
Abstract
The small intestine is a complex organ system that is vital to the life of the individual. There are a number of congenital anomalies that occur and present most commonly in infancy; however, some may not present until adulthood. Most congenital anomalies of the small intestine will present with obstructive symptoms while some may present with vomiting, abdominal pain, and/or gastrointestinal bleeding. Various radiologic procedures can aid in the diagnosis of these lesions that vary depending on the particular anomaly. Definitive therapy for these congenial anomalies is surgical, and in some cases, surgery needs to be performed urgently. The overall prognosis of congenital anomalies of the small intestine is very good and has improved with improved medical management and the advent of newer surgical modalities. The congenital anomalies of the small intestine reviewed in this article include malrotation, Meckel’s diverticulum, duodenal web, duodenal atresia, jejunoileal atresia, and duplications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Congenital anomalies of the gastrointestinal tract can affect any portion of the gastrointestinal tract from the esophagus to the anus. These include tracheoesophageal atresia, gastroschisis, omphalocele, colonic duplication, Hirschsprung’s disease, and imperforate anus, as well as a number of anomalies of the small intestine. The major congenital anomalies of the small intestine include malrotation, Meckel’s diverticulum, duodenal web, duodenal atresia, jejunoileal atresia, and duplications. Many of these will present in the neonatal period with obstruction. One can also present later on in childhood or adulthood with vomiting, abdominal pain, abdominal mass, or with gastrointestinal bleeding. Some of these entities can be life threatening making it imperative that health care providers be aware of these so as to be able to rapidly make the correct diagnosis and provide appropriate therapy. It is important to realize that many of these anomalies of the small intestine are also associated with other congenital anomalies. Some of these anomalies were associated with high mortality rates in the first half of the 1900s but now, due to improved medical therapy including total parenteral nutrition and newer surgical modalities, the prognosis of congenital anomalies of the small intestine is very good. This article will review the major congenital anomalies of the small intestine including malrotation, Meckel’s diverticulum, duodenal web, duodenal atresia, jejunoileal atresia, and duplications.
Malrotation
Malrotation is a broad term that encompasses a number of rotational and fixation abnormalities of the intestines. Mall, in 1898, described the embryonic origins of malrotation [1]. Proper development of the midgut involves a complex process of herniation of the gut out of the abdominal cavity prior to rotating 270° counterclockwise around the axis of the superior mesenteric artery. The midgut then reenters the abdomen around 10–12 weeks gestation, undergoing fixation with the duodenojejunal junction being fixed to the posterior abdominal wall to the left of the spine, the ligament of Treitz, while the cecum becomes fixed in the right lower quadrant. Fixation to the posterior abdominal wall is present from the ligament of Treitz to the cecum and prevents intestinal torsion around the vascular supply. Absence of this fixation predisposes one to a midgut volvulus. Malrotation describes any abnormal rotation or fixation in the above process and can be further characterized as non-rotation, incomplete (mixed) rotation, reversed rotation, or mesocolic hernia [2]. It is estimated that up to 1 in 200 live births have an asymptomatic rotational anomaly [3]; however, symptomatic malrotation occurs less frequently, 1 in 6000 live births [3, 4]. The male to female ratio is two to one. Over a third of cases are associated with a congenital anomaly. These include intestinal atresia or web, Meckel diverticulum, intussusception, Hirschsprung disease, mesenteric cyst, anomalies of the extrahepatic biliary system, and congenital heart disease [5]. Those with congenital diaphragmatic hernia, gastroschisis, and omphalocele all have malrotation. A recent retrospective review noted the association of malrotation in patients with an anorectal malformation and two or more VACTERL (vertebral, anorectal, cardiac, tracheoesophageal fistula, renal, radial, limb) anomalies [6].
While one can present at any age with a malrotation, it is most common in the first several years of life with the majority in the first month of life [7•]. Approximately 40 % of cases present in adulthood [8]. Infants typically present with bilious emesis secondary to a midgut volvulus. Due to the narrow mesenteric base, the intestine twists around the superior mesenteric artery resulting in vascular compromise and ensuing necrosis of the gut if it is not surgically corrected rapidly. Therefore, bilious emesis in an infant is a medical emergency that demands prompt attention. Other infants may present with gastroesophageal reflux. Older children can present acutely or with recurrent abdominal pain, vomiting, and/or poor weight gain [9]. Some cases with be asymptomatic and be an incidental diagnosis on upper gastrointestinal contrast studies done for other clinical reasons.
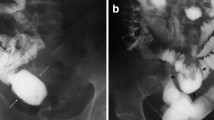
The definitive imaging for malrotation is an upper gastrointestinal series (UGI). Normally, the duodenojejunal junction is located at the level of the ligament of Treitz to the left of the spinal pedicle [10]. A rotational disorder is present when the duodenojejunal junction is not in this location. If a volvulus is present, a plain abdominal radiography may reveal distention of the stomach and proximal duodenum along with minimal or absent small bowel gas due to complete or partial obstruction. However, the most common finding on plain film in this situation is a normal bowel gas configuration. A “double bubble sign” is the classic hallmark of a duodenal obstruction denoting dilated proximal duodenum and stomach. These findings are similar to other etiologies of proximal obstruction, such as duodenal atresia or stenosis. Performance of an UGI in the presence of malrotation with a volvulus may reveal a “corkscrew” appearance or “coiled spring” appearance as the contrast passes into the distal duodenum and proximal jejunum [11] (Fig. 1). There may also be a “bird-beaked” appearance at the level of the obstruction from luminal narrowing.

Upper gastrointestinal series in a newborn infant with malrotation and midgut volvulus. Note absence of C-loop.
Studies have investigated the use of ultrasonography to diagnose malrotation, looking for retromesenteric localization of the third portion of the duodenum [12] or reversed mesenteric vessel position and “whirlpool sign” [13]. Esposito et al. suggest reversed position of the superior mesenteric artery and vein has low sensitivity in isolated malrotation without volvulus. However, the whirlpool sign seems to have high sensitivity (81 %) and specificity for the diagnosis of volvulus. A recent prospective study compared sonography to UGI to diagnose malrotation, focusing on a combination of three sonographic findings: inversion of the superior mesenteric artery and vein, the whirlpool sign, and an intraperitoneal transverse duodenum [14]. They concluded that the presence of all three of these findings may make sonography superior to UGI to evaluate patients with suspected malrotation. However, there has been at least one case reported with normal retroperitoneal positioning of the third part of the duodenum; thus, this finding is not a definitive way to rule out malrotation [15]. Ultimately, the use of sonography depends on the availability of an experienced radiologist or technician, which may not be available at all centers or at all hours of the day. One of the most important factors in managing these patients is close communication between the radiologist and surgeon [16]. For now, UGI remains the gold standard to diagnosis malrotation with or without volvulus [17•]. A barium enema may be helpful in equivocal cases. Normally, the cecum is located in the right lower quadrant. In the majority of those with malrotation, the cecum will be malpositioned too, but finding the cecum in the normal location does not rule out a malrotation.
Once symptomatic malrotation is diagnosed it must be surgically corrected. If there is confirmed or suspected volvulus, then emergent surgery is indicated to prevent further injury to the intestine. The Ladd procedure is the standard surgical operation used to repair malrotation with or without volvulus. This was first described by William Ladd in 1936 which reduces the volvulus if present, divides peritoneal bands causing the obstruction, repositions the small and large intestines to the right and left side of the abdomen, respectively, and excises the appendix [18].
When malrotation is found incidentally in an asymptomatic child, one should consider pursuing a Ladd procedure given the potentially devastating outcome of a volvulus; however, this is controversial. Recently, the American Pediatric Surgical Association published recommendations, based on a systematic review, for the diagnosis and management of asymptomatic malrotation [17•]. They concluded that there is minimal evidence to support screening asymptomatic patients with congenital heart disease or patients with heterotaxy syndrome. In addition, one should consider operating on younger patients who are asymptomatic, while it may be appropriate to observe older asymptomatic patients. Regarding patients with congenital heart disease with asymptomatic malrotation, it was recommended to treat the heart defect prior to performing a Ladd procedure.
The Ladd procedure can be performed laparoscopically or as an open procedure. Performance of the Ladd’s procedure laparoscopically for malrotation without volvulus may decrease the time until enteral nutrition is re-introduced and reduce the length of stay compared to an open procedure [19–21]. Ooms noted that patients who underwent laparotomy had more complications, including small bowel obstructions, and longer hospital stays compared to laparoscopy, suggesting that a laparoscopic approach should be considered first in an otherwise stable patient [22]. This may not be the case in neonates due to concerns of difficult visualization from the limited intraperitoneal working space [23].
Meckel’s Diverticulum
Meckel’s diverticulum is the most common congenital anomaly of the gastrointestinal tract. This is named after Johann Meckel who first described its embryonic origin in the 1800s [24]. It is found in approximately 2 % of the population with a male to female ratio of two to four to one [25]. This abnormality develops due to incomplete obliteration of the vitelline, or omphalomesenteric, duct that results in an out pouching of ileum on the anti-mesenteric side (Fig. 2), 40–100 cm proximal to the ileocecal valve. A Meckel’s is a true diverticulum, containing all three layers of the bowel wall, mucosa, muscularis, and serosa. The diverticula may contain ectopic mucosa, with gastric mucosa being the most common which is present in 50–60 % of the cases, but they may also contain pancreatic, duodenal, or rarely colonic or hepatobiliary mucosa [26]. Ectopic gastric mucosa secretes acid that can lead to ulceration of adjacent ileal mucosa and subsequently result in bleeding. The “rule of 2 s” has been associated with Meckel’s diverticulum; it occurs in 2 % of the population; it is located about 2 ft from the ileocecal valve; it is typically about 2 in. long and presents in those less than 2 years of age 50 % of the time.

Meckel’s diverticulum. Reprinted by permission from Batool, A, Gastrointestinal bleeding, in Coppola, CP, Kennedy, AP, and Scorpio, RJ, Pediatric surgery: Diagnosis and treatment, p. 270 (Fig. 1), Copyright 2014, Springer International Publishing.
The majority, 85–95 %, of patients with a Meckel’s diverticulum are asymptomatic [27]. The most common presentation of a Meckel’s in children is painless rectal bleeding that can be acute or chronic [26]. The bleeding is secondary to peptic ulceration due to secretion of acid from the ectopic gastric mucosa. The ulcer is located adjacent to or slightly distal to the diverticulum. Typically, the bleeding, which can be brisk, consists of dark red or maroon blood but melena is also possible.
Small bowel obstruction is the second most common presentation of a child born with Meckel’s diverticulum [27]. There are five sources of obstruction. (1) A Meckel’s diverticulum may be the lead point for ileocolic intussusception. In fact, Meckel’s diverticulum is the most common anatomic lead point for such intussusception and may lead to recurrent obstruction following successful hydrostatic reduction. (2) Prolapse of the diverticulum through a persistent omphalomesenteric effect may result in complete intestinal obstruction. (3) Volvulus may occur involving the associated ileum around a persistent fibrous band emanating from the tip of the diverticulum and ending at the umbilicus, following the path of the omphalomesenteric duct. (4) A loop of small intestine may become involved within an internal hernia produced by an aberrant right vitelline artery or fibrous band arising from the associated mesentery. (5) Lastly, a Meckel’s diverticulum may become incarcerated within an inguinal hernia (Littre’s hernia). All of these entities require prompt diagnosis and surgical repair including resection of the diverticulum and possibly the associated small intestine should there be vascular compromise. Meckel’s diverticulitis can also perforate resulting in peritonitis and may mimic an acute appendicitis. Children with trisomy 18 are at higher risk for this [28].
The diagnostic approach depends on the presenting symptoms. For the patient presenting with bleeding, the most sensitive test to diagnose a Meckel’s diverticulum is a technetium-99 m pertechnetate scintigraphy, Meckel’s scan. This test has an 85 to 95 % sensitivity and specificity in children [29]. The sensitivity and specificity appear to be lower in adults [30]. Technetium-99 m pertechnetate is administered intravenously and serial abdominal images are obtained using a gamma camera over 60 min. Parietal cells of the gastric mucosa take up technetium-99 m pertechnetate, revealing ectopic gastric mucosa in the right lower quadrant. There is excretion of technetium-99 m pertechnetate by the kidneys resulting in ureters and bladder being visualized as well. Administration of histamine-2 receptor antagonists increases the uptake and retention of the pertechnetate by the gastric mucosa enhancing the detection of a Meckel’s diverticulum [31, 32]. This pharmacologic enhancement may be beneficial for patients with an initial negative scan with a high index of clinical suspicion. False positive results have been noted in patients with intussusception, hydronephrosis, arteriovenous malformation, inflammatory bowel disease, and intestinal duplication due to heterotopic gastric mucosa. False-negative scans can be due to suboptimal examination technique, impaired blood supply to the bowel, or insufficient mass of ectopic gastric tissue to take up the isotope. Patients with a Meckel’s who do not have ectopic gastric mucosa will have a negative scan.
Additional alternatives to investigate gastrointestinal bleeding include capsule endoscopy and double balloon enteroscopy. Capsule endoscopy has identified a Meckel’s in patients with unexplained gastrointestinal bleeding [33, 34], although one case did report retention of the capsule with co-occurring enteroliths [35]. Retrograde (per rectum) and prograde (per os) double balloon enteroscopy have been described as a means of visualizing a Meckel’s diverticulum in patients with gastrointestinal bleeding. In addition, this has also aided in therapy [36, 37]. Double balloon enteroscopy has been demonstrated to be relatively safe in the hands of experienced professionals and can be considered if other diagnostic procedures have failed to reveal a suspected Meckel’s [38]. A retrospective study found diagnostic yield for double balloon enteroscopy to be significantly greater than capsule endoscopy [39]. Arteriography can be utilized to identify the source of bleeding if the Meckel’s scan is negative but it requires fairly brisk bleeding, 1 ml/min. The upper GI and small bowel follow through series is typically not very helpful in this situation. Finally, in the setting of a negative work up and a high clinical suspicion, some patients may be considered for a diagnostic laparoscopy given that this is minimally invasive and safe in the hands of an experienced pediatric surgeon. In those presenting with obstructive symptoms, conventional radiographic approach is undertaken.
Once a symptomatic Meckel’s diverticulum has been identified, the standard of care is laparoscopic removal to prevent further bleeding episodes. Conventional laparoscopy is often used, but reports of single-incision laparoscopic surgery are emerging [40]. If a Meckel’s is found incidentally, the management is controversial. In asymptomatic patients where it is noted radiographically, many would recommend close follow-up without surgical intervention [41]. If there is discovery of an incidental Meckel’s during an operative procedure for another condition, some would recommend its removal due to low risk of complications and future risk of complications [42]. Others, however, recommend leaving it in [43]. The macroscopic appearance of a Meckel’s does not indicate if heterotopic gastric mucosa is present or not and thus does not aid in the decision to remove it [44].
Duodenal Web
Duodenal webs occur when there is incomplete bowel lumen recanalization during the 8th to 10th week of gestation. This results in a thin web of the mucosa and submucosa layers, with the muscular layer being absent, causing some degree of obstruction [45]. The web is located in the second portion of the duodenum 85–90 % of the time [46]. Much less frequently, they are located in the third or fourth portion of the duodenum [47]. The incidence of duodenal web is estimated to be 1 in 10,000 to 1 in 40,000 live births [48]. Duodenal webs are most often congenital, however is has been described that they may be a rare complication of long-term non-steroidal anti-inflammatory use [49, 50]. Duodenal webs are frequently associated with other congenital anomalies including Down’s syndrome, malrotation, congenital heart disease, and annular pancreas [45, 51].
Duodenal webs can present prenatally with growth failure and/or polyhydramnios. Most other cases present in infancy with vomiting, which can be bilious, food refusal, and/or failure to thrive [52]. Additionally, duodenal web has been reported as causing upper GI bleeding in infants [53, 54]. Webs are not exclusive to children as they can also present at older ages [51, 52, 55, 56].
Duodenal webs are often more difficult to diagnose than atresias due to the obstruction being partial. The diagnosis of a duodenal web can be considered prenatally by noting polyhydramnios and a dilated stomach on ultrasound. A plain abdominal radiograph may be normal or it could reveal a double bubble from a dilated proximal duodenum and stomach. An upper GI series may reveal the classic windsock sign. This consists of a thin radiolucent line spanning across the duodenal lumen with a dilated proximal duodenum that gradually tapers prior too ending abruptly further down the duodenum mimicking a windsock [47, 57].
Therapeutic options are duodenoduodenostomy or duodenotomy with excision/lysis of the web. In cases where there is an enlarged proximal duodenum (duodenal diameter greater than or equal to 5 cm), imbrications (the operative overlapping of layers of tissue in the closure of wounds or the repair of defects) or tapering duodenoplasty may be required [58]. During any attempt at resection of a duodenal web, care should be given to identify the ampulla of Vater as it may be injured due to its proximity to the obstructing web. In most cases, the web is located at or just distal to the ampulla. The ampulla may be identified by gentle compression of the gallbladder while viewing within the medial wall of the duodenal lumen for expression of bile. Surgery can be performed as an open procedure or laparoscopically. Some have noted that either approach was equally effective with no significant differences in outcome [59]. Some have noted that the laparoscopic approach was associated with a shorter length of stay and more rapid advancement to full feeding [60], while others felt that the open procedure was preferable [61]. Ultimately larger studies are needed to provide better evidence. More recently, there are reports of advanced endoscopy utilizing a number of techniques to treat duodenal webs [48, 51, 62–68]. To date, there have not been any studies published comparing surgical verses endoscopic approaches. Both approaches are associated with complications, including bleeding and pancreatitis, while endoscopic approaches may be associated with incomplete obliteration of the web [63]. For patients that have prolonged duodenal ileus, there may be a benefit to using TPN or a trans-anastomotic tube for enteral nutrition [58]. If malrotation is present, a Ladd procedure should be performed. Long-term prognosis is very good and is primarily dependent on any associated congenital anomalies [69].
Duodenal Atresia
Duodenal atresia has a reported incidence of approximately 1 in 5000 to 10,000 live births [3, 4], with the most recent data at 0.9 per 10,000 [70]. There are four major types of duodenal atresia [71] (Fig. 3). Type 1 is complete mucosal membrane or diaphragm with the muscularis and serosa remaining intact such that there is no discontinuity of the bowel. Type 2 consists of a fibrous cord connecting the two segments of duodenum that are discontinuous. This differs from type 3 where there is no fibrous connection between the proximal and distal segments of duodenum. Type 4 consists of several atretic segments such that it appears like a string of sausages. Duodenal atresia results from failure of recanalization of the duodenum after the seventh week of gestation, perhaps from an ischemic event or genetic factors may also play a role [72]. Duodenal atresia, unlike other intestinal atresias, is commonly associated with other congenital anomalies such as Down’s syndrome, which is present in 25–40 % of cases [73]. Other associated anomalies include VATER (vertebral defects, anal anomalies, esophageal atresia, and renal abnormalities), malrotation, annular pancreas, biliary tract abnormalities, cardiac, and mandibulofacial anomalies [70].

Types of duodenal atresia. Reprinted by permission from Coppola, CP, “Duodenal atresia,” in Coppola, CP, Kennedy, AP, and Scorpio, RJ, Pediatric surgery: Diagnosis and treatment, p. 142 (Fig. 1), Copyright 2014, Springer International Publishing.
The initial manifestation of duodenal atresia or other intestinal atresia could be polyhydramnios due to the infant’s inability to swallow and absorb the amniotic fluid. About half of infants with duodenal atresia will have polyhydramnios [74]. The postnatal presentation is typically within the first day or two of life with obstructive symptoms, such as persistent emesis, bilious emesis, gastric distention, and/or feeding difficulties [72]. The physical examination differs from jejunal or ileal atresia in that the abdomen is typically not distended due to the proximal obstruction in duodenal atresia.
Perinatal ultrasound may be the first diagnostic test in the evaluation of duodenal atresia. One should consider intestinal atresia in an infant with maternal polyhydramnios since about 15 % of infants with this will have gastrointestinal tract abnormalities [75] and up to 80 % of duodenal atresia cases will have polyhydramnios [3]. Other ultrasonic findings may include a “double bubble” that is the classic finding on a plain radiograph of the abdomen due to dilated proximal duodenum and stomach associated with lack of bowel gas in the distal intestine [76]. If the double bubble sign is noted, most feel that no other radiographic studies are required. Administration of contrast into the upper GI tract could lead to aspiration. Due to the fact that the double bubble sign can also occur with malrotation, some authors recommend performing a contrast enema to look for a microcolon of disuse as would be found in one with intestinal atresia [77] versus a malpositioned colon and cecum as may be present in malrotation [78].
Once duodenal atresia is identified, a naso- or orogastric tube should be placed to decompress the stomach and minimize aspiration along with routine supportive management such as intravenous fluids. Once clinically stable, the patient needs surgical repair via laparotomy or laparoscopy. Options for surgical therapy include a side-to-side or end-to-side duodenoduodenostomy or duodenojejunostomy. Prior performing the anastomosis, a small rubber catheter should be passed distally to investigate for any additional intraluminal obstruction [58]. As with duodenal web, there is some controversy regarding performing surgery as an open procedure or laparoscopically [59–61, 79]. One review concluded duodenal atresia should only undergo laparoscopic repair at designated centers of expertise [80]. Delayed transition to full enteral nutrition is more likely to occur in patients with co-morbid congenital heart disease or malrotation and prematurity [81]; however, the majority of these cases were repaired via laparotomy. Intraoperatively, it is important to exclude any associated malrotation, other small bowel atresia, or annular pancreas.
Long-term prognosis for duodenal atresia is very good with survival rates approximately 90 % [69]. The major causes of morbidity and mortality from duodenal atresia are related to associated anomalies and ultra-short bowel syndrome requiring long-term total parenteral nutrition [82]. Infants with a birth weight of less than 2 kg are also at higher risk of mortality [83].
Duodenal stenosis is less frequent than duodenal atresia and can present later in life due to only a partial obstruction and may be found on endoscopy for work up of persistent vomiting, failure to thrive, or hematemesis [84].
Jejunoileal Atresia
Jejunoileal atresias are discussed separately from duodenal atresia due to differences in etiology, associated anomalies, as well as treatment and outcome. Jejunoileal atresias occur as a result of an ischemic insult during pregnancy [85]. The ischemic insult can be due to intussusception, perforation, volvulus, intestinal strangulation via a hernia, or thromboembolism. Maternal smoking and cocaine use have been associated with intestinal atresia [86]. There is an estimated incidence of approximately 1 to 3 per 10,000 live births [70]. This disorder affects both sexes equally. Jejunoileal atresias are equally distributed between the jejunum and ileum. Associated congenital anomalies are less common with jejunoileal atresia than duodenal atresia. The most common associated conditions are cystic fibrosis, malrotation, and gastroschisis, all of which are present in about 10 % of cases [82]. Intestinal atresia is associated with low birth weight and multiparity.
There is a rare disorder of multiple intestinal atresias that can occur anywhere in the gastrointestinal tract and is almost always fatal [87]. Hereditary multiple intestinal atresia (HMIA) is an autosomal recessive disorder that consists of multiple atretic segments that occurs most commonly in French Canadians and may be associated with combined immune deficiency. This is due to mutations of tetratricopeptide repeat domain–7A (TTC7A) gene. The TTC7A protein is important for the development and function of the thymus and intestinal epithelium [88]. This mutation has also been associated with very early onset inflammatory bowel disease [89].
There are four types of intestinal atresia based on the anatomic characteristics [71] (Fig. 4). Type I is an intraluminal web consisting of mucosa and submucosa with continuity of the proximal and distal muscular layers without a mesenteric defect. Type II atresia is when the bowel is discontinuous but without a mesenteric defect. Type III has two subtypes. In Type III A, the bowel is discontinuous and there is also a mesenteric defect. Type III B has discontinuous bowel but with an extensive mesenteric defect with the bowel wrapped around a single artery such that it looks like a Christmas tree or apple peel (Fig. 5). Type IV consists of multiple atretic segments that appear like a string of sausage.

Types of intestinal atresia. Reprinted by permission from Coppola, CP, “Intestinal atresia,” in Coppola, CP, Kennedy, AP, and Scorpio, RJ, Pediatric surgery: Diagnosis and treatment, p. 148 (Fig. 1), Copyright 2014, Springer International Publishing.

Type III(b) Jejunal atresia with associated “apple peel” or “Christmas tree” deformity of the mesentery. Note the single vessel within the center of the coils of bowel responsible for perfusion of the distal intestinal segment.
The typical presentation is an infant in the first 1–2 days of life with bilious vomiting, a history of maternal polyhydramnios, and abdominal distention depending on the level of atresia with more distal lesions having more distention. The infant may also have feeding difficulties and hyperbilirubinemia. With more distal lesions, there may be failure to pass meconium. Infants with more proximal lesions may pass meconium due to the generation of succus entericus.
The diagnosis of jejunoileal atresia may be detected by prenatal ultrasound. Findings suggestive of atresia include dilated, echogenic bowel and maternal polyhydramnios that is seen in about one third of cases [90]. These findings, however, have a poor predictive value for bowel abnormalities and when questionable, fetal magnetic resonance imaging can be considered [90]. Postnatally, the first step in the work up is a plain abdominal radiograph that often reveals multiple dilated loops of intestine with air fluid levels and at times a triple bubble sign; dilated stomach, duodenum, and proximal jejunum [91]. Peritoneal calcifications suggest the presence of meconium peritonitis, which is a sign of intrauterine intestinal perforation and can be seen in about 12 % of cases [90]. The presence of meconium peritonitis should raise suspicion of a meconium ileus and cystic fibrosis. The obstructive findings noted above can also be seen in other disorders such as Hirschsprung’s disease. A barium enema may help to distinguish atresia from other obstructive disorders. Infants with jejunoileal atresia typically have a microcolon. If meconium ileus is present, one may consider meglumine diatrizoate (Gastrografin) enema that is hypertonic and can help evacuate the meconium and well as make a diagnosis.
Once diagnosed, surgical therapy should be undertaken expeditiously. Pre-operatively, the neonate needs to be stabilized and have fluid and electrolyte abnormalities corrected. A nasogastric tube should be placed to decompress the stomach and minimize aspiration. Broad-spectrum antibiotics are indicated to help decrease risk of infection which is a major cause of mortality. Surgery can be done via an open approach or laparoscopically, but the latter can be challenging [92, 93]. Intraoperatively, the entire bowel is closely examined for sites of obstruction and the presence of other atresias. It is also important to assess patency of the colon either by pre-operative contrast study or intraoperative irrigation of the distal atretic limb. If malrotation or gastroschisis is present, it must also be corrected.
Post-operative mortality is related to prematurity, associated anomalies, infection, and short gut syndrome. Post-operative complications include anastomotic leak, stenosis at the site of anastomosis, and short gut syndrome. Additionally, these patients may have oral feeding intolerance, which is more likely if any of the following are present: meconium peritonitis, luminal discrepancy, number of anastomoses, presence of immature ganglion, and short bowel syndrome [94]. The complexity of jejunoileal atresia (based on Grosfeld’s classification), when there are not any other congenital malformations, is not associated with a worse prognosis in terms of initiation of enteral nutrition, post-operative complications, duration of post-operative TPN, and percentage of short bowel syndrome [95].
The prognosis for infants with jejunoileal atresia is very good with over 90 % survival [82]. The prognosis for those with short gut syndrome is dependent on the length of the remaining small bowel, the presence of the ileocecal valve, and the dependence on long-term parenteral nutrition [96]. Wilmore, in 1972, published data on prognosis of short gut syndrome in infants [97]. Those with greater than 40 cm of small bowel had a 95 % survival rate. This decreased to 50 % in those with 15–40 cm and an intact ileocecal valve. Those with less than 40 cm and no ileocecal valve and those with less than 15 cm with the ileocecal valve did not survive. Things have improved since then due to newer surgical techniques including bowel lengthening procedures (STEP procedure), improved medical care, and the ability to perform small bowel transplantation [98]. There are case reports of infants surviving with only 10 cm [99]. For those with short bowel syndrome, the overall prognosis is that 47 % will get off of TPN, 26 % will have a small bowel transplant, and 27 % will not survive [100].
Small Bowel Duplications
Gastrointestinal duplications are rare, estimated to occur in 1 per 100,000 births [101]. Calder is given credit for the first report of intestinal duplications in 1733. These were known by a number of terms such as giant diverticuli, enteric cysts, intestinal duplex, and “unusual Meckel’s diverticulum” until Ladd popularized the term intestinal duplication in 1930s [102]. Males appear to be more commonly affected, 60–80 % of cases, and about one third have associated congenital anomalies [103, 104]. It is estimated that 2 to 12 % are found in the duodenum, about 44 % in the ileum, and about 50 % in the jejunum [105•]. Multiple duplications are noted in 15–20 % of cases.
The etiology of duplications is unknown [106]. Theories include abnormalities in recanalization, a vascular insult, persistence of embryonic diverticuli, and partial twining. Duplications can be cystic or tubular depending on their length [107]. They consist of an epithelial lining from some portion of the GI track and a smooth muscle wall that are located on the mesenteric side of the intestine. Most duplications do not communicate with the adjacent bowel [104].
Duplications can present at any age; however, 60–80 % present in the first 2 years of life [103, 108, 109]. The presentation depends on the size and the epithelial type of the duplication. Small cystic duplications can be the lead point of an intussusception. Larger tubular duplications can accumulate secretions, dilate and cause obstructive type symptoms. Those that are lined with gastric epithelium will secrete acid which can result in ulceration and present with GI bleeding or if it perforates with an acute abdomen. Other modes of presentation include chronic abdominal pain, nausea and vomiting, jaundice, pancreatitis, and as an abdominal mass [107, 110, 111].
Small bowel duplications may be detected prenatally by ultrasound [104, 112–114]. Ultrasound is commonly used as part of the evaluation of an acute abdomen or a mass and may detect a duplication. Enteric duplications on ultrasonography often have an inner hyperechoic rim with an outer surrounding hypoechoic layer (“double-wall” sign) along with peristalsis being present [112]. Other diagnostic imaging considerations include CT scan, MRI, contrast studies, and radionuclide scans [105•, 109, 115]. Contrast studies can be helpful in those cases where there is communication of the duplication with the native GI track or by demonstrating a mass effect. Technetium-99 m pertechnetate scanning can be useful in those duplications containing ectopic gastric mucosa, but this is the case in only 15–25 % of cases [107]. Endoscopic ultrasound has been used more recently to diagnose duplications in the upper GI tract including the duodenum [105•]. Many duplications are detected incidentally during surgery for another reason.
Treatment for duplication, in general, is excision of the lesion to avoid or correct complications including bleeding, volvulus, intestinal necrosis, and cancer development [107, 116]. The adjacent normal bowel is frequently removed as well due to both structures having a common blood supply. The surgery can be performed via open or laparoscopic approach [112]. Duplications near the ampulla of Vater pose a challenge and may not be able to be removed. Those that cannot be excised should be drained and the mucosa stripped. The cyst may then be drained into the adjacent intestine to prevent recurrent collection of fluid. For proximal lesions, an endoscopic approach can be considered when a skilled advanced endoscopist is available [110, 111]. For lesions with gastric mucosa that cannot be removed, one can use acid suppressants to minimize GI bleeding. In cases of enteric duplications being detected prenatally, prenatal surgery is not required; however, serial ultrasound surveillance is recommended to monitor size [104].
Conclusions
Congenital anomalies of the small bowel are varied in type, but other than a Meckel’s diverticulum, the major presenting manifestation of these is that of obstruction. Some, such as malrotation and duplications, may present as abdominal pain and vomiting. Meckel’s diverticulum typically presents as painless gastrointestinal bleeding. Duplications can also present as gastrointestinal bleeding or as a mass. These anomalies present most commonly early on in life but they may not present until adulthood. Radiographic studies are the mainstay in the diagnosis of congenital anomalies of the small intestine. Upon making the diagnosis of a congenital anomaly of the small intestine, one also needs to assess them for the presence of another associated disorder since about a third of cases will have an associated anomaly. Once diagnosed, operative therapy is required for congenital anomalies of the small intestine. The overall prognosis of these disorders is very good due to improved medical and surgical therapies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Mall FP. Development of the human intestine and its position in the adult. Johns Hopkins Hospital Bulletin. 1898;9:197–208.
Gosche JR, Boulanger SC, Islam S. Midgut abnormalities. Surg Clin North Am. 2006;86(2):285–99.
Hartman GE. Intestinal obstruction. In: Stevenson DK, Cohen RS, Sunshine P, editors. Neonatology: clinical practice and procedures. New York, NY: McGraw-Hill; 2015. http://accesspediatrics.mhmedical.com/content.aspx?bookid=1462&Sectionid=85592341. Accessed January 12, 2016.
Song C, Upperman JS, Niklas V. Structural anomalies of the gastrointestinal tract. In: Gleason CA, Devaskar SU, editors. Avery’s diseases of the newborn. 9th ed. Philadelphia, PA: Elsevier Saunders; 2012. https://www.clinicalkey.com/#!/content/book/3-s2.0-B9781437701340100691. Accessed January 12, 2016.
Christison-Lagay E, Langer JC. Intestinal rotation abnormalities. In: Ziegler MM, Azizkhan RG, Allmen D, Weber TR, editors. Operative pediatric surgery. 2nd ed. New York, NY: McGraw-Hill; 2014. http://accesssurgery.mhmedical.com/content.aspx?bookid=959&Sectionid=53539609. Accessed January 12, 2016.
Chesley PM, Melzer L, Bradford MC, Avansino JR. Association of anorectal malformation and intestinal malrotation. Am J Surg. 2015;209(5):907–12.
Aboagye J, Goldstein SD, Salazar JH, Papandria D, Okoye MT, Al-Omar K, et al. Age at presentation of common pediatric surgical conditions: reexamining dogma. J Pediatr Surg. 2014;49:995–9. This article highlights the most common age of presentation for several pediatric surgical pathologies and includes graphics to convey this information.
Durkin ET, Lund DP, Shaaban AF, Schurr MJ, Weber SM. Age-related differences in diagnosis and morbidity of intestinal malrotation. J Am Coll Surg. 2008;206(4):658–63.
Wanjari AK, Deshmukh AJ, Tayde PS, Lonkar Y. Midgut malrotation with chronic abdominal pain. N Am J Med Sci. 2012;4(4):196–8.
Sizemore AW, Rabbani KZ, Ladd A, Applegate KE. Diagnostic performance of the upper gastrointestinal series in the evaluation of children with clinically suspected malrotation. Pediatr Radiol. 2008;38(5):518–28.
Lampl B, Levin TL, Berdon WE, Cowles RA. Malrotation and midgut volvulus: a historical review and current controversies in diagnosis and management. Pediatr Radiol. 2009;39(4):359–66.
Yousefzadeh DK, Kang L, Tessicini L. Assessment of retromesenteric position of the third portion of the duodenum: an US feasibility study in 33 newborns. Pediatr Radiol. 2010;40(9):1476–84.
Esposito F, Vitale V, Noviello D, Di Serafino M, Vallone G, Salvatore M, et al. Ultrasonographic diagnosis of midgut volvulus with malrotation in children. J Pediatr Gastroenterol Nutr. 2014;59(6):786–8.
Zhou LY, Li SR, Wang W, Shan QY, Pan FS, Liu JC, et al. Usefulness of sonography in evaluating children suspected of malrotation: comparison with an upper gastrointestinal contrast study. J Ultrasound Med. 2015;34(10):1825–32.
Taylor GA. CT appearance of the duodenum and mesenteric vessels in children with normal and abnormal bowel rotation. Pediatr Radiol. 2011;41(11):1378–83.
Tackett JJ, Muise ED, Cowles RA. Malrotation: current strategies navigating the radiologic diagnosis of a surgical emergency. World J Radiol. 2014;6(9):730–6.
Graziano K, Islam S, Dasgupta R, Lopez ME, Austin M, Chen LE, et al. Asymptomatic malrotation: diagnosis and surgical management. An American Pediatric Surgical Association outcomes and evidence based practice committee systematic review. J Pediatr Surg. 2015;50(10):1783–90. This article provides a great reference on how to approach and manage patients with asymptomatic malrotation.
Ladd WE. Surgical diseases of the alimentary tract in infants. N Engl J Med. 1936;215:705–8.
Bass KD, Rothenberg SS, Chang JH. Laparoscopic Ladd’s procedure in infants with malrotation. J Pediatr Surg. 1998;33(2):279–81.
Draus Jr JM, Foley DS, Bond SJ. Laparoscopic Ladd procedure: a minimally invasive approach to malrotation without midgut volvulus. Am Surg. 2007;73(7):693–6.
Fraser JD, Aguayo P, Sharp SW, Ostlie DJ, St Peter SD. The role of laparoscopy in the management of malrotation. J Surg Res. 2009;156(1):80–2.
Ooms N, Matthyssens LE, Draaisma JM, de Blaauw I, Wijnen MH. Laparoscopic treatment of intestinal malrotation in children. Eur J Pediatr Surg. 2015. [Epub ahead of print].
Miyano G, Fukuzawa H, Morita K, Kaneshiro M, Miyake H, Nouso H, et al. Laparoscopic repair of malrotation: what are the indications in neonates and children. J Laparoendosc Adv Surg Tech A. 2015;25(2):155–8.
Opitz JM, Schultka R, Gobbel L. Meckel on developmental pathology. Am J Med Genet A. 2006;140(2):115–28.
Sagar J, Kumar V, Shah DK. Meckel’s diverticulum: a systematic review. J R Soc Med. 2006;99(10):501–5.
St-Vil D, Brandt ML, Panic S, Bensoussan AL, Blanchard H. Meckel’s diverticulum in children: a 20-year review. J Pediatr Surg. 1991;26(11):1289–92.
Elsayes KM, Menias CO, Harvin HJ, Francis IR. Imaging manifestations of Meckel's diverticulum. AJR Am J Roentgenol. 2007;189(1):81–8.
Hayashi A, Kumada T, Furukawa O, Nozaki F, Hiejima I, Shibata M, et al. Severe acute abdomen caused by symptomatic Meckel’s diverticulum in three children with trisomy 18. Am J Med Genet A. 2015;167A(10):2447–50.
Sinha CK, Pallewatte A, Easty M, De Coppi P, Pierro A, Misra D, et al. Meckel’s scan in children: a review of 183 cases referred to two paediatric surgery specialist centres over 18 years. Pediatr Surg Int. 2013;29(5):511–7.
Lin S, Suhocki PV, Ludwig KA, Shetzline MA. Gastrointestinal bleeding in adult patients with Meckel’s diverticulum: the role of technetium 99m pertechnetate scan. South Med J. 2002;95(11):1338–41.
Petrokubi RJ, Baum S, Rohrer GV. Cimetidine administration resulting in improved pertechnetate imaging of Meckel’s diverticulum. Clin Nucl Med. 1978;3(10):385–8.
Rerksuppaphol S, Hutson JM, Oliver MR. Ranitidine-enhanced 99mtechnetium pertechnetate imaging in children improves the sensitivity of identifying heterotopic gastric mucosa in Meckel's diverticulum. Pediatr Surg Int. 2004;20(5):323–5.
Xinias I, Mavroudi A, Fotoulaki M, Tsikopoulos G, Kalampakas A, Imvrios G. Wireless capsule endoscopy detects Meckel’s diverticulum in a child with unexplained intestinal blood loss. Case Rep Gastroenterol. 2012;6(3):650–9.
Desai SS, Alkhouri R, Baker SS. Identification of Meckel diverticulum by capsule endoscopy. J Pediatr Gastroenterol Nutr. 2012;54(2):161.
Courcoutsakis N, Pitiakoudis M, Mimidis K, Vradelis S, Astrinakis E, Prassopoulos P. Capsule retention in a giant Meckel’s diverticulum containing multiple enteroliths. Endoscopy. 2011;43:E308–9.
Qi S, Huang H, Wei D, Lv C, Yang Y. Diagnosis and minimally invasive surgical treatment of bleeding Meckel’s diverticulum in children using double-balloon enteroscopy. J Pediatr Surg. 2015;50(9):1610–2.
Fukushima M, Kawanami C, Inoue S, Okada A, Imai Y, Inokuma T. A case series of Meckel’s diverticulum: usefulness of double-balloon enteroscopy for diagnosis. BMC Gastroenterol. 2014;14:155.
Zheng CF, Huang Y, Tang ZF, Chen L, Leung YK. Double-balloon enteroscopy for the diagnosis of Meckel’s diverticulum in pediatric patients with obscure GI bleeding. Gastrointest Endosc. 2014;79(2):354–8.
He Q, Zhang Y, Xiao B, Jiang B, Bai Y, Zhi F. Double-balloon enteroscopy for diagnosis of Meckel’s diverticulum: comparison with operative findings and capsule endoscopy. Surgery. 2013;153(4):549–54.
Chan KW, Lee KH, Wong HY, Tsui SY, Wong YS, Pang KY, et al. Laparoscopic excision of Meckel’s diverticulum in children: what is the current evidence? World J Gastroenterol. 2014;20(41):15158–62.
Zani A, Eaton S, Rees CM, Pierro A. Incidentally detected Meckel diverticulum: to resect or not to resect? Ann Surg. 2008;247(2):276–81.
Bani-Hani KE, Shatnawi NJ. Meckel’s diverticulum: comparison of incidental and symptomatic cases. World J Surg. 2004;28(9):917–20.
Soltero MJ, Bill AH. The natural history of Meckel’s diverticulum and its relation to incidental removal. A study of 202 cases of diseased Meckel’s diverticulum found in King County, Washington, over a fifteen year period. Ann J Surg. 1976;132(2):168–73.
Gezer, HO, Temiz A, Ince E, Ezer SS, Hasbay B, Hicsonmez A. Meckel diverticulum in children: evaluation of macroscopic appearance for guidance in subsequent surgery. J Pediatr Surg. 2015. [Epub ahead of print].
Eksarko P, Nazir S, Kessler E, LeBlanc P, Zeidman M, Asarian AP, et al. Duodenal web associated with malrotation and review of literature. J Surg Case Rep. 2013;2013(12).
Melek M, Edirne YE. Two cases of duodenal obstruction due to a congenital web. World J Gastroenterol. 2008;14(8):1305–7.
Materne R. The duodenal wind sock sign. Radiology. 2001;218(3):749–50.
Beeks A, Gosche J, Giles H, Nowicki M. Endoscopic dilation and partial resection of a duodenal web in an infant. J Pediatr Gastroenterol Nutr. 2009;48(3):378–81.
Serracino-Inglott F, Smith GH, Anderson DN. Duodenal webs—no age limit. HBO (Oxford). 2003;5(3):186–7.
Rha SE, Lee JH, Lee SY, Park SM. Duodenal diaphragm associated with long-term use of nonsteroidal anti-inflammatory drugs: a rare cause of duodenal obstruction in an adult. AJR Am J Roentgenol. 2000;175(3):920–1.
Lee SS, Hwang ST, Jang NG, Tchah H, Choi DY, Kim HY, et al. A case of congenital duodenal web causing duodenal stenosis in a down syndrome child: endoscopic resection with an insulated-tip knife. Gut Liver. 2011;5(1):105–9.
Karnsakul W, Gillespie S, Cannon ML, Kumar T. Food refusal as an unusual presentation in a toddler with duodenal web. Clin Pediatr (Phila). 2009;48(1):81–3.
Nagpal R, Schnaufer L, Altschuler SM. Duodenal web presenting with gastrointestinal bleeding in a seven-month-old infant. J Pediatr Gastroenterol Nutri. 1993;16(1):90–2.
Al Shahwani N, Mandhan P, Elkadhi A, Ali MJ, Latif A. Congenital duodenal obstruction associated with Down’s syndrome presenting with hematemesis. J Surg Case Rep. 2013;2013(12).
Ladd AP, Madura JA. Congenital duodenal anomalies in the adult. Arch Surg. 2001;136(5):576–84.
Sarkar S, Apte A, Sarkar N, Sarker D, Longia S. Vomiting and food refusal causing failure to thrive in a 2 year old: an unusual and late manifestation of congenital duodenal web. BMJ Case Rep. 2011.
Eisenberg RL, Levine MS. Miscellaneous abnormalities of the stomach and duodenum. In: Gore RM, Levine MS, editors. Textbook of gastrointestinal radiology. 4tth ed. Philadelphia, PA: Elsevier Saunders; 2015. https://www.clinicalkey.com/#!/content/book/3-s2.0-B9781455751174000349. Accessed January 12, 2016.
Sarin YK, Sharma A, Sinha S, Deshpande VP. Duodenal webs: an experience with 18 patients. J Neonatal Surg. 2012;1(2):20.
Jensen AR, Short SS, Anselmo DM, Torres MB, Frykman OK Shin CE, et al. Laparoscopic verses open treatment of congenital duodenal obstruction: multicenter short-term outcomes analysis. J Laparoendosc Adv Surg Tech A. 2013;23(10):876–80.
Spilde TL, St. Peter SD, Keckler SJ, Holcomb 3rd GW, Snyder CL, Ostlie DJ. Open vs laparoscopic repair of congenital duodenal obstructions: a concurrent series. J Pediatr Surg. 2008;43(6):1002–5.
Parmentier B, Peycelon M, Muller CO, El Ghoneimi A, Bonnard A. Laparoscopic management of congenital duodenal atresia or stenosis. A single-center early experience. J Pediatr Surg. 2015;50(11):1833–6.
Barabino A, Gandullia P, Arrigo S, Vignola S, Mattioli G, Grattarola C. Successful endoscopic treatment of a double duodenal web in an infant. Gastrointest Endosc. 2011;73(2):401–3.
Barabino A, Arrigo S, Gandullia P, Vignola A. Duodenal web: complications and failure of endoscopic treatment. Gastrointest Endosc. 2012;75(5):1123–4.
Bleve C, Costa L, Bertoncello V, Ferrara F, Zolpi E, Chiarenza SF. Endoscopic resection of a duodenal web in an 11-month-old infant with multiple malformations. Endoscopy. 2015;47(S ssye):E210–1.
Huang MH, Bian HQ, Liang C, Wei WQ, Duan XF, Yang J. Gastroscopic treatment of membranous duodenal stenosis in infants and children: report of 6 cases. J Pediatr Surg. 2015;50(3):413–6.
Kay GA, Lobe TE, Custer MD, Hollabaugh RS. Endoscopic laser ablation of obstructing congenital duodenal webs in the newborn: a case report of limited success with criteria for patient selection. J Pediatr Surg. 1992;27(3):279–81.
Kay S, Yoder S, Rothenberg S. Laparoscopic duodenoduodenostomy in the neonate. J Pediatr Surg. 2009;44(5):906–8.
Torroni F, De Angelis P, Caldaro T, di Abriola GF, Ponticelli A, Bergami G, et al. Endoscopic membranectomy of duodenal diaphragm: pediatric experience. Gastrointest Endosc. 2006;63(3):530–1.
Escobar MA, Ladd AP, Grosfeld JL, West KW, Rescorla FJ, Scherer LR 3rd, et al. Duodenal atresia and stenosis: long-term follow-up over 30 years. J Pediatr Surg. 2004;39(6):867–71.
Best KE, Tennant PW, Addor MC, Bianchi F, Boyd P, Calzolari E, et al. Epidemiology of small intestinal atresia in Europe: a register-based study. Arch Dis Child Fetal Neonatal Ed. 2012;97(5):F353–8.
Grosfeld JL, Ballantine TV, Shoemaker R. Operative management of intestinal atresia and stenosis based on pathologic findings. J Pediatr Surg. 1979;14(3):368–75.
Lloyd DA, Kenny SE. anomalies including hernias. In: Kleinman R, Sanderson I, Goulet O, Sherman P, Mieli-Vergani G, Shneider B, editors. Walker’s pediatric gastrointestinal disease. 5th ed. Hamilton, Ontario: B.C. Decker Inc; 2008. http://www.r2library.com/Resource/detail/1550093649/ch0013s0509. Accessed January 12, 2016.
Freeman SB, Torfs CP, Romitti PA, Royle MH, Druschel C, Hobbs CA, et al. Congenital gastrointestinal defects in Down syndrome: a report from the Atlanta and National Down Syndrome Projects. Clin Genet. 2009;75(2):180–4.
Brantberg A, Blaas HG, Salvesen KA, Haugen SE, Mollerlokken G, Eik-Nes SH. Fetal duodenal obstructions: increased risk of prenatal sudden death. Ultrasound Obstet Gynecol. 2002;20(5):439–46.
Pauer HU, Viereck V, Krauss V, Osmers R, Krauss T. Incidence of fetal malformations in pregnancies complicated by oligo- and polyhydramnios. Arch Gynecol Obstet. 2003;268(1):52–6.
Correia-Pinto J, Ribeiro A. Congenital duodenal obstruction and double-bubble sign. N Engl J Med. 2014;371(11), e16.
Laya BF, Andres MM, Conception NDP, Dizon RH. Patterns of microcolon: imaging strategies for diagnosis of lower intestinal obstruction in neonates. J Am Osteopath Coll Radiol. 2015;4(1):1–11.
Strouse PJ. Malrotation. Semin Roentgenol. 2008;43(1):7–14.
Son TN, Liem NT, Kien HH. Laparoscopic simple oblique duodenoduodenostomy in management of congenital duodenal obstruction in children. J Laparoendosc Adv Surg Tech A. 2015;25(2):163–6.
van der Zee DC. Laparoscopic repair of duodenal atresia: revisited. World J Surg. 2011;35(8):1781–4.
Bairdain S, Yu DC, Lien C, Khan FA, Pathak B, Grabowski MJ, et al. A modern cohort of duodenal obstruction patients: predictors of delayed transition to full enteral nutrition. J Nutr Metab. 2014.
Dalla Vecchia LK, Grosfeld JL, West KW, Rescorla FJ, Scherer LR, Engum SA. Intestinal atresia and stenosis: a 25-year experience with 277 cases. Arch Surg. 1998;133(5):490–6.
Piper HG, Alesbury J, Waterford SD, Zurakowski D, Jaksic T. Intestinal atresias: factors affecting clinical outcomes. J Pediatr Surg. 2008;43(7):1244–8.
Nicholson MR, Acra SA, Chung DH, Rosen MJ. Endoscopic diagnosis of duodenal stenosis in a 5-month-old male infant. Clin Endosc. 2014;47(6):568–70.
Louw JH, Barnard CN. Congenital intestinal atresia; observations on its origin. Lancet. 1955;269(6899):1065–7.
Werler MM, Sheehan JE, Mitchell AA. Maternal medication use and risks of gastroschisis and small intestinal atresia. Am J Epidemiol. 2002;155(1):26–31.
Guttman FM, Braun P, Garance PH, Blanchard H, Collin PP, Dallaire L, et al. Multiple atresias and a new syndrome of hereditary multiple atresias involving the gastrointestinal tract from stomach to rectum. J Pediatr Surg. 1973;8(5):633–40.
Fernandez I, Patey N, Marchand V, Birles M, Maranda B, Haddad E, et al. Multiple intestinal atresia with combined immune deficiency related to TTC7A defect is a multiorgan pathology: study of a French-Canadian-based cohort. Medicine (Baltimore). 2014;93(29), e327.
Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology. 2014;146(4):1028–39.
Frischer JS, Azizkhan RG. Jejunoileal atresia and stenosis. In: Coran AG, editor. Pediatric surgery. 7th ed. Philadelphia, PA: Elsevier Saunders; 2012. https://www.clinicalkey.com/#!/content/book/3-s2.0-B9780323072557000829. Accessed January 12, 2016.
Vinocur DN, Lee EY, Eisenberg RL. Neonatal intestinal obstruction. AJR Am J Roentgenol. 2012;198(1):W1–10.
Juang D, Snyder CL. Neonatal bowel obstruction. Surg Clin North Am. 2012;92(3):685–711.
Tajiri T, Ieiri S, Kinoshita Y, Masumoto K, Nishimoto Y, Taguchi T. Transumbilical approach for neonatal surgical diseases: woundless operation. Pediatr Surg Int. 2008;24(10):1123–6.
Wang J, Du L, Cai W, Pan W, Yan W. Prolonged feeding difficulties after surgical correction on intestinal atresia: a 13-year experience. J Pediatr Surg. 2014;49(11):1593–7.
Federici S, Sabatino MD, Domenichelli V, Straziuso S. Worst prognosis in the “complex” jejunoileal atresia: is it real? European J Pediatr Surg Rep. 2015;3(1):7–11.
Calisti A, Olivieri C, Coletta R, Briganti V, Oriolo L, Giannino G. Jejunoileal atresia: factors affecting the outcome and long-term sequelae. J Clin Neonatol. 2012;1(1):38–41.
Wilmore DW. Factors correlating with a successful outcome following extensive intestinal resection in newborn infants. J Pediatr. 1972;80(1):88–95.
Thompson JS, Rochling FA, Weseman RA, Mercer DF. Current management of short bowel syndrome. Curr Probl Surg. 2012;49(2):52–115.
Infantino BJ, Mercer DF, Hobson BD, Fischer RT, Gerhardt BK, Grant WJ, et al. Successful rehabilitation in pediatric ultrashort small bowel syndrome. J Pediatr. 2013;163(5):1361–6.
Squires RH, Duggan C, Teitelbaum DH, Wales PW, Balint J, Venick R, et al . Natural history of pediatric intestinal failure: initial report from the pediatric intestinal failure consortium. J Pediatr. 2012;161(4):723–8.
Tsai SD, Sopha SC, Fishman EK. Isolated duodenal duplication cyst presenting as a complex solid and cystic mass in the upper abdomen. J Radiol Case Rep. 2013;7(11):32–7.
Ladd WE. Duplications of the alimentary tract. South Med J. 1937;30:363–71.
Ildstad ST, Tollerud DJ, Weiss RG, Ryan DP, McGowan MA, Martin LW. Duplications of the alimentary tract. Clinical characteristics, preferred treatment, and associated malformations. Ann Surg. 1988;208(2):184–9.
Laje P, Flake AW, Adzick NS. Prenatal diagnosis and postnatal resection of intraabdominal enteric duplications. J Pediatr Surg. 2010;45(7):1554–8.
Liu R, Adler DG. Duplication cysts: diagnosis, management, and the role of endoscopic ultrasound. Endosc Ultrasound. 2014;3(3):152–60. This article reviews the literature on duplication cysts and discusses the role of endoscopic ultrasound and fine needle aspiration in management.
Stern LE, Warner BW. Gastrointestinal duplications. Semin Pediatr Surg. 2000;9(3):135–40.
Niu BB, Bai YZ. Ileal tubular duplication in a 4-year-old girl. Surgery. 2015;157(1):166–7.
Karkera PJ, Bendre P, D’souza F, Ramchandra M, Nage A, Palse N. Tubular colonic duplication presenting as rectovestibular fistula. Pediatr Gastroenterol Hepatol Nutr. 2015;18(3):197–201.
Li BL, Huang X, Zheng CJ, Zhou JL, Zhao YP. Ileal duplication mimicking intestinal intussusception: a congenital condition rarely reported in adult. World J Gastroenterol. 2013;19(38):6500–4.
Arantes V, Nery SR, Starling SV, Albuquerque W, Alberti LR. Duodenal duplication cyst causing acute recurrent pancreatitis managed curatively by endoscopic marsuplialization. Endoscopy. 2012;44(S 02):E117-118.
Meier AH, Mellinger JD. Endoscopic management of a duodenal duplication cyst. J Pediatr Surg. 2012;47(11):e33–5.
Ballehaninna UK, Nguyen T, Burjonrappa SC. Laparoscopic resection of antenataly identified duodenal duplication cyst. JSLS. 2013;17(3):454–8.
Palacios A, De Vera M, Martinez-Escoriza JC. Prenatal sonographic findings of duodenal duplication: case report. J Clin Ultrasound. 2013;41(S 1):1–5.
Vivier PH, Beurdeley M, Bachy B, Aguilella C, Ickowicz V, Lemoine F, et al. Ileal duplication. Diagn Interv Imaging. 2013;94(1):98–100.
Hur J, Yoon CS, Kim MJ, Kim OH. Imaging features of gastrointestinal tract duplications in infants and children: from oesophagus to rectum. Pediatr Radiol. 2007;37(7):691–9.
Chen JJ, Lee HC, Yeung CY, Chan WT, Jiang CB, Sheu JC. Meta-analysis: the clinical features of the duodenal duplication cyst. J Pediatr Surg. 2010;45:1598–606.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Grant Morris and Alfred Kennedy Jr. declare that they have no conflicts of interest. William Cochran reports he is on the speaker’s bureau for Nestle Nutrition, outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Small Intestine
Rights and permissions
About this article

Cite this article
Morris, G., Kennedy, A. & Cochran, W. Small Bowel Congenital Anomalies: a Review and Update. Curr Gastroenterol Rep 18, 16 (2016). https://doi.org/10.1007/s11894-016-0490-4
Published:
DOI: https://doi.org/10.1007/s11894-016-0490-4