
Abstract
Purpose of Review
This review seeks to detail the clinical and pathologic features specific to BRAFV600E colorectal cancer. Application of novel preclinical findings translated into the clinic for the development of new therapeutic options for patients with BRAFV600E metastatic colorectal cancer will be detailed.
Recent Findings
While BRAF inhibitors as monotherapy do not have the same clinical activity for colorectal cancer relative to other solid tumors harboring an oncogenic BRAFV600E mutation, combination approaches targeting BRAF + MEK + EGFR hold promise for patients with BRAFV600E colorectal cancer.
Summary
Simultaneous targeting of multiple drivers along the MAPK pathway improves clinical outcomes for patients with BRAFV600E colorectal cancer. Targeted therapies and immunotherapy hold great promise in the years to come for patients with this subtype of colorectal cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2018, it is estimated that almost 50,000 Americans will succumb to colorectal cancer (CRC) [1]. For the majority of patients presenting with metastatic CRC, systemic chemotherapy remains the mainstay of treatment. However, targeted therapies, personalized to the genomic profile of a given colorectal tumor, have demonstrated clinical benefit for patients whose tumors are wild-type at the KRAS and NRAS loci (anti-EGFR antibodies) [2], harbor microsatellite instability (immune checkpoint blockade therapies) [3•], or contain an NTRK fusion (Trk inhibitor) [4].
BRAF mutations define another distinct molecular subentity of the CRC population in whom remarkable treatment advances have occurred within recent years. These alterations are present in approximately 8–10% of metastatic CRC cases and are most frequently characterized by valine-to-glutamic acid substitutions at codon 600 of the BRAF gene [5]. These BRAFV600E mutations activate oncogenic signaling of the MAPK pathway via eventual downstream phosphorylation of ERK and result in heightened proliferative and anti-apoptotic behavior for the tumor cell [6]. This review highlights the relevant clinical advances in the context of the unique underlying tumor biology for patients with metastatic BRAFV600E CRC.
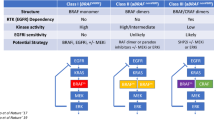
Characterization of BRAFV600E CRC Tumors
Clinically, BRAFV600E mutations have been linked with female gender, history of tobacco exposure, and advancing age [7,8,9]. Pathologically, they occur more commonly as proximal (right-sided), poorly differentiated, mucinous CRC tumors [9,10,11]. In addition, BRAFV600E CRC tumors are more likely to have deficient mismatch repair (dMMR) and be classified as MSI-high (MSI-H) [12]. Here, microsatellite instability arises not from a germline mutation in the dMMR genes associated with hereditary non-polyposis colorectal cancer (HNPCC) syndrome but rather from epigenetic silencing derived from promoter hypermethylation of the MLH1 gene [13, 14]. In general, BRAFV600E CRC tumors are characterized by extensive methylation across the genome not typically observed across other molecular subpopulations of CRC [15].
Hypermethylation across promoter regions of specific gene foci enriched with cytosine-guanine regions, termed CpG-island methylator phenotype (CIMP)–high tumors, drives tumorigenesis in precancerous, dysplastic cells along the sessile serrated adenoma pathway [16, 17]. This pattern of colorectal tumor development, to which BRAFV600E mutations are linked [18,19,20], is distinct from the traditional adenoma pathway from which the majority of colorectal cancers arise. While an oncogenic driver in many other tumors (including, but not limited to, melanoma, thyroid cancer, and non-small cell lung cancer), the introduction of a BRAFV600E mutation into a normal colorectal cell does not transform the cell into a cancer [21, 22]. Capitalizing on the CIMP-high biology, methylation (and subsequent loss of function) of tumor suppressor genes in a sessile serrated polyp with a preexisting BRAFV600E mutation creates the synergistic interaction necessary to generate a malignant colorectal lesion [23]. Here, the interplay between the complex, coexisting pathogenic drivers parlays into the aggressive clinical phenotype seen with BRAFV600E CRC tumors relative to their BRAF wild-type (BRAFWT) counterparts.
With regard to genomic alterations, BRAFV600E mutations occur mutually exclusively in colorectal cancer to activating mutations in the KRAS and NRAS oncogenes [24, 25], which also drive pathogenic signaling of the MAPK pathway. Analysis of 276 CRC specimens by the Cancer Genome Atlas (TCGA) project found that BRAFV600E mutations co-occur with hypermutated tumors that carry higher somatic mutation burdens intratumorally [26]. This association is likely driven by the MLH1 hypermethylation and reinforces the interplay between the epigenome and corresponding genomic characteristics unique to this subtype. Certainly, an understanding of the biology relevant and specific to BRAFV600E CRC can inform the oncologist on the clinical manifestations in order to optimize treatment planning.
BRAF V600E as a Clinical Biomarker in CRC
BRAFV600E mutations carry a poor prognostic implication for patients with CRC, regardless of the stage at presentation. In the PETACC-3 trial [27], 3278 patients with stage II or stage III CRC were randomized to receive adjuvant chemotherapy with 5-fluorouracil with or without irinotecan. Nearly half of trial participants (1403, or 43%) had archival tissue available for genomic profiling, including BRAF mutation status. Overall survival (OS) for patients with microsatellite stable tumors was lower when accompanied by a BRAFV600E mutation for patients with stage II and with stage III disease alike.
Worsened clinical outcomes for patients with BRAFV600E CRC also extend to patients with stage IV tumors [28,29,30,31]. For example, one series of 524 patients with metastatic CRC demonstrated a significantly inferior OS for patients with BRAFV600E tumors when compared to patients with BRAFWT tumors (10 versus 35 months, respectively) [7]. When limited to microsatellite stable CRC, one series showed that the risk of cancer-specific mortality was higher in the BRAF-mutated group (hazard ratio (HR) 2.3, 95% confidence interval (CI) 1.3–4.0) [32]. Another series, however, demonstrated that, even for patients with MSI-H metastatic CRC, progression-free survival (PFS) with standard chemotherapy (3 versus 10 months, P < 0.001) and OS (14 versus 30 months, P < 0.001) is shortened for patients with BRAFV600E CRC tumors [33]. Therefore, the worsened prognostic implications linked to BRAFV600E mutations for patients with advanced CRC appear to be consistent regardless of microsatellite status.
Despite these tumors lacking driver mutations in KRAS or NRAS, there has not been benefit demonstrated with use of anti-EGFR antibodies as a lone targeted therapy for BRAFV600E metastatic CRC. For example, in the PRIME study [34], patients with untreated, advanced CRC were treated with FOLFOX chemotherapy with or without the anti-EGFR antibody panitumumab. Here, there was no benefit with the addition of panitumumab in PFS or in OS despite the patients having RASwild-type tumors. A separate study examined tumors from 773 patients with metastatic CRC treated with cetuximab (another anti-EGFR antibody) as part of their treatment for mutations in KRAS, BRAF, and PIK3CA, in order to assess for a correlation with clinical outcomes [35]. Compared to their wild-type counterparts, those with BRAFV600E tumors treated with cetuximab had lower disease control rates (odds ratio (OR) 0.15, P = 0.001), shorter PFS (HR 3.7, P < 0.001), and shorter OS (HR 3.0, P < 0.001). Other series have likewise confirmed that a BRAFV600E mutation does not serve as a predictive biomarker for response to anti-EGFR therapies for patients with metastatic CRC [36•, 37,38,39,40,41]. Given this lack of data demonstrating a clinical benefit, use of anti-EGFR antibodies as a single targeted therapy for RASwild-type, BRAFV600E metastatic CRC is not recommended.
Because patients with BRAFV600E CRC are expected to fare poorly in the metastatic setting, systemic chemotherapy options have remained limited. The randomized phase III TRIBE trial compared FOLFIRI/bevacizumab to FOLFOXIRI/bevacizumab as frontline therapy for metastatic CRC, where response rates were improved in the overall population by the addition of oxaliplatin to FOLFIRI/bevacizumab [42]. Post hoc stratification reported a non-significant trend in PFS benefit with FOLFOXIRI/bevacizumab uniquely for the BRAFV600E patients relative to the BRAFWT patients (HR 0.57, 95% CI 0.27–1.2) [43]. While encouraging, the numbers of BRAF-mutated patients here treated with FOLFIRI/bevacizumab (N = 12) or with FOLFOXIRI/bevacizumab (N = 16) remain small and limit definitive interpretation accordingly. In addition, administration of triplet cytotoxic chemotherapy with FOLFOXIRI is associated with greater toxicity. Therefore, these findings should be interpreted with caution and should not yet be generalized to the entire BRAFV600E CRC population, given the rapid clinical deterioration often inherent to their underlying aggressive disease that would not allow tolerance of this regimen. Following loss of response to frontline systemic chemotherapy, outcomes for patients with BRAFV600E metastatic CRC have been reported to be especially poor. In a single-institution, retrospective series of 72 patients with BRAFV600E metastatic CRC [44], median PFS with systemic therapy in the second-line and third-line settings were 2.5 months and 2.6 months, respectively. Alternatively stated, patients with BRAFV600E metastatic CRC developed disease progression even by the time of first restaging when treated with standard options beyond the first line of chemotherapy.
Targeted Therapies Against MAPK Signaling in BRAF V600E Advanced Cancers
The advent of targeted therapies against MAPK signaling in recent years has heralded in promising new options for patients with BRAFV600E advanced cancers. Vemurafenib is a selective inhibitor specific to the mutated BRAFV600E kinase domain [45] which first demonstrated promising clinical activity in patients with advanced BRAFV600E melanoma [46], with rapid reductions of tumor burden observed even within 2 weeks of single-agent therapy [47]. The response rate for BRAFV600E metastatic melanoma was reported in a large phase III trial [48] at 48%, higher than the 5% of patients with response to dacarbazine in the control arm of the same trial. Survival outcomes here were likewise improved with vemurafenib, a result which led to FDA approval for this BRAF inhibitor. Similarly, patients with BRAFV600E or BRAFV600K unresectable melanoma participating in a phase III trial of the reversible BRAFV600E kinase inhibitor dabrafenib (versus dacarbazine) showed responses to the former agent in 50% of cases [49]. BRAF inhibitors as monotherapy have anti-tumor activity in other BRAFV600E tumors besides melanoma, such as non-small cell lung cancer, thyroid cancer (papillary and anaplastic), hairy cell leukemia, and Langerhans cell histiocytosis [50,51,52,53]. Collectively, for various solid tumors, the presence of a BRAFV600E mutation serves as a predictive biomarker for clinical benefit with targeted therapies against the BRAF kinase.
While inhibition of the kinase domain of the downstream MEK has also demonstrated clinical efficacy as a single agent in these tumors, the combination of targeted therapies against BRAF and MEK together delays the onset of acquired resistance in preclinical models of BRAFV600E tumors [54], relative to BRAF inhibitors as monotherapies. Here, inactivation of mutated BRAF pharmacologically can generate resistant clones by activating mutations in MAPK1/2 and other drivers in MAPK signaling. These findings have likewise translated into the clinical setting for patients with BRAFV600E malignancies. BRAF/MEK combinations with vemurafenib/cobimetinib, dabrafenib/trametinib, and encorafenib/binimetinib have demonstrated clinical benefit in patients with BRAFV600E melanoma [55,56,57], non-small cell lung cancer [58], and anaplastic thyroid cancer [59]. In BRAFV600E melanoma, this combination approach has clinical superiority over single-agent BRAF inhibitors and has been associated with response rates in the 55–70% range. Therefore, sustained anti-tumor activity is promoted by dual targeting of BRAF and MEK in order to deepen blockade of the pivotal MAPK signaling and translates to improved clinical outcomes in patients with advanced cancers harboring BRAFV600E mutations.
Targeting MAPK Signaling in BRAF V600E CRC
Pivoting upon the successes of targeted therapies for BRAF with or without MEK inhibitors, it would be expected that a similar pattern of clinical activity would be observed for patients with BRAFV600E metastatic CRC. However, a study of single-agent vemurafenib in 21 patients resulted in a single patient with a radiographic partial response (response rate (RR) 5%, 95% CI 0–26%) [60•]. Median PFS here was 2.1 months among a pretreated population and did not appear to prolong survival outcomes relative to historical controls. Similarly, low response rates with BRAF inhibitors were observed with dabrafenib (RR 11%, 95% CI 0–48%) [61] and with encorafenib (RR 0%, 95% CI 0–23%) [62]. Seemingly, the response rates are lower for patients with colorectal cancer in this monotherapy approach than in other cancers despite harboring the same oncogenic BRAFV600E mutation.
Clinical outcomes likewise do not appear to improve with the addition of a MEK inhibitor to a BRAF inhibitor for BRAFV600E metastatic CRC. In one trial [63], 43 patients were treated with the combination of dabrafenib and trametinib. Responses were noted in 5% of patients (RR 12%, 95% CI 4–26%), and median PFS was 3.5 months. Again, patients receiving these two agents overall did not fare as well as other patients with BRAFV600E tumors receiving the same treatment.
Subsequent basic science work helped to elucidate the lack of response to these agents in a mechanism unique to BRAFV600E CRC. In vitro, BRAFV600E CRC cell lines are inherently resistant to vemurafenib, but a siRNA screen revealed that sensitivity to this drug could be restored with knockdown of EGFR [64••]. This finding implicated EGFR activation as a culprit responsible for de novo resistance to targeted therapies against BRAF in this setting. Additional work in xenograft models of BRAFV600E CRC confirmed an anti-tumor response preclinically with a combination of agents against BRAF and EGFR simultaneously that was not observed with either protein was targeted alone [65, 66].
These promising preclinical findings led to a next generation of clinical trials testing BRAF and EGFR combination approaches for patients with metastatic BRAFV600E CRC. In a phase I trial of vemurafenib, cetuximab, and irinotecan [67], an initial signal was observed, with radiographic responses reported in 6 of 17 patients (RR 37%). In order to build upon these promising early clinical findings, the randomized phase II SWOG 1406 clinical trial was conducted [68••]. Here, 99 patients with pretreated BRAFV600E CRC were treated with irinotecan/cetuximab with or without vemurafenib so that the benefit of the addition of a BRAF inhibitor to an anti-EGFR targeted therapy could be tested prospectively in this setting. This study met successfully its primary endpoint, with a prolongation in PFS (4.3 months versus 2.0 months, HR 0.48, P = 0.001) for those receiving vemurafenib, irinotecan, and cetuximab. Disease control rates (DCR, defined as the proportion of patients with stable disease or partial/complete response as the best radiographic assessment according to RECIST 1.1 criteria) were also more favorable in this group (67% versus 22%, P = 0.001). Subgroup analysis here failed to demonstrate preferential clinical activity based on microsatellite status, sidedness (left versus right) of the primary tumor, or PIK3CA mutation status. Overall, this trial provided the first randomized prospective data confirming that the previously detailed preclinical data that targeted approaches against BRAF and EGFR are effective in this population. As a result, the combination of vemurafenib, irinotecan, and cetuximab was updated in the 2018 NCCN Guidelines [69] as a recommended therapy for patients with BRAFV600E metastatic CRC.
Other studies investigating this approach in parallel have corroborated this strategy, with similar clinical findings as observed with the SWOG 1406 study. In a single-arm study, 15 patients with BRAFV600E metastatic CRC were treated with vemurafenib and panitumumab (no cytotoxic exposure here) [70]. This dual combination was safe and well tolerated. For the 12 patients assessable for response, two patients (RR 13%) had radiographic responses, with one of these participants experiencing a 100% reduction in tumor volume from baseline. Eleven patients (89%) had a radiographic response or stable disease by RECIST 1.1 criteria as their best-measured assessment on the study. Another trial of 20 patients with BRAFV600E metastatic CRC treated with dabrafenib and panitumumab observed radiographic responses in 2 patients (RR 10%) [71].
Encorafenib and cetuximab were examined in 54 patients with BRAFV600E metastatic CRC treated with (N = 28) or without (N = 26) alpelisib, a class I PI3K inhibitor [72]. Use of alpelisib here was supported by preclinical data demonstrating in xenograft models of BRAFV600E CRC that PI3K/mTOR signaling may drive de novo resistance to BRAF inhibition, and that sensitivity to targeted therapies can be restored by a PI3K inhibitor [66]. Response rates were 18% and 19% for those treated with encorafenib and cetuximab with or without alpelisib, respectively. While this study was not designed statistically to compare the two treatment arms, there appeared to be no added clinical benefit by the addition of a PI3K inhibitor here. Although the total numbers were small, alterations in PI3K/mTOR genes did not correlate with anti-tumor activity.
Extending Clinical Benefit for Patients With BRAF V600E Metastatic CRC
Despite the practice-changing successes noted with BRAF and EGFR targeted therapies here, acquired resistance to these drugs invariably develops, a theme unfortunately common across solid tumors. Understanding of resistance mechanisms has led however to further advancements in the management of BRAFV600E metastatic CRC. In the aforementioned phase I study of vemurafenib, cetuximab, and irinotecan [67], post-progression blood samples were analyzed for genomic profiling by circulating tumor DNA in order to identify acquired alterations not present in baseline samples which could be implicated in resistance to treatment. Oncogenic aberrations in KRAS, EGFR, ARAF, MAP2K1, GNAS, and ERBB2 were all observed in this series of patients and implicated reactivation of MAPK signaling as a driver of tumor progression of targeted therapies against BRAF and EGFR. Addition of inhibitors to downstream effectors of MAPK signaling like MEK and ERK have restored sensitivity in preclinical models of BRAFV600E metastatic CRC resistant to BRAF and EGFR targeted therapies [73•, 74] and have provided rationale for triple combination approaches deepening efforts to thwart this oncogenic pathway in this subpopulation of CRC.
In a trial of dabrafenib, panitumumab, and trametinib for 91 patients with BRAFV600E metastatic CRC, complete or partial responses were seen in 19 (21%) patients, with a DCR of 86% [71]. Grade 3/4 toxicity profiles were worse than for those receiving dabrafenib and panitumumab without a MEK inhibitor. Median PFS (both ~ 4 months) was likewise similar with the triple combination than dabrafenib and panitumumab. Nonetheless, these findings suggested, based upon the notable DCR, that simultaneous targeting of BRAF, EGFR, and MEK holds promise for this population with poor tumor biology.
Perhaps the most encouraging result to date for the treatment of BRAFV600E metastatic CRC has been from the recent report of efficacy from the safety lead-in analysis [75••] of the BEACON trial, a randomized phase III study of irinotecan/cetuximab versus encorafenib/cetuximab with or without binimetinib. In the initial pilot in which 29 patients with BRAFV600E metastatic CRC were treated with the BRAF/EGFR/MEK combination, radiographic responses were seen in 14 patients (RR 48%), with all 29 patients having disease control (i.e., no de novo progression at first restaging according to RECIST 1.1 criteria) as the best response. Median PFS here was 8.0 months. Final analyses of the phase III trial are highly anticipated. However, these very encouraging early results led to FDA breakthrough therapy designation for the combination of encorafenib, cetuximab, and binimetinib for patients with BRAFV600E metastatic CRC. The development of improved response rates with subsequent generations of clinical trials testing MAPK-targeting agents in this setting are detailed in Fig. 1.

Evolution of targeted therapies in BRAFV600E metastatic colorectal cancer: response rate (95% confidence interval)
Immunotherapy in BRAF V600E Metastatic CRC
Immune checkpoint blockade agents targeting PD-1/PD-L1 and CTLA-4 have demonstrated durable clinical activity for patients with MSI-H metastatic CRC [3•]. Given the association between microsatellite instability and BRAF mutations in patients with CRC, these agents are relevant in this population as well. Indeed, although the numbers of patients treated are small, objective responses to the anti-PD-1 antibody nivolumab as a single agent (RR 25%) [76] and in combination with the anti-CTLA-4 antibody ipilimumab (RR 55%) [77••] for patients with MSI-H, BRAFV600E metastatic CRC. Therefore, immunotherapy is an attractive option with safety and efficacy alike for those patients with MSI-H tumors and coexisting BRAFV600E mutations.
Conclusions
Unique tumor biology specific to BRAFV600E colorectal tumors generate a clinical phenotype vastly different to their BRAFWT counterparts and cause hastened clinical deterioration and poor survival outcomes. That traditional agents for metastatic CRC are futile from an efficacy standpoint in the BRAFV600E CRC setting prompted preclinical efforts implicating activity from targeted therapies targeting MAPK signaling drivers. Most recently, inhibitors of BRAF, EGFR, and MEK have revolutionized how clinicians will approach BRAFV600E metastatic CRC in the years to come. In doing so, the evolving treatment of BRAFV600E CRC provides a real-life success story of translational, bench-to-bedside science.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65.
• Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20 First report of microsatellite instability as a predictive biomarker for benefit to immune checkpoint blockade therapy.
Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–9.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54.
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67.
Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117(20):4623–32.
Tie J, Gibbs P, Lipton L, Christie M, Jorissen RN, Burgess AW, et al. Optimizing targeted therapeutic development: analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer. 2011;128(9):2075–84.
Gonsalves WI, Mahoney MR, Sargent DJ, et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst. 2014;106(7).
Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B. BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer. 2006;5:2.
Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58(1):90–6.
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):934.
Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–11.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96(15):8681–6.
Kambara T, Simms LA, Whitehall VL, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53(8):1137–44.
O’Brien MJ, Yang S, Clebanoff JL, et al. Hyperplastic (serrated) polyps of the colorectum: relationship of CpG island methylator phenotype and K-ras mutation to location and histologic subtype. Am J Surg Pathol. 2004;28(4):423–34.
Yang S, Farraye FA, Mack C, Posnik O, O’Brien MJ. BRAF and KRAS mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol. 2004;28(11):1452–9.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–93.
O’Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol. 2006;30(12):1491–501.
Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007;104(47):18654–9.
Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309(5736):886–7.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602.
Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–74.
Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361(1):98–9.
Van Cutsem E, Kohne CH, Lang I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011–9.
Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28(3):466–74.
Kalady MF, Dejulius KL, Sanchez JA, et al. BRAF mutations in colorectal cancer are associated with distinct clinical characteristics and worse prognosis. Dis Colon Rectum. 2012;55(2):128–33.
Price TJ, Hardingham JE, Lee CK, Weickhardt A, Townsend AR, Wrin JW, et al. Impact of KRAS and BRAF gene mutation status on outcomes from the phase III AGITG MAX trial of capecitabine alone or in combination with bevacizumab and Mitomycin in advanced colorectal Cancer. J Clin Oncol. 2011;29(19):2675–82.
Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A, Li G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PLoS One. 2012;7(10):e47054.
Chen D, Huang JF, Liu K, Zhang LQ, Yang Z, Chuai ZR, et al. BRAFV600E mutation and its association with clinicopathological features of colorectal cancer: a systematic review and meta-analysis. PLoS One. 2014;9(3):e90607.
Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65(14):6063–9.
Saridaki Z, Papadatos-Pastos D, Tzardi M, Mavroudis D, Bairaktari E, Arvanity H, et al. BRAF mutations, microsatellite instability status and cyclin D1 expression predict metastatic colorectal patients’ outcome. Br J Cancer. 2010;102(12):1762–8.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–34.
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–62.
• Rowland A, Dias MM, Wiese MD, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112(12):1888–94 BRAF V600E mutation is not predictive for benefit to anti-EGFR therapies in patients with metastatic colorectal cancer.
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–12.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–30.
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101(4):715–21.
Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14(8):749–59.
Pietrantonio F, Petrelli F, Coinu A, di Bartolomeo M, Borgonovo K, Maggi C, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51(5):587–94.
Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371(17):1609–18.
Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306–15.
Morris V, Overman MJ, Jiang ZQ, Garrett C, Agarwal S, Eng C, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13(3):164–71.
Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–9.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36.
Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366(21):2038–40.
Rosove MH, Peddi PF, Glaspy JA. BRAF V600E inhibition in anaplastic thyroid cancer. N Engl J Med. 2013;368(7):684–5.
Brose MS, Cabanillas ME, Cohen EE, et al. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(9):1272–82.
Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102(12):1724–30.
Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9.
Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):603–15.
Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid Cancer. J Clin Oncol. 2018;36(1):7–13.
• Kopetz S, Desai J, Chan E, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol. 2015;33(34):4032–8 Unlike other advanced malignancies, BRAF inhibitor as a monotherapy is not an effective treatment approach for patients with BRAF V600E metastatic colorectal cancer.
Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–901.
JD CAG-R, Robert C, Hidalgo M, von Moos R, Arance A, Elez E, et al. Encorafenib (LGX818), an oral BRAF inhibitor, in patients (pts) with BRAF V600E metastatic colorectal cancer (mCRC): results of dose expansion. Ann Oncol. 2014;25(suppl 4):iv167-iv209.
Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal Cancer. J Clin Oncol. 2015;33(34):4023–31.
•• Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3 In preclinical models of BRAF V600E colorectal cancer, blockade of BRAF V600E generates reflexive activation of EGFR and downstream MAPK signaling.
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–35.
Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–67.
Hong DS, Morris VK, El Osta B, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6(12):1352–65.
•• Kopetz S, McDonough SL, Morris VK, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406). J Clin Oncol. 2017;35(4_suppl):520–520 The addition of cetuximab to the BRAF inhibitor vemurafenib improves survival outcomes in patients with BRAF V600E metastatic colorectal cancer.
Network. NCC. NCCN Guidelines Colon Cancer, version 4.2018. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed 23 Oct 2018.
Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21(6):1313–20.
Corcoran RB, Andre T, Atreya CE, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov. 2018;8(4):428–43.
van Geel R, Tabernero J, Elez E, et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017;7(6):610–9.
• Ahronian LG, Sennott EM, Van Allen EM, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015;5(4):358–67 Resistance to targeted therapies against BRAF and EGFR in patients with BRAF V600E metastatic colorectal cancer may be driven by acquired activating mutations in genes relevant to propagating MAPK signaling.
Hazar-Rethinam M, Kleyman M, Han GC, Liu D, Ahronian LG, Shahzade HA, et al. Convergent therapeutic strategies to overcome the heterogeneity of acquired resistance in BRAF(V600E) colorectal cancer. Cancer Discov. 2018;8(4):417–27.
•• Van Cutsem E, Cuyle P, Huijberts S, et al. O-027BEACON CRC study safety lead-in: assessment of the BRAF inhibitor encorafenib + MEK inhibitor binimetinib + anti–epidermal growth factor receptor antibody cetuximab for BRAFV600E metastatic colorectal cancer. Ann Oncol. 2018;29(suppl_5):mdy149.026-mdy149.026 Targeted therapies against BRAF, MEK, and EGFR are safe and active for patients with BRAF V600E metastatic colorectal cancer.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
•• Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–9 Patients with BRAF V600E metastatic colorectal cancer and microsatellite instability demonstrate sustained clinical response to immune checkpoint blockade therapy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Systemic Therapies in Colorectal Cancer
Rights and permissions
About this article

Cite this article
Morris, V.K. Systemic Therapy in BRAF V600E-Mutant Metastatic Colorectal Cancer: Recent Advances and Future Strategies. Curr Colorectal Cancer Rep 15, 53–60 (2019). https://doi.org/10.1007/s11888-019-00429-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-019-00429-z