
Abstract
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm (MPN) characterized by an overactive Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway through mutations in JAK2 exons 12 or 14 (JAK2 V617F). The dominant clinical characteristics include erythrocytosis (with or without leukocytosis/thrombocytosis), thrombotic events, and symptoms. Increased risk of mortality is mainly caused by thrombotic events and progression to post-polycythemia vera myelofibrosis (PPV-MF) or secondary acute myeloid leukemia (sAML). The most important prognostic factors include age and a history of thrombotic events, although recent evidence has indicated that leukocytosis and additional cytogenetic aberrations may also be of significant prognostic value. First-line therapies include aspirin and phlebotomies, which significantly reduce the incidence of thrombotic events and prolong survival. Cytoreductive treatment with hydroxyurea (approved) and conventional or pegylated interferon-α (effective, but not approved in many countries) is initiated for high-risk or inadequately controlled disease, e.g., uncontrolled hematocrit, leukocytosis, thrombocytosis, thrombotic events, splenomegaly, or symptoms. However, some patients may not receive initial benefit from first-line therapy or may become resistant or intolerant in due course. Although second-line treatment options are limited, clinical trials have shown the efficacy of ruxolitinib toward improving blood counts, enlarged spleen, and symptoms and potentially reducing thrombotic events. Identification of patients with uncontrolled PV is important for clinical care, as such patients have a high risk of complications, and future studies with JAK inhibitors or other agents alone or in combination are needed to test their potential to reduce rates of thrombotic events and transformation to PPV-MF or sAML.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm (MPN) primarily characterized by erythrocytosis [1–5], and often leukocytosis and/or thrombocytosis [2, 3, 5]. Elevated blood counts result from a trilineage proliferation of hematopoiesis in the bone marrow (BM), with most patients (≈98–99 %) having mutations in the Janus kinase 2 (JAK2) gene [6–8]. The point mutation JAK2 V617F in exon 14 appears to be the driver mutation for PV [9–14] and is present in ≈96 % of patients [6, 7], while mutations (small deletions or insertions) in exon 12 of JAK2 are found in approximately 3 % of patients with PV [6–8].
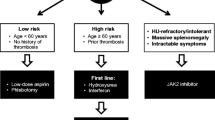
Polycythemia vera is diagnosed slightly more often in men than women, with the median age at diagnosis being 60 years [15]; however, ≈10 % of patients are <40 years at diagnosis [16•]. Thrombotic events are a major complication of the disease, accounting for 45 % of all deaths [4] and may be found at atypical sites (e.g., splanchnic and cerebral veins) [17, 18]. The course of PV (Fig. 1) begins with an erythrocytotic phase [19] during which patients may be asymptomatic or experience symptoms (e.g., fatigue, headache, paresthesia, arthralgia; Fig. 2) [18]. Over time, the disease commonly evolves to an advanced phase, with patients exhibiting disease features that may include fatigue, severe pruritus, bone pain, night sweats, and in some cases, splenomegaly [16•, 18, 22]. Additionally, patients with a long disease duration (usually >10 years) may progress to post-polycythemia vera myelofibrosis (PPV-MF; HR, 15.24; 95 % CI 4.22–55.06 [23]; 10- and 15-year cumulative risk, 2.3 and 6 %, respectively [16•]), with a median time to progression of 13 years [24]. In this phase, patients have increased BM fibrosis and worsening splenomegaly, with some patients exhibiting progressive cytopenias [25]. Furthermore, patients are at risk of evolution to secondary acute myeloid leukemia (sAML) with a 15-year cumulative risk of 7 % [4, 15, 23, 24].

Progression of polycythemia vera. AML acute myeloid leukemia, HU hydroxyurea, IWG-MRT International Working Group-Myeloproliferative Neoplasms Research and Treatment, JAK2 Janus kinase 2, MF myelofibrosis, PPV-MF post-polycythemia vera myelofibrosis, WHO World Health Organization

For many patients, the disease is easily managed. However, some patients have a suboptimal response to available therapies, which can manifest as uncontrolled hematocrit (HCT), thrombotic events, leukocytosis, thrombocytosis, symptoms, or increasing spleen size and lead to what we have termed “inadequately controlled PV.” This can occur at any phase of the disease and can ultimately result in reduced survival. In this review, we will describe how we identify these patients and discuss potential therapeutic strategies to improve management of their PV.
Current Risk Stratification and Initial Treatment Strategies
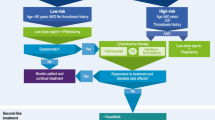
Traditionally, risk classification in PV is determined by age and history of thrombotic events [26, 27]. Patients <60 years with no history of thrombotic events are considered low risk, whereas patients aged ≥60 years and/or who have prior occurrences of thrombotic events are considered high risk. An intermediate-risk category that includes patients <60 years of age with no history of thrombotic events but with cardiovascular risk factors (e.g., hypertension, hypercholesterolemia, diabetes, obesity, smoking) has been proposed but not formally defined [28, 29]. Higher risk patients have reduced survival compared with low-risk patients [16•]. An analysis of overall survival (OS) in a large cohort of treated patients with PV (N = 1545) found median survival to be 28, 19, and 11 years for low-, intermediate-, and high-risk patients, respectively, based on a prognostic model that used age, leukocytosis, and venous thrombosis as variables (Fig. 3) [16•]. However, the impact of arterial thrombosis on survival was not assessed in this model and will require further clarification.

Risk-stratified survival in patients with PV (N = 1545) [16•]. Adverse points were assigned to age ≥67 years (5 points), age 57–66 years (2 points), white blood cell count ≥15 × 109/L (1 point), and venous thrombosis (1 point): low-risk (0 points), intermediate-risk (1 or 2 points), and high-risk (≥3 points). Using mature survival data from a subgroup of patients seen at the Mayo Clinic (n = 337), median survival was calculated to be 26, 15, and 8.3 years for low-, intermediate-, and high-risk patients, respectively [16•]; the model was subsequently validated in the entire study cohort (N = 1545). Reprinted by permission from Macmillan Publishers Ltd: Tefferi et al. [16•]
Given that thrombotic events are such a major complication of the disease, reducing thrombotic risk is a main goal of therapy. Treatment strategies target HCT <45 %, a treatment goal that was recently supported by the CYTO-PV (Cytoreductive Therapy in Polycythemia Vera) study (N = 365), in which aggressively targeting HCT <45 % was shown to reduce the risk of major thrombotic events and death from cardiovascular causes [30••]. In this study, patients randomized to the high-HCT group (target HCT 45–50 %) had a fourfold increase in the rate of death from cardiovascular causes or major thrombosis compared with those in the low-HCT group (target HCT <45 %), as well as a higher incidence of thrombosis. Treatment recommendations are based on the patient’s risk stratification. All patients are usually treated for optimization of cardiovascular risk factors with low-dose aspirin and phlebotomy [31], while hydroxyurea (HU) and interferon-α (IFN-α) [25, 27] are the recommended first-line treatments for high-risk patients [25, 27]. Currently, there are no recommendations for PV-specific therapies for intermediate-risk patients, making adequate treatment of this patient subgroup difficult.
Identifying Inadequately Controlled PV
For a large proportion of patients, following the recommendations just described results in well-managed PV, with controlled HCT, hematologic parameters, spleen, and symptoms. Other patients, however, develop resistance or intolerance to treatment (Table 1). These patients have suboptimal responses which can manifest in different ways, including elevated HCT, the occurrence of thrombotic events, persistent leukocytosis/thrombocytosis, increasing spleen size, and burdensome symptoms. These indicators of inadequately controlled disease are not confined to any one phase of treatment and may occur at any time during the course of the disease. These patients are at an increased risk of thrombosis [32–35], reduced quality of life (QOL) [22, 36–38], and shortened survival [16•, 33, 39, 40•, 41], and thus are usually considered high-risk. Patients with intolerance require a change of therapy if the intolerance is sufficiently severe; however, intolerance itself has not been shown to result in the same increased risk of thrombosis or shortened survival.
Given the results of the CYTO-PV study [30••], the two most important indicators of inadequately controlled PV are the presence of uncontrolled HCT and the occurrence of thrombotic events. As discussed above, tight control of HCT is imperative for reducing the risk of thrombotic events and, therefore, the risk of death. Other clinical features, such as persistent leukocytosis (leukocyte count > 10 × 109/L) and/or thrombocytosis (platelet count > 400 × 109/L), are also indicative of inadequately controlled disease [27]. Leukocytosis at PV diagnosis has been found to be prognostic for thrombosis [34, 35], leukemic transformation [16•, 42, 43], and survival [16•, 33, 39, 41, 44]. Similarly, studies have found a correlation between persistence of leukocytosis and adverse outcomes in PV, e.g., thrombosis [30••], including arterial [32, 35] and venous thrombosis [33], and hematologic transformation and shorter survival [40•], regardless of a patient’s risk category. In the CYTO-PV study, patients in the high-HCT group had significantly higher leukocyte counts than patients in the low-HCT group, suggesting that leukocytosis despite treatment (which in this study was a mixture of phlebotomy only or cytoreductive therapies such as HU) could have contributed to the higher rates of thrombosis seen in the high-HCT group [30••]. A subsequent multivariable, time-dependent analysis by Barbui and colleagues [45] found that the risk of thrombosis increased significantly in patients with a leukocyte count > 11 × 109/L. Interestingly, an increase in the risk of thrombosis was also seen in patients with a leukocyte count > 7 × 109/L; however, this increase was not statistically significant. These results, while interesting, need to be interpreted bearing in mind that patients differed not only in leukocyte count but also in HCT values. Similarly, findings from the ECLAP study showed that leukocytosis was significantly associated with vascular risk in patients, regardless of treatment [32]. Furthermore, leukocytosis despite treatment with HU was associated with a higher risk of hematologic transformation (P = .004) and reduced survival (P = .007) in a study evaluating the utility of the European LeukemiaNet (ELN) response criteria in patients treated with HU (median follow-up, 7.2 years) [40•]. This last study also showed that, although there is no clear association between elevated platelet counts and major thrombotic events, failing to achieve an ELN-defined platelet response with treatment is associated with a higher risk of thrombosis and bleeding in patients with PV [40•]. These findings suggest that successful management of PV should include a response in leukocyte and platelet counts as well as achieving HCT < 45 %.
Another sign of inadequately controlled disease is that of burdensome and intractable symptoms, but these have not been linked to reduced survival or increased risk of thrombosis. Several studies have shown that patients with PV have a considerable symptom burden (Fig. 2) [20, 21, 46]. Fatigue is the most frequently reported symptom, based on studies using the Myeloproliferative Neoplasm Self-Assessment Form (MPN-SAF) [20–22], a questionnaire designed to capture patient-reported symptoms in patients with MPN [21] that has been used in several trials to measure symptomatic response to treatment. Pruritus, another common symptom, is characterized by strong, and in some cases, unbearable itching, stinging, tingling, or burning sensations, usually following contact with water, and is considered by many to be the most troublesome symptom [21, 38, 47]. Additionally, approximately 30 to 40 % of patients present with splenomegaly and splenomegaly associated symptoms during the course of the disease [16•].
Conventional Treatment Options for Patients with Inadequately Controlled PV
First-Line Treatment Options
Patients who are inadequately controlled do not always have many treatment options. Patients who show elevated HCT can increase the frequency of phlebotomies. However, frequent phlebotomies are not always well tolerated and may lead to iron deficiency, which has been associated with other complications such as restless legs syndrome [48] and cognitive dysfunction [49]. Alternatively, patients may also begin treatment with HU or IFN-α. In some instances, however, patients already being treated with HU and/or IFN-α may become resistant to or intolerant of these therapies. Approximately one fourth of patients with PV develop ELN-defined resistance (11 %) or intolerance (13 %) to HU (Table 2) [40•, 50], and are consequently at a high-risk of thrombosis and bleeding (34.7 and 16.8 per 1000 person-years, respectively) compared with those responding to HU per ELN criteria (28.3 vs 12.8 per 1000 person-years, respectively) [40•]. Furthermore, HU-resistance is associated with an increased risk of death and transformation to PPV-MF or sAML (P < .001) [40•]. There are currently no ELN criteria for IFN-resistance or -intolerance.
Patients resistant to or intolerant of HU or IFN-α require increasing treatment doses and/or more frequent phlebotomies (>1/month) to achieve HCT control. Other patients achieve HCT control but have uncontrolled leukocytosis or thrombocytosis and/or symptoms, or experience toxicity (e.g., mucositis, leg ulcers, and skin cancer with HU, or flu-like symptoms, neuropsychiatric symptoms, and autoimmune problems with IFN [51]). However, it is sometimes difficult to discern whether these toxicities are a consequence of disease progression and/or cytoreductive treatment.
Second-Line Treatment Options
IFN-α is the ELN-recommended second-line therapy in patients who have become resistant to or intolerant of HU [27] in part because it is not considered leukemogenic [43], an important characteristic considering that some drugs administered after HU may increase the risk of patients developing sAML [43], although the leukemogenic effect of HU itself remains unproven. However, IFN-α has never been formally assessed in a randomized phase 3 study as second-line therapy in HU-resistant or HU-intolerant patients. Studies have shown that IFN-α is very effective in reducing rates of thrombosis [52, 53], controlling symptoms [54, 55], and eliciting complete hematologic response [55, 56] or substantial decreases in JAK2 V617F allele burden [56]. However, IFN-α is associated with significant treatment-related toxicities (e.g., flu-like symptoms, depression, autoimmune disorders) [18] leading to permanent discontinuation in 20 to 40 % of patients [18, 57] and preventing its use in elderly patients and in those with some preexisting psychological and immune disorders [4, 58].
A newer pegylated formulation (PEG-IFN-α; Pegasys) has been reported to be more tolerable, to result in high rates of hematologic responses, and to reduce the JAK2 V617F allele burden in phase 2 trials of patients with PV who are either treatment naive or have been previously treated with phlebotomies or cytoreductive treatment [58, 59]. In one study, all evaluable patients who were treated first line (37/40) had a hematologic response, with 95 % achieving complete clinicohematologic response; complete molecular response was achieved in seven patients [58]. Overall, 24 % of patients discontinued treatment due to toxicity. In a second study in the second-line setting (PV, n = 40; essential thrombocythemia [ET], n = 39), the complete hematologic response was 76 % and the complete molecular response was 18 % in patients treated with PEG-IFN-α-2a [59]; 20 % of patients discontinued due to treatment-related adverse events. PEG-IFN-α is currently being tested in two phase 3 trials for the treatment of PV and, in the future, may become a viable option for some patients [60]. A randomized, open-label trial through the Myeloproliferative Disorders Research Consortium is evaluating the safety, toxicity, and tolerability of PEG-IFN-α–2a vs HU in high-risk patients with PV or ET (ClinicalTrials.gov, NCT01259856). The primary outcome will be hematologic response rates in the two study arms. PROUD-PV (Pegylated Interferon Alpha-2b Versus Hydroxyurea in Polycythemia Vera; ClinicalTrials.gov, NCT01949805), the second phase 3 study, is comparing the efficacy and safety of the novel monopegylated IFN-α-2b against HU in high-risk JAK2 V617F-positive PV. The primary outcome is peripheral blood count remission and normal spleen size after 1 year of treatment. However, neither of these studies is evaluating the use of IFN-α in patients who are HU-resistant or HU-intolerant.
Anagrelide is another recommended second-line therapy for the treatment of PV and is also considered nonleukemogenic. Anagrelide, however, only has platelet-reducing activity [27], and its combination with HU may be necessary in patients with progressive disease or those with uncontrolled HCT, leukocytes, symptoms, or platelet counts. The longest follow-up study of HU plus anagrelide found that a low-dose combination in patients with PV or ET whose disease was resistant or refractory to single-agent HU or anagrelide led to complete remission in 8 of 12 and partial responses in 3 of 12 patients, with a median platelet count reduction of 45 % [61]. Additionally, the low-dose combination therapy was associated with few adverse events. However, anagrelide is not approved in the EU as combination therapy and is only approved as treatment for MPN in patients with ET. Additionally, in patients with ET, anagrelide treatment has been associated with cardiac toxicity, increased bleeding when combined with aspirin, and an increased risk of transformation to secondary MF [62].
Busulfan has also been recommended as a second-line therapy in patients who are HU refractory [25] because it may lead to durable hematologic responses and was shown to reduce the JAK2 V617F allele burden, albeit in a small patient cohort [63]. Busulfan was recently evaluated retrospectively as second-line therapy in patients with PV or ET showing signs of resistance to HU (n = 36) [64]. Complete clinicohematologic response was achieved in 83 % of patients with an 87 % probability of sustained CR at 1 year and 62 % at 2 years. Fifty percent of patients discontinued because they achieved clinicohematologic response; others discontinued due to hematologic toxicity (n = 8) and transformation to sAML (n = 1). The rate of partial molecular response in evaluable patients with PV was 60 % (n = 3/5); no patient achieved a complete molecular response. At 2 years, the probability of survival was 85 % and the probability of thrombosis 11 %. However, although transformation to sAML was only seen in 2 of these 36 patients, there is significant concern regarding leukemogenicity when busulfan is given following treatment with HU [43]; thus, busulfan continues to be recommended only in patients >70 years of age [27].
Pipobroman or phosphorus 32 (32P) might also be considered [27], although both agents are not widely available in all countries and only a few patients are treated with these agents. A recent, large multicenter study analyzing survival and leukemic transformation in 1545 patients with PV reported that approximately 11 % of assessed patients had been treated with pipobroman as a single agent or in combination with other therapies; 4.2 % had been treated with alkylating drugs, including 32P [16•]. Like busulfan, these agents may be leukemogenic [39, 40•, 43, 64], and in the case of pipobroman, patients treated with this agent have a cumulative incidence of treatment-related myelodysplastic syndrome/sAML of 13, 34, and 52 % at 10, 15, and 20 years (P = .004), respectively [65]. Therefore, these therapies are usually reserved for older patients or those with a short life expectancy [39, 40•, 43, 64].
Patients with persistent leukocytosis may be difficult to treat, especially those who are also HU-resistant or HU-intolerant. Although a response definition for patients with leukocytosis and/or thrombocytosis has been established by the ELN [66], there are no clear recommendations for which therapy to use when leukocytosis or thrombocytosis persists despite treatment. Options include IFN-α, busulfan, and pipobroman, as discussed earlier. Most patients should continue taking low-dose aspirin as antiplatelet therapy, based on the findings from the European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study [31]. However, aspirin should probably be withdrawn if a patient’s platelet count exceeds 1500 × 109/L [27] because patients with extreme thrombocytosis may be at risk of developing an acquired von Willebrand defect and in turn, aspirin-associated bleeding [25]. In patients with extremely elevated platelet counts, treatment with cytoreductive agents (e.g., anagrelide plus HU) may be required before aspirin is instituted, but it is important to note that the concomitant use of anagrelide and aspirin may be associated with increased bleeding events [67]. For those patients with severe PV-related symptoms, there is little and conflicting evidence regarding efficacy of current treatment options and their ability to impact symptom burden [20, 21, 36, 38, 56, 68]. However, very few of these studies prospectively tested the efficacy of standard therapies in treating PV-associated symptoms or evaluated disease burden in the same patients before and after treatment, making it difficult to accurately determine treatment-related changes in symptom burden.
Ruxolitinib
Although not currently a part of the ELN guidelines, the JAK1/JAK2 inhibitor ruxolitinib was recently approved for the treatment of patients with PV who are resistant to or intolerant of HU. This approval was based on the findings of the phase 2 Study 256 [69] and the phase 3 RESPONSE (Randomized Study of Efficacy and Safety in Polycythemia Vera with JAK Inhibitor INCB018424 versus Best Supportive Care) study [70••]. Results from Study 256 (n = 34; ClinicalTrials.gov, NCT00726232) showed that ruxolitinib led to responses in blood counts, including HCT, meaningful reductions in spleen size, and improvements in symptoms in patients with PV who were HU-resistant or HU-intolerant [69]. Most patients (97 %) achieved HCT <45 % without phlebotomy and normalization of leukocytosis (73 and 76 % of patients with baseline leukocyte count >15 × 109 and >10 × 109/L, respectively) [69]. Additionally, 74 % of patients with elevated platelets achieved a sustained platelet response (<400 × 109/L). Thrombocytopenia and anemia were the most common adverse events, with grade 3/4 events occurring in 9 % of patients each; both adverse events were managed with dose modifications.
Based on these results, the phase 3 RESPONSE study (N = 222; Clinicaltrials.gov, NCT01243944) was initiated [70••]. RESPONSE assessed the efficacy and safety of ruxolitinib vs standard therapy in patients with PV who had an inadequate response to or had unacceptable side effects from HU (i.e., HU-resistant or HU-intolerant) and who had splenomegaly. The primary endpoint, a composite of the percentage of patients who achieved both HCT control without phlebotomy between weeks 8 and 32 and a ≥35 % reduction in spleen volume from baseline at week 32, was achieved in 21 % of patients in the ruxolitinib group vs 1 % of those receiving standard therapy (P < .001). Higher proportions of patients in the ruxolitinib arm achieved HCT control (60 vs 20 %). Additionally, patients in the ruxolitinib arm required fewer phlebotomies to maintain HCT control compared with those who received standard therapy (i.e., the “best available therapy” arm of the trial). A total of 19.8 and 62.4 % of patients receiving ruxolitinib or standard therapy, respectively, underwent ≥1 phlebotomy; 2.8 and 20.2 %, respectively, underwent ≥3 phlebotomies. Furthermore, patients receiving ruxolitinib had fewer occurrences of thrombotic events (1 [portal vein thrombosis] vs 6 [myocardial infarction, deep vein thrombosis, pulmonary embolism, splenic infarction, thrombophlebitis, and thrombosis], respectively). However, neither the number of phlebotomies nor the rate of thrombotic events were predefined primary endpoints, limiting their interpretation.
Ruxolitinib was well tolerated, and the most common adverse events were anemia (grade 3/4, 2 %) and thrombocytopenia (grade 3/4, 5 %), consistent with findings from Study 256; the corresponding rates in the standard therapy arm were 0 and 4 %. Herpes zoster infection (all cases were grade 1 or 2) was reported in 6.4 % of patients randomized to ruxolitinib and 0 % of patients in the standard therapy arm. Overall, the rate of infections was 41.8 % in the ruxolitinib group and 36.9 % in the standard therapy arm. Nonmelanoma skin cancer was diagnosed in four patients in the ruxolitinib group and in two patients in the standard therapy arm; the majority of patients (all but 1) had a history of nonmelanoma skin cancer or precancerous skin lesions. Three patients randomized to ruxolitinib developed PPV-MF and 1 patient received a diagnosis of sAML. PPV-MF developed in one patient assigned to standard therapy; additionally, two patients received a diagnosis of PPV-MF after crossover, one of whom had progression to sAML. This is perhaps disappointing, as these are events that ideally would have been prevented or reduced. Overall, ruxolitinib proved superior to standard therapy in inducing HCT control, reducing spleen size, and improving symptoms and QOL in this patient population.
Despite these intriguing results, there were some limitations in the design of the study. Eligible patients were required to have splenomegaly, a disease characteristic that is infrequent and is usually associated with advanced disease. Additionally, 59 % of patients in the standard therapy arm were treated with HU although most patients (54 %) were considered HU-intolerant. Furthermore, clinicohematologic response according to ELN criteria (HCT < 45 % without phlebotomy, response in platelet and leukocyte count, normal spleen size, and no disease-related symptoms) [71] was not reported, although more patients treated with ruxolitinib than those treated with standard therapy achieved a response in HCT, platelet count, and leukocyte count (24 vs 9 %). Moreover, BM biopsies were not requested at study entry and were used only to confirm progression to PPV-MF, preventing the reporting of normalization of BM features as well as the identification of those who may have developed PPV-MF (although PPV-MF that required phlebotomy) at or shortly after study enrollment. In addition, the impact of study endpoints, in particular reduction of splenomegaly and freedom from phlebotomy, could be questioned in terms of their disease modification.
Additional trials evaluating ruxolitinib in this patient population include the RESPONSE2 study (Clinicaltrials.gov, NCT02038036) and the MAJIC (Randomised Study of Best Available Therapy Versus JAK Inhibition in Patients With High Risk Polycythaemia Vera or Essential Thrombocythaemia Who Are Resistant or Intolerant to Hydroxycarbamide) study (EudraCT, 2011-005279-18). RESPONSE2 is a phase 3 trial that will study the efficacy and safety of ruxolitinib in patients without splenomegaly who are HU-resistant or HU-intolerant. The MAJIC study is a UK-specific, randomized, phase 2 study that will assess the efficacy and safety of ruxolitinib vs best available therapy in patients with high-risk PV (or ET) who are HU-resistant or HU-intolerant and who have an enlarged spleen. Alternatively, patients may be candidates for investigational studies with JAK inhibitors other than ruxolitinib, as well as deacetylase (HDAC) inhibitors.
The RESPONSE study suggests that ruxolitinib may also be an effective treatment for persistent leukocytosis and thrombocytosis. At week 32, more patients randomized to ruxolitinib had a leukocyte count ≤15 × 109/L than patients randomized to standard therapy (70 vs 43 %, respectively) [72]. Similarly, more patients receiving ruxolitinib had a platelet count ≤600 × 109/L (82 vs 64 %) [72]. Complete hematologic remission (CHR; normalization in HCT, leukocytes, and platelets) was achieved in 24 % of ruxolitinib-treated patients; in contrast, only 9 % of patients treated with standard therapy achieved this result (P = .003). Given these data, JAK inhibitors could also play an important role in the treatment strategy for patients with persistent leukocytosis and/or thrombocytosis. However, the role of leukocytosis in PV remains unclear, and it is still unknown whether therapies that control leukocytosis and thrombocytosis, including JAK inhibitors, will reduce the risk of thrombotic events, progression to PPV-MF and sAML, and consequently, the risk of death. Controlled studies are still needed to determine if current management of PV requires modification based on leukocyte and platelet count. Nonetheless, given that leukocytosis is directly associated with an increased risk of thrombosis [45], a response in leukocyte count is an important aspect of the response to therapy in patients with PV.
Likewise, the use of ruxolitinib to treat patients with PV who have severe symptoms was supported by findings from Study 256 [69] and the RESPONSE study [70••]. In Study 256, symptom analyses included the proportion of patients with a 50 % reduction in symptoms from baseline and those patients with complete resolution of pruritus, night sweats, and bone pain. Clinically meaningful improvements in symptoms were seen within 4 weeks of receiving ruxolitinib and were maintained through week 144 [69]. In RESPONSE, ruxolitinib-treated patients had significant improvements in the 14-item MPN-SAF total symptom score (TSS) compared with those treated with conventional therapy [70••]. Patients receiving ruxolitinib reported improvements in all individual symptoms, especially in those belonging to the cytokine symptom cluster (fatigue, itching, muscle ache, night sweats, and sweating while awake). In contrast, patients treated with standard therapy experienced no change or worsening of their symptoms.
Despite these encouraging results, the phase 3 RELIEF (Randomized Switch Study From Hydroxyurea to Ruxolitinib for RELIEF of Polycythemia Vera Symptoms) study (ClinicalTrials.gov, NCT01632904) showed no significant difference between ruxolitinib and HU for the treatment of persistent symptoms [73]. In RELIEF, patients were randomized to receive ruxolitinib 10 mg twice a day plus placebo (HU; n = 54) or HU plus placebo (ruxolitinib; n = 56). More patients in the ruxolitinib arm compared with the HU arm (43.4 vs 29.3 %) achieved the study’s primary endpoint (a ≥50 % reduction in the MPN-SAF TSS cytokine cluster at week 16). However, despite this positive trend in favor of ruxolitinib, it was not statistically significant (P = .139). Although unclear, it is possible that differences in patient populations may have led to such differences in the RELIEF and RESPONSE study results. For example, patients in RESPONSE were HU-resistant or HU-intolerant and were required to have splenomegaly, whereas patients in RELIEF reported symptoms despite treatment with HU and had no splenomegaly. Additionally, differences in trial design (open vs blinded) may have led to the observed differences in study results. Findings from the RESPONSE2 study may help provide answers to some of these unresolved questions.
Other Experimental Strategies
Other JAK inhibitors currently being evaluated in clinical trials for the treatment of MPNs include momelotinib (CYT387) and pacritinib (SB1518). At the time of writing pacritinib has been put onto a full clinical hold by the FDA for bleeding and potential cardiac toxicities. Both have proved encouraging in phase 2 trials, and phase 3 studies in MF are ongoing. Momelotinib is being assessed in a randomized phase 2 study in patients with PV or ET who are JAK-inhibitor naive (Clinicaltrials.gov, NCT01998828); however, its clinical efficacy in PV is unknown. There are currently no studies evaluating pacritinib in this setting.
HDAC inhibitors prevent proliferation of tumor cells by inducing cell-cycle arrest, differentiation, and/or apoptosis, and are therefore desirable candidates in treating malignancies [74]. At least two HDAC inhibitors (vorinostat and givinostat) have been evaluated in patients with PV. However, vorinostat was not well tolerated (44 % of patients discontinued due to adverse events) and is no longer being evaluated [75]. Givinostat has specificity for JAK2 V617F-mutated cells and was tested in a phase 2 study in patients with JAK2 V617F-positive PV who were HU-resistant or HU-intolerant (n = 12) [76]. Overall, givinostat was well tolerated; 75 % of patients had a reduction in splenomegaly, 54 % had a clinical response after 12 weeks on treatment, and pruritus disappeared in all but one patient. Givinostat was later evaluated in a phase 2 study of patients with PV (n = 44) who showed no response when treated with maximum tolerated doses of HU [77]. Patients received givinostat (50 or 100 mg/day) in combination with HU at the maximum tolerated dose. The combination was well tolerated; only 18 % of patients discontinued treatment: 11 % during the first 12 weeks of treatment and 7 % between weeks 12 and 24. After 12 weeks of treatment, complete or partial response according to ELN criteria was observed in 55 and 50 % of patients receiving 50 or 100 mg givinostat, respectively. Improvements in pruritus were also observed [77]: 58 % of patients with grade 3/4 pruritus showed an improvement or symptom resolution (grade ≤ 1).
Two additional studies with givinostat are currently recruiting. The first study is a dose-finding phase 1/2 nonrandomized study assessing the safety and efficacy in patients with JAK2 V617F-positive PV (ClinicalTrials.gov, NCT01901432). The primary outcomes will be determination of the maximum tolerated dose, preliminary efficacy, safety, and tolerability. The second study is a multicenter, open-label study evaluating the safety, tolerability, and efficacy of givinostat in patients with PV, ET, or MF who have completed givinostat treatment in a study of chronic MPN or are participating in a compassionate use program with givinostat (ClinicalTrials.gov, NCT01761968). The primary outcomes will be long-term safety and efficacy. Efficacy will be determined based on achievement of complete and partial response rate according to ELN response criteria.
Uncovering the Molecular Underpinnings of PV
Despite such great advancements, there is still more to be accomplished, specifically in further unraveling the genetic basis of PV, preventing thrombosis in patients on treatment, and preventing progression to PPV-MF and sAML. To help achieve these goals, many groups are working on determining the genetic underpinnings of PV, and at least 102 genes outside the JAK2 pathway that have differential regulation in patients with PV have already been identified [78]. Furthermore, clonal dominance at the progenitor level seems to be present in those patients carrying mutations other than those in JAK2 and is characteristic of evolution of the disease [79]. In one report of a patient with TET2/JAK2-positive disease, PEG-IFN-α-2a therapy led to complete molecular response of the JAK2 clone without reduction of the TET2 clone [80]. This suggests that other signaling pathways are playing an additional (or complementary) pathogenetic role in PV and that targeted therapies against genes involved in these other pathways, as monotherapies or in combination with ruxolitinib or standard therapies, may aid in the treatment of PV. Additionally, a recent study [81] showed that in patients with PV (and other MPN), the order in which mutations are acquired may have a substantial effect on disease features and the response to therapy. Ortmann and colleagues showed that in patients with PV who carry both TET2 and JAK2 mutations, those who acquired JAK2 mutations first had a more proliferative response to JAK2 V617F but had JAK2-mutant progenitor cells that were more responsive to ruxolitinib in vitro. Interestingly, these patients were also at an increased risk of thrombosis. Further studies will help guide development of molecularly targeted therapies as these become available.
Conclusion
In a proportion of patients with PV, it is important to recognize that their response to current therapies is inadequate and alternative therapeutic options are needed. We identified these patients as those who, despite treatment, have thrombotic events, elevated HCT, leukocytosis/thrombocytosis, splenomegaly, and burdensome symptoms, and discussed evidence for these being associated with significant events as well as different treatment options available. The discovery of JAK2 mutations as driver mutations for this disease paved the way for the development of targeted therapies, and now, the first JAK1/JAK2 inhibitor, ruxolitinib, has been approved for the treatment of patients with PV and inadequate response to or intolerance of HU. Its approval may soon lead to the inclusion of JAK inhibitors in the treatment strategy for PV (Table 3). Conventional therapy is less costly than JAK inhibitors, but as discussed, it is not always an optimal treatment. It is clear that for some patients, JAK inhibitors may be more efficacious in alleviating disease burden and improving QOL, and these benefits may ultimately outweigh the cost of therapy. Overall, as new therapies are developed, it will become imperative to identify the appropriate agent for each patient population that is able to prevent thrombotic events without risk of transformation to PPV-MF and sAML.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the world health organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
Stuart BJ, Viera AJ. Polycythemia vera. Am Fam Physician. 2004;69:2139–44.
Hensley B, Geyer H, Mesa R. Polycythemia vera: current pharmacotherapy and future directions. Expert Opin Pharmacother. 2013;14:609–17.
Passamonti F. How I, treat polycythemia vera. Blood. 2012;120:275–84.
Vannucchi AM. Insights into the pathogenesis and management of thrombosis in polycythemia vera and essential thrombocythemia. Intern Emerg Med. 2010;5:177–84.
Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83.
Tefferi A. Mutations galore in myeloproliferative neoplasms: would the real Spartacus please stand up? Leukemia. 2011;25:1059–63.
Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68.
James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8.
Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61.
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90.
Kralovics R, Teo SS, Buser AS, et al. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood. 2005;106:3374–6.
Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–92.
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97.
Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117:755–61.
Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27:1874–81. This large multicenter retrospective study showed that life expectancy in patients with PV is significantly shorter than in patients without the disease. It identified predictors of poor survival and presented a prognostic model for patients with PV.
Martinelli I, De Stefano V. Rare thromboses of cerebral, splanchnic and upper-extremity veins. A narrative review. Thromb Haemost. 2010;103:1136–44.
Vannucchi AM. How I, treat polycythemia vera. Blood. 2014;124:3212–20.
Boiocchi L, Mathew S, Gianelli U, et al. Morphologic and cytogenetic differences between post-polycythemic myelofibrosis and primary myelofibrosis in fibrotic stage. Mod Pathol. 2013;26:1577–85.
Johansson P, Mesa R, Scherber R, et al. Association between quality of life and clinical parameters in patients with myeloproliferative neoplasms. Leuk Lymphoma. 2012;53:441–4.
Scherber R, Dueck AC, Johansson P, et al. The myeloproliferative neoplasm symptom assessment form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401–8.
Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international internet-based survey of 1179 MPD patients. Cancer. 2007;109:68–76.
Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23:2224–32.
Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood. 2008;111:3383–7.
Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am J Hematol. 2015;90:162–73.
Cervantes F, Passamonti F, Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia. 2008;22:905–14.
Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761–70.
Finazzi G, Barbui T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia. 2008;22:1494–502.
Barbui T, Finazzi MC, Finazzi G. Front-line therapy in polycythemia vera and essential thrombocythemia. Blood Rev. 2012;26:205–11.
Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368:22–33. This article details findings from the CYTO-PV clinical trial, the first randomized study to evaluate the recommended hematocrit target of < 45% in patients with PV. This study demonstrated that aggressively treating to a hematocrit target of < 45% was associated with a significantly lower rate of major thrombosis or death from cardiovascular causes , compared with a hematocrit target of 45% to 50%.
Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114–24.
Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109:2446–52.
Gangat N, Strand J, Li CY, Wu W, Pardanani A, Tefferi A. Leucocytosis in polycythaemia vera predicts both inferior survival and leukaemic transformation. Br J Haematol. 2007;138:354–8.
Caramazza D, Caracciolo C, Barone R, et al. Correlation between leukocytosis and thrombosis in philadelphia-negative chronic myeloproliferative neoplasms. Ann Hematol. 2009;88:967–71.
De Stefano V, Za T, Rossi E, et al. Leukocytosis is a risk factor for recurrent arterial thrombosis in young patients with polycythemia vera and essential thrombocythemia. Am J Hematol. 2010;85:97–100.
Scherber R, Dueck A, Kiladjian JJ, et al. Conventional therapeutic options have limited impact on MPN symptoms: insights from a prospective analysis of the MPN-SAF TSS. Haematologica. 2012;97(suppl 1)[abstract 366].
Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30:4098–103.
Siegel FP, Tauscher J, Petrides PE. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am J Hematol. 2013;88:665–9.
Kiladjian JJ, Gardin C, Renoux M, Bruno F, Bernard JF. Long-term outcomes of polycythemia vera patients treated with pipobroman as initial therapy. Hematol J. 2003;4:198–207.
Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119:1363–9. This study demonstrated that patients meeting the ELN criteria for resistance to hydroxyurea have a substantial increase in the risk of death and the risk of hematologic transformation. These findings highlight the need for other therapeutic options for patients who are resistant to hydroxyurea.
Bonicelli G, Abdulkarim K, Mounier M, et al. Leucocytosis and thrombosis at diagnosis are associated with poor survival in polycythaemia vera: a population-based study of 327 patients. Br J Haematol. 2013;160:251–4.
Gangat N, Wolanskyj AP, McClure RF, et al. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia. 2007;21:270–6.
Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105:2664–70.
Alvarez-Larran A, Bellosillo B, Martinez-Aviles L, et al. Postpolycythaemic myelofibrosis: frequency and risk factors for this complication in 116 patients. Br J Haematol. 2009;146:504–9.
Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126:560–1.
Abelsson J, Andreasson B, Samuelsson J, et al. Patients with polycythemia vera have the worst impairment of quality of life among patients with newly diagnosed myeloproliferative neoplasms. Leuk Lymphoma. 2013;54:2226–30.
Saini KS, Patnaik MM, Tefferi A. Polycythemia vera-associated pruritus and its management. Eur J Clin Invest. 2010;40:828–34.
Tobiasson M, Alyass B, Soderlund S, Birgegard G. High prevalence of restless legs syndrome among patients with polycytemia vera treated with venesectio. Med Oncol. 2010;27:105–7.
Kim J, Wessling-Resnick M. Iron and mechanisms of emotional behavior. J Nutr Biochem. 2014;25:1101–7.
Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148:961–3.
Sever M, Newberry KJ, Verstovsek S. Therapeutic options for patients with polycythemia vera and essential thrombocythemia refractory/resistant to hydroxyurea. Leuk Lymphoma. 2014;55:2685–90.
Zhang ZR, Duan YC. Interferon alpha 2b for treating patients with JAK2V617F positive polycythemia vera and essential thrombocytosis. Asian Pac J Cancer Prev. 2014;15:1681–4.
Heis N, Rintelen C, Gisslinger B, Knobl P, Lechner K, Gisslinger H. The effect of interferon alpha on myeloproliferation and vascular complications in polycythemia vera. Eur J Haematol. 1999;62:27–31.
Hasselbalch HC, Larsen TS, Riley CH, Jensen MK, Kiladjian JJ. Interferon alpha in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Status and Perspectives. Curr Drug Targets. 2011;12:392–419.
Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer. 2006;107:451–8.
Larsen TS, Iversen KF, Hansen E, et al. Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res. 2013;37:1041–5.
Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer. 2006;106:2397–405.
Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112:3065–72.
Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon alpha-2a. Blood. 2013;122:893–901.
The PROUD-PV study. In: Proud-PV website. AOP Orphan Pharmaceuticals AG. 2015. http://www.proud-pv.com/proud-pv-study.html. Accessed 24 Aug 2015.
Ahn IE, Natelson E, Rice L. Successful long-term treatment of Philadelphia chromosome-negative myeloproliferative neoplasms with combination of hydroxyurea and anagrelide. Clin Lymphoma Myeloma Leuk. 2013;13 suppl 2:S300–4.
Birgegard G, Besses C, Griesshammer M, et al. Treatment of essential thrombocythemia in europe: an observational study of 3649 high-risk patients in EXELS. Blood. 2014;124 [abstract 1846].
Kuriakose ET, Gjoni S, Wang YL, et al. JAK2V617F allele burden is reduced by busulfan therapy: a new observation using an old drug. Haematologica. 2013;98:e135–7.
Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol. 2014;93:2037–43.
Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29:3907–13.
Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: a ELN and IWG-MRT consensus project. Blood. 2013;121:4778–81.
Wagstaff AJ, Keating GM. Anagrelide: a review of its use in the management of essential thrombocythaemia. Drugs. 2006;66:111–31.
Silver RT. Recombinant interferon-alpha for treatment of polycythaemia vera. Lancet. 1988;2:403.
Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120:513–20.
Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426–35. This article reports the primary findings from the RESPONSE study, the first phase 3 randomized study evaluating the efficacy and safety of a JAK inhibitor (ruxolitinib) vs standard therapy in patients with PV who had an inadequate response to or had unacceptable side effects from hydroxyurea. Ruxolitinib was superior to standard therapy in controlling hematocrit, reducing spleen volume, and improving disease-associated symptoms, findings which formed the basis for the approval of ruxolitinib for the treatment of patients with PV who are resistant to or intolerant of HU.
Barosi G, Birgegard G, Finazzi G, et al. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference. Blood. 2009;113:4829–33.
Harrison CN, Masszi T, Zachee P, et al. Complete hematologic control with ruxolitinib in patients with polycythemia vera (PV) resistant to or intolerant of hydroxyurea. European Hematology Association 20th Congress; 11–14 Jun 2015; Vienna, Austria [abstract E1353].
Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxyurea therapy versus switching to ruxolitinib in patients with polycythemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Poster presented at: 56th ASH Annual Meeting and Exposition; 6–9 Dec 2014; San Francisco, CA [abstract 3168].
Vigushin DM, Coombes RC. Targeted histone deacetylase inhibition for cancer therapy. Curr Cancer Drug Targets. 2004;4:205–18.
Andersen C, Mortensen N, Vestergaard H, Bjerrum O, Klausen T, Hasselbalch H. A phase II study of vorinostat (MK-0683) in patients with primary myelofibrosis (PMF) and post-polycythemia vera myelofibrosis (PPV-MF). Haematologica. 2013;98(suppl 1) [abstract P279].
Rambaldi A, Dellacasa CM, Finazzi G, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150:446–55.
Finazzi G, Vannucchi AM, Martinelli V, et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol. 2013;161:688–94.
Spivak JL, Considine M, Williams DM, et al. Two clinical phenotypes in polycythemia vera. N Engl J Med. 2014;371:808–17.
Angona A, Alvarez-Larran A, Bellosillo B, et al. Hematopoietic clonal dominance, stem cell mutations, and evolutionary pattern of JAK2V617F allele burden in polycythemia vera. Eur J Haematol. 2014;94:251–7.
Kiladjian JJ, Masse A, Cassinat B, et al. Clonal analysis of erythroid progenitors suggests that pegylated interferon alpha-2a treatment targets JAK2V617F clones without affecting TET2 mutant cells. Leukemia. 2010;24:1519–23.
Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372:601–12.
Acknowledgments
Editorial assistance was provided by Karen Chinchilla, PhD, and was supported by Novartis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Andreas Reiter has participated as an advisory board member, has provided expert testimony, and has received consultancy fees, honoraria, and travel support from Novartis Pharma outside of the submitted work.
Claire Harrison has participated as an advisory board member and has received research funding, consultancy fees, honoraria, and travel support from Novartis Pharma outside of the submitted work. Dr. Harrison is a section editor for Current Hematologic Malignancy Reports.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Myeloproliferative Disorders
Rights and permissions
About this article

Cite this article
Reiter, A., Harrison, C. How We Identify and Manage Patients with Inadequately Controlled Polycythemia Vera. Curr Hematol Malig Rep 11, 356–367 (2016). https://doi.org/10.1007/s11899-016-0311-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-016-0311-8