell Biol. 2008;87:291–303.
Ognibene A, Sabatelli P, Petrini S, et al. Nuclear changes in a case of X-linked Emery-Dreifuss muscular dystrophy. Muscle Nerve. 1999;22:864–9.
Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–63.
Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–5.
Bruston F, Delbarre E, Ostlund C, Worman HJ, Buendia B, Duband-Goulet I. Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 2010;584:2999–3004.
Taniura H, Glass C, Gerace L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J Cell Biol. 1995;131:33–44.
Shoeman RL, Traub P. The in vitro DNA-binding properties of purified nuclear lamin proteins and vimentin. J Biol Chem. 1990;265:9055–61.
Ludérus ME, den Blaauwen JL, de Smit OJ, Compton DA, van Driel R. Binding of matrix attachment regions to lamin polymers involves single-stranded regions and the minor groove. Mol Cell Biol. 1994;14:6297–305.
Guelen L, Pagie L, Brasset E, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–51.
Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11(7):780–5.
Manju K, Muralikrishna B, Parnaik VK. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci. 2006;119:2704–14.
Gnocchi VF, Ellis JA, Zammit PS. Does satellite cell dysfunction contribute to disease progression in Emery-Dreifuss muscular dystrophy? Biochem Soc Trans. 2008;36:1344–9.
Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–7.
Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol. 2009;184:45–55.
Spann TP, Goldman AE, Wang C, Huang S, Goldman RD. Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J Cell Biol. 2002;156:603–8.
Kumaran RI, Muralikrishna B, Parnaik VK. Lamin A/C speckles mediate spatial organization of splicing factor compartments and RNA polymerase II transcription. J Cell Biol. 2002;159:783–93.
González JM, Navarro-Puche A, Casar B, Crespo P, Andrés V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol. 2008;183:653–66.
Crisp M, Liu Q, Roux K, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172:41–53.
Shumaker DK, Kuczmarski ER, Goldman RD. The nucleoskeleton: lamins and actin are major players in essential nuclear functions. Curr Opin Cell Biol. 2003;15:358–66.
Stierlé V, Couprie J, Ostlund C, et al. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry. 2003;42:4819–28.
Tzur YB, Wilson KL, Gruenbaum Y. SUN-domain proteins: ‘Velcro’ that links the nucleoskeleton to the cytoskeleton. Nat Rev Mol Cell Biol. 2006;7:782–8.
Hodzic DM, Yeater DB, Bengtsson L, Otto H, Stahl PD. Sun2 is a novel mammalian inner nuclear membrane protein. J Biol Chem. 2004;279:25805–12.
Malone CJ, Fixsen WD, Horvitz HR, Han M. UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development. 1999;126:3171–81.
Starr DA, Han M. ANChors away: an actin based mechanism of nuclear positioning. J Cell Sci. 2003;116:211–6.
Haque F, Mazzeo D, Patel JT, et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285(5):3487–98.
Meinke P, Nguyen TD, Wehnert MS. The LINC complex and human disease. Biochem Soc Trans. 2011;39(6):1693–7.
Chen CY, Chi YH, Mutalif RA, et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149:565–77.
Khatau SB, Kusuma S, Hanjaya-Putra D, et al. The differential formation of the LINC-mediated perinuclear actin cap in pluripotent and somatic cells. PLoS One. 2012;7:e36689.
Gundersen GG, Worman HJ. Nuclear positioning. Cell. 2013;152:1376–89.
Méjat A, Misteli T. LINC complexes in health and disease. Nucleus. 2010;1:40–52.
Folker ES, Ostlund C, Luxton GW, Worman HJ, Gundersen GG. Lamin A variants that cause striated muscle disease are defective in anchoring transmembrane actin-associated nuclear lines for nuclear movement. Proc Natl Acad Sci U S A. 2011;108:131–6.
Luxton GW, Gomes ER, Folker ES, Vintinner E, Gundersen GG. Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science. 2010;329:956–9.
Luxton GW, Gomes ER, Folker ES, Worman HJ, Gundersen GG. TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus. 2011;2:173–81.
Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol. 2013;23(24):R1113–21.
Kubben N, Voncken JW, Misteli T. Mapping of protein- and chromatin-interactions at the nuclear lamina. Nucleus. 2010;1:460–71.
Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol. 2009;577:67–79.
Kavanagh DM, Powell WE, Malik P, Lazou V, Schirmer EC. Organelle proteome variation among different cell types: lessons from nuclear membrane proteins. Subcell Biochem. 2007;43:51–76.
Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16:1884–95.
Muchir A, Pavlidis P, Decostre V, et al. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–93.
Bakay M, Wang Z, Melcon G, et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129(Pt 4):996–1013.
Mejat A, Decostre V, Li J, et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J Cell Biol. 2009;184(1):31–44.
Dialynas G, Flannery KM, Zirbel LN, et al. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Hum Mol Genet. 2012;21(7):1544–56.
Lin F, Morrison JM, Wu W, Worman HJ. MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet. 2005;14(3):437–45.
Van Berlo JH, Voncken JW, Kubben N, et al. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet. 2005;14(19):2839–49.
van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59(5):493–500.
van Rijsingen IA, Nannenberg EA, Arbustini E, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15(4):376–84.
Sylvius N, Tesson F. Lamin A/C and cardiac diseases. Curr Opin Cardiol. 2006;21(3):159–65.
van Berlo JH. When lamin A/C fails, the heart suffers. Neth Hear J. 2006;14(10):354.
Larsen MK, Nissen PH, Berge KE, et al. Molecular autopsy in young sudden cardiac death victims with suspected cardiomyopathy. Forensic Sci Int. 2012;219(1–3):33–8.
Quarta G, Syrris P, Ashworth M, et al. Mutations in the lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33(9):1128–36.
Hermida-Prieto M, Monserrat L, Castro-Beiras A, et al. Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. Am J Cardiol. 2004;94(1):50–4.
van Rijsingen IA, Bakker A, Azim D, et al. Lamin A/C mutation is independently associated with an increased risk of arterial and venous thromboembolic complications. Int J Cardiol. 2013;168(1):472–7.
Frock RL, Chen SC, Da DF, et al. Cardiomyocyte-specific expression of lamin A improves cardiac function in Lmna−/− mice. PLoS One. 2012;7(8):e42918.
Arimura T, Helbling-Leclerc A, Massart C, et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14(1):155–69.
Anselme F, Moubarak G, Savoure A, et al. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm. 2013;10(10):1492–8.
Benedetti S, Menditto I, Degano M, et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology. 2007;69(12):1285–92.
van der Kooi AJ, Bonne G, Eymard B, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59(4):620–3.
Goizet C, Yaou RB, Demay L, et al. A new mutation of the lamin A/C gene leading to autosomal dominant axonal neuropathy, muscular dystrophy, cardiac disease, and leuconychia. J Med Genet. 2004;41(3):e29.
Stewart CL, Kozlov S, Fong LG, Young SG. Mouse models of the laminopathies. Exp Cell Res. 2007;313(10):2144–56.
Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15(8):369–79.
• Ramos FJ, Chen SC, Garelick MG, et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012;4(144):144ra103. Novel insights into mTORC1 inhibition as possible therapeutic strategy.
Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18(2):241–7.
• Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation. 2011;123(1):53–61. Feasibility for MAPK inhibitors in cardioprotection for LMNA cardiomyopathy.
Wu W, Shan J, Bonne G, Worman HJ, Muchir A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim Biophys Acta. 2010;1802(7–8):632–8.
Muchir A, Kim YJ, Reilly SA, Wu W, Choi JC, Worman HJ. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet Muscle. 2013;3(1):17.
Muchir A, Reilly SA, Wu W, et al. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res. 2012;93(2):311–9.
Wu W, Iwata S, Homma S, Worman HJ, Muchir A. Depletion of extracellular signal-regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum Mol Genet. 2014;23(1):1–11.
Brunet A, Pouyssegur J. Mammalian MAP kinase modules: how to transduce specific signals. Essays Biochem. 1997;32:1–16.
Wang CM, Cigliano A, Delogu S, et al. Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis Implications for the treatment of human liver cancer. Cell Cycle. 2013;12(13):1999–2010.
Dowling RJO, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta, Proteins and Proteomics. 2010;1804(3):433–9.
Lisi L, Aceto P, Navarra P, Dello Russo C. mTOR kinase: a possible pharmacological target in the management of chronic pain. Biomed Res Int. 2015;2015:394257.
Page C, Doubell AF. Mitogen-activated protein kinase (MAPK) in cardiac tissues. Mol Cell Biochem. 1996;157(1–2):49–57.
Chen SC, Kennedy BK, Lampe PD. Phosphorylation of connexin43 on S279/282 may contribute to laminopathy-associated conduction defects. Exp Cell Res. 2013;319(6):888–96.
Muchir A, Wu W, Choi JC, et al. Abnormal p38 mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet. 2012;21(19):4325–33.
Chatzifrangkeskou M, Le Dour C, Wu W, et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum Mol Genet. 2016;25:2220–33.
Choi JC, Muchir A, Wu W, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012;4(144):144ra102.
Choi JC, Wu W, Muchir A, Iwata S, Homma S, Worman HJ. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J Biol Chem. 2012;287(48):40513–24.
Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497(7450):507–11.
Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426–31.
Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A. 2007;104(12):4949–54.
Young SG, Meta M, Yang SH, Fong LG. Prelamin A farnesylation and progeroid syndromes. J Biol Chem. 2006;281(52):39741–5.
Liu BH, Wang ZM, Zhang L, Ghosh S, Zheng HL, Zhou ZJ. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat Commun. 2013;4:1868.
Fong LG, Frost D, Meta M, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311(5767):1621–3.
Toth JI, Yang SH, Qiao X, et al. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102(36):12873–8.
Capell BC, Erdos MR, Madigan JP, et al. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102(36):12879–84.
Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666–71.
Acknowledgments
This work is supported by the National Institutes of Health (R01HL103931) and the Collins Family Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Xi Wang, Allyson Zabell, and Wonshill Koh each declare no potential conflicts of interest.
W. H. Wilson Tang reports grants from the National Institutes of Health. Dr. Tang is a section editor for Current Treatment Options in Cardiovascular Medicine.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Heart Failure
Rights and permissions
About this article

Cite this article
Wang, X., Zabell, A., Koh, W. et al. Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies. Curr Treat Options Cardio Med 19, 21 (2017). https://doi.org/10.1007/s11936-017-0520-z
Published:
DOI: https://doi.org/10.1007/s11936-017-0520-z