
Abstract
Purpose of Review
This review provides a brief history of newborn screening (NBS) for severe combined immunodeficiency (SCID), discusses the theoretical basis for the T cell receptor excision circle (TREC) assay, highlights the results of recent studies using the TREC, and provides practical advice for the evaluation of infants with an abnormal TREC assay.
Recent Findings
Currently, all but three states perform NBS for SCID in the USA. NBS using the TREC assay is highly sensitive in identifying infants with SCID and may also identify infants with T cell lymphopenia due to other causes such as congenital syndromes, multiple congenital anamolies, and some combined immunodeficiencies.
Summary
Regardless of the genetic etiology, all forms of SCID are characterized by a severe deficiency of naïve T cells. TRECs are a biomarker of newly formed, naïve T cells that have recently left the thymus. Consequently, the TREC assay identifies infants with SCID and other causes of severe T cell lymphopenia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe combined immunodeficiency (SCID) is a genetically heterogeneous set of diseases caused by mutations in one of several genes involved in T cell receptor (TCR) DNA rearrangement/repair (RAG1, RAG2, LIG4, DCLRE1C, NHEJ, PRKCD), DNA synthesis/metabolism (ADA, AK2, PNP), or TCR signaling (PTPRC, IL2RG, JAK3, IL7RA, CDE, CDZ, CORO1A) [1]. In all forms of SCID, there is a marked decrease in the production of naïve T cells, which leads to a combined humoral and cellular immunodeficiency. Most babies with SCID appear phenotypically normal and, therefore, are not diagnosed at birth. Maternal antibodies in the infant may delay the onset of serious infections in infants with SCID. However, once maternal antibodies wane, the infants suffer from recurrent, severe infections and die if not diagnosed and treated early in life.
Newborn screening (NBS) of infants for SCID using the T cell receptor excision circle (TREC) assay has proven to be a sensitive test for the early diagnosis of infants with SCID [2••]. NBS using the TREC assay is widely used in the USA and increasingly throughout the world. In this review, we will discuss a brief history of NBS in general and for SCID, the theoretic basis for the TREC assay and practical considerations regarding the initial evaluation of infants with an abnormal TREC screen for SCID.
History of NBS for SCID
Beginning in 1963, population-based NBS tests were performed to identify infants with serious diseases that were amenable to treatment if identified early. As technology improved and more disorders could be identified by NBS, Wilson and Junger in 1968 proposed specific criteria for the addition of specific diseases that should be considered for NBS [3]. Although controversies exist, these criteria continue to serve as an important conceptual framework for population-based screening throughout the world.
SCID has been shown to meet the fundamental criteria of Wilson and Junger [4•]. For example, SCID is a significant health problem with a frequency of approximately 1:46,000 live births. The natural history of SCID is well understood and there is an accepted treatment for the disease that can lead to a cure. There is an economical, feasible, sensitive assay (TREC assay) that identifies infants prior to the onset of disease. Cost-benefit analysis to date indicates that the diagnosis and treatment of SCID is cost-effective [5, 6]. Due to the fatal nature of SCID and the ability to identify and successfully treat this disorder, NBS for SCID has been widely accepted with little controversy.
In the USA, the federal government publishes the recommended diseases for NBS in the Recommended Uniform Screening Panel (RUSP). The decision to implement NBS for a specific disease, however, is made by an individual state with the federal government recommendations serving in only an advisory capacity. To nominate a specific disease for NBS by the federal government, a standard form is submitted to a committee of experts (Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children, SACHDNC), which acts under the direction of the Secretary of Health and Human Services (HHS). The SACHDNC evaluates the appropriateness of the disease in question and if this is an inexpensive, sensitive screening test for the disorder. The SACHDNC subsequently makes recommendations which are subject to approval by the Secretary of HHS. The addition of a specific disease to the RUSP hastens the adaption of the NBS for this disorder throughout the USA. However, as noted previously, each state has the ultimate discretionary power to add a specific disease for statewide NBS.
Douek et al. in 1998 developed the TREC assay and used it to demonstrate that effective anti-retroviral therapy in patients with AIDs led to immunological reconstitution of the T cell compartment through the production of newly formed, naïve T cells not the expansion of the small pool of memory T cells present prior to anti-retroviral therapy [7]. In 2005, Chan and Puck demonstrated that the TREC assay performed on standard NBS cards and could identify babies with SCID regardless of genetic cause [8]. Based on this advance, SCID was nominated to the SACHDNC in 2007 for addition to the RUSP. However, due to the lack of a field-tested, economically feasible screening test to identify infants with SCID, the nomination was denied. The small sample size (n = 239 NBS cards) and high false positive rate of 2.9% reported in the initial study were also likely important factors in the decision.
In 2006, investigators from the Medical College of Wisconsin obtained funding from the Jeffrey Modell Foundation and Children’s Hospital of Wisconsin Foundation to support a pilot study for statewide NBS for SCID in Wisconsin. In collaboration with the Wisconsin State Hygiene Laboratory, these investigators optimized the TREC assay for use in population-based, high-throughput NBS and decreased the false positive rate to 0.02% using over 5600 stored NBS cards [9]. In 2008, Wisconsin became the first state to institute NBS for SCID using the TREC assay. Results of the first year of NBS for SCID in Wisconsin demonstrated the ability of the TREC assay to identify infants with SCID [10••]. These results were presented to the SACHDNC, which unanimously recommended addition of SCID to the RUSP. In May 2010, HHS Secretary Kathleen Sebelius approved the SACHDNC’s recommendation and SCID was added to the RUSP. The addition of SCID to the RUSP resulted in an acceleration in the number of states screening for SCID over the following years (Fig. 1). In 2018, all but the three states have implemented NBS for SCID and these states have plans to do so in the near future.

States participating in NBS for SCID (a) and the year of implementation (b). Currently, approximately 94% of all infants in the USA are screened for SCID. Pilot studies are planned in the 3 remaining states in 2018 or 2019. Reproduced with permission from the Immune Deficiency Foundation
Theoretical Basis of the TREC Assay
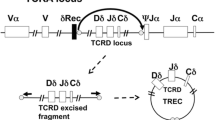
The ability to recognize diverse antigens by the TCR is essential to provide an adequate defense against a multitude of pathogens. T cell diversity is based on the germline rearrangement of genes encoding the T cell receptor (TCR) during maturation of the T cells in the thymus (VDJ rearrangement). TCR rearrangement and subsequent expression of a TCR are essential for T cells to emigrate from the thymus.
The TCR is a protein heterodimer composed of two chains (αβ or γδ) that pair during the maturation process. Consequently, there are two types of T cells: α/β T cells, which account for approximately 95% of T cells in humans, and γδ T cells. During T cell receptor rearrangement, double-strand breaks are made in the DNA and different segments of the TCR are joined. Intervening pieces of DNA are generated during the recombination process, which are called T cell receptor excision circles (TRECs). In humans, a common DNA rearrangement in the TCR α chain occurs in approximately 80% of all α/β T cells [11]. This common TCR α chain rearrangement generates a specific TREC that can be enumerated by quantitative, real-time polymerase chain reaction (qRT-PCR) and is the basis of the TREC assay [7]. Importantly, TRECs do not replicate during T cell division. Therefore, TREC values are low in cases of engraftment of maternal lymphocytes or in leaky SCID (see below), where the expansion of a few T cells in the periphery of infants may lead to normal or near normal T cell numbers.
In the TREC assay, DNA is extracted from a 3.2-mm punch of the dried blood spot on a NBS card and qRT-PCR is then performed to enumerate the number of TRECs present [9]. The blood spotted on a NBS card is ideally obtained from a heel stick on a newborn. However, in some situations, blood is spotted from a peripheral line, which may be diluted by an IV solution or contain substances that may inhibit the PCR. Additionally, the NBS card may be inappropriately stored or transported leading to degradation of DNA. For these reasons, qRT-PCR is also performed to ensure DNA template integrity using a second “housekeeping” gene (β-actin or RNAse P). This latter PCR reaction may be done simultaneously with the qRT-PCR for TRECs or only if the initial TRECs are low, which is the protocol followed in the state of Wisconsin. Therefore, a TREC assay can only be considered “positive” if the qRT-PCR for TRECs is low but the qRT-PCR for the housekeeping gene is normal.
Results of NBS Using the TREC Assay
The TREC assay for NBS was originally designed to detect infants with SCID or leaky SCID (primary targets). SCID is defined as a T cell count < 300/mm and T cell proliferation assay (mitogen assay) less than 10% of control. Leaky SCID is T cell lymphopenia caused by hypomorphic mutations (mutations of a gene leading to a gene product with abnormal but not absent function) in SCID-causing genes associated with a T cell count > 300/mm and abnormal T cell function. Because TRECs serve as a biomarker of low naïve T cell counts, other causes of an abnormal TREC assay (secondary targets) are much more common and include congenital syndromes, secondary causes of T cell lymphopenia, thymic defects, some combined immunodeficiencies, and idiopathic T cell lymphopenia (Table 1) [2••]. Idiopathic T cell lymphopenia, which is also known as variant SCID, refers to T cell lymphopenia with a T cell count greater than 300/mm [2••] without a known cause. We prefer the term idiopathic T cell lymphopenia because variant SCID implies a much more ominous prognosis and is confusing to both physicians and patients.
Depending on the state’s established normal cutoff value, T cell lymphopenia will be found in approximately 50% of infants with an abnormal TREC assay [12]. The cutoff value for an abnormal TREC must be empirically determined by each state as this value will affect the false positive rate. Congenital syndromes and secondary causes of T cell lymphopenia, not SCID, are the most common causes of abnormal TREC assay on NBS (Fig. 2). DiGeorge syndrome (22q11.2 deletion syndrome) and trisomy 21 (Down syndrome) are responsible for approximately 60 and 15% of T cell lymphopenia respectively in infants with congenital syndromes (Fig. 2) [2•• 13, 14]. Infants with complete DiGeorge syndrome, which accounts for less than 0.1% of these patients, have congenital athymia and profound T cell lymphopenia whereas in incomplete DiGeorge syndrome the extent of T cell lymphopenia is variable [15]. Results from NBS indicate that the TREC assay identifies only a minority infants with DiGeorge syndrome, which is dependent on the extent of lymphopenia [16]. Several other congenital syndromes are also associated with T cell lymphopenia including trisomy 18, CHARGE syndrome, Jacobsen’s syndrome, and ataxia telangiectasia. Egress of lymphocytes from the intravascular space due to cardiac defects, GI tract abnormalities, or multiple congenital defects comprise the majority of secondary causes of T cell lymphopenia (Fig. 3).

Congenital syndromes associated with T cell lymphopenia. DiGeorge and trisomy 21 are the most common causes of a low TREC assay in infants with congenital syndromes. Adapted from [2••]

Secondary causes of T cell lymphopenia. Secondary causes of T cell lymphopenia are most commonly caused by egress of lymphocytes into the extravascular space. Adapted from [2••]
Infants with SCID and leaky SCID are found in a small percentage (~ 10%) of abnormal TREC assays (Fig. 4) [2••, 13, 14]. Mutations in the IL2RG are the most common cause of SCID, although the percentage of SCID caused by IL2RG is far less than prior studies indicated. Surprisingly, mutations in RAG1, not IL2RG, are the most common cause of leaky SCID. The frequency of idiopathic T cell lymphopenia as a cause of abnormal TREC assays has been reported as approximately 3%, although this entity is likely underdiagnosed.

Genetic etiologies of SCID and leaky SCID. Mutations in IL2RG are the most common cause of SCID (a). Mutations in RAG1 are the most common cause of leaky SCID (b). Adapted from [2••]
Idiopathic T cell lymphopenia appears to have better overall prognosis than SCID or leaky SCID and is reversible in over one third of cases [17]. However, the frequency, etiology, and natural history of idiopathic T cell lymphopenia remain poorly defined. The US Immunodeficiency Network currently has an ongoing registry on this disorder (https://usidnet.org/fill/). To date, the prognosis of infants diagnosed with SCID or leaky SCID by NBS using the TREC assay is excellent. HSCT in infants with SCID without infection results in 2-year survival rate of 95% whereas in infants with infection survival is reduced to 81% [18, 19].
Initial Evaluation of Infants with Abnormal TREC Results
States differ in the standard operating protocols in the performance of the TREC assay and reporting of abnormal results. In the state of Wisconsin, TREC assays are repeated and not considered abnormal when both the numbers of TRECs and β-actin are low. Similarly, the TREC assay is reflexively performed on premature infants, which are known to have a high false positive rate, until they have reached an adjusted gestational age over 36 weeks [10••]. In Wisconsin and several other states, the abnormal TREC assay is directly reported to the physician of record and a specialist (clinical immunologist) who will work with the primary care provider and the family to facilitate an evaluation of the infant.
When the clinical immunologist receives the result of the abnormal TREC, it is imperative to first determine the absolute value of the TREC test. For example, a TREC value of 0 or severely reduced TREC value (< 20% of the cutoff value) requires more immediate attention as they are more likely to have SCID or severe T cell lymphopenia. Some states do not repeat the TREC assay on premature infants; therefore, it is essential for the immunologist to be aware of the specific protocols that are followed in their state. If possible, prior to the initial flow cytometry, the clinical immunologist should discuss with the local provider or family member to determine if there were issues prenatally (e.g., medications, exposures) or perinatal conditions (e.g., prematurity, infections, congenital anomalies) that may be contributing to the abnormal TREC assay. If there are other medical conditions that could result in T cell lymphopenia, it is sometimes economical to wait until the condition is treated or improved to perform a full immunologic investigation on these infants.
Lymphocyte enumeration by flow cytometry is the initial study performed on infants with an abnormal TREC assay and is usually done prior to a formal evaluation by the clinical immunologist [20•]. Flow cytometric studies should include enumeration of total numbers of T cells (CD4 and CD8), B cells, and NK cells. Importantly, the percentage of naïve (CD45RA+) and memory (CD45RO+) T cells must be measured. In normal neonates, ~90% of all T cells are naïve. In contrast, maternal engraftment of T cells or infants with hypomorphic mutations of SCID-causing genes may lead to a normal total number of T cells; however, these cells are CD45RO+ memory T cells. If the numbers of naïve T cells are normal, no further evaluation is required. However, if T cells numbers are low or T cells are markedly skewed to a memory phenotype, we generally proceed with a more formal evaluation. Our protocol somewhat contrasts with what is done in some other states in which infants are not evaluated if the T cell count is > 1500/mm [2••]. However, T cell lymphopenia may not be pronounced in some combined immunodeficiencies or in partial DiGeorge syndrome and we have made the diagnosis of DiGeorge Syndrome in infants with an abnormal TREC assay and a T cell count > 1500/mm. Finally, T cell proliferation (mitogen) assay should be performed in infants with a moderately or severely reduced T cell count.
If the results of the above studies indicate possible SCID or leaky SCID, and if expertise is not available locally, the infant should be referred to a center skilled in the diagnosis and treatment of patients with SCID. Additionally, measures to prevent the acquisition of infections should be immediately implemented, including the avoidance of public places or sick contacts, the use of boiled water only, withholding breast feeding until the CMV status of the mother is known, avoidance of live vaccines, and transfusion precautions with CMV-negative, irradiated blood products [20•]. The specialist should perform a detailed history that includes a family history (early childhood deaths or history of immunodeficiency in family members), personal history (acute illness, diarrhea, failure to thrive), and physical examination specifically noting the presence of tonsils or other lymphoid tissue, cardiac abnormalities, rashes, or dysmorphic features.
In infants that are acutely ill, or whom exhibit features that suggest congenital syndromes or have conditions associated with secondary T cell lymphopenia, repeat lymphocyte enumeration studies should be performed when the infant is clinically stable. Longitudinal immunological evaluation of these infants is of utmost importance to determine if the T cell lymphopenia resolves. DNA deletion, duplication arrays should be performed in infants with multiple congenital anomalies or with dysmorphic features. For infants with T cell counts greater than 300/mm, it is imperative to determine if the child is immune deficient. Therefore, longitudinal assessment of numbers of T cells, B cells and NK cells should be performed as well as serum IgG, IgA, and IgM, levels, vaccine titers and T cell mitogen studies.
The decision to perform HSCT in an infant with SCID is based on the clinical and immunological findings and does not require a genetic diagnosis. However, a genetic diagnosis should be pursued in every case. The management of an infant may be influenced by the specific genetic cause of severe T cell lymphopenia. For example, infants with complete DiGeorge syndrome lack a thymus. Therefore, HSCT is not feasible and the infant should be treated by thymic transplantation [21]. SCID caused by DNA repair defects (e.g., LIG4, NHEJ) are highly sensitive to radiation and chemotherapeutic agents, which could influence diagnostic studies and conditioning regimens. High-throughput DNA sequencing targeting the genetic causes of SCID as well genes that lead to combined immunodeficiencies associated with significant T cell lymphopenia is the preferred approach and can lead to specific diagnosis in a rapid and cost-efficient manner [22]. Currently, reimbursement for genetic studies by private insurance companies for patients with SCID or other primary immunodeficiencies is problematic, which can complicate obtaining a genetic diagnosis.
Conclusions
Currently, over 90% of infants born in the USA undergo NBS for SCID. Results to date demonstrate the TREC assay has a predictive value for T cell lymphopenia of approximately 50% and is virtually 100% sensitive for the diagnosis of SCID and leaky SCID. Infants with SCID detected by NBS have an excellent prognosis following treatment by HSCT.
TRECs are a biomarker of the numbers of naïve T cells that have recently left the thymus. Consequently, the TREC assay will detect severe T cell lymphopenia regardless of the cause. The vast majority of abnormal TREC assays are due to congenital syndromes and secondary causes of T cell lymphopenia. DiGeorge syndrome is the most common primary immunodeficiency detected by NBS, but the TREC assay will not identify all of these patients. Similarly, infants with combined primary immunodeficiencies that are characterized by variably low numbers of naïve T cells cannot be reliably identified by NBS using the TREC assay. Therefore, the clinician should be diligent in the evaluation of infants for primary immunodeficiencies regardless of the result of the TREC assay.
There remain several significant challenges in the diagnosis and treatment of infants with primary immunodeficiencies initially detected by NBS. The implementation of the NBS for SCID needs to be performed uniformly in the USA and adapted by other countries. There is a lack of specialists with expertise in the diagnosis and treatment of infants with SCID and other primary immunodeficiencies. Complex laboratory testing including the enumeration of naïve T cells by flow cytometry, T cell mitogen studies, and advanced genetic testing may not be available in some areas. Furthermore, obtaining insurance approval for genetic testing in the USA in many cases currently is very difficult. Community outreach programs are also needed to further educate clinicians and families on NBS for SCID. Despite these challenges, the institution of NBS using TREC assay in USA represents a fundamental advance in the early diagnosis and treatment of infants with SCID and other forms of T cell lymphopenia.
Abbreviations
- NBS:
-
Newborn screening
- HHS:
-
Health and Human Services
- HSCT:
-
Hematopoietic stem cell transplantation
- SCID:
-
Severe combined immunodeficiency
- TREC:
-
T cell receptor excision circle
- SACHDNC:
-
Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children
- TCR:
-
T cell receptor
- qRT-PCR:
-
Quantitative, real-time polymerase chain reaction
- RUSP:
-
Recommended Uniform Screening Panel
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Chinn IK, Shearer WT. Severe combined immunodeficiency disorders. Immunol Allergy Clin N Am. 2015;35:671–94.
•• Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312:729–38. This study shows the results of NBS of for SCID in over 3 million infants in ten states and the Navajo nation using the TREC assay. A must read for specialists that evaluate infants identified by NBS for SCID.
Wilson JM, Jungner YG. Principles and Practice of Screening for Disease. Public Health Papers. Geneva Switzerland, volume 34. 1968:7–151. http://www.who.int/iris/handle/10665/37650
• Accetta Pedersen DJ, Verbsky J, Routes JM. Screening newborns for primary T-cell immunodeficiencies: consensus and controversy. Expert Rev Clin Immunol. 2011;7:761–8. This article discusses NBS from a public health perspective and reviews some of the controversies involving statewide NBS for SCID and other disorders.
Ding Y, Thompson JD, Kobrynski L, Ojodu J, Zarbalian G, Grosse SD. Cost-effectiveness/cost-benefit analysis of newborn screening for severe combined immune deficiency in Washington State. J Pediatr. 2016;172:127–35.
Chan K, Davis J, Pai SY, Bonilla FA, Puck JM, Apkon M. A Markov model to analyze cost-effectiveness of screening for severe combined immunodeficiency (SCID). Mol Genet Metab. 2011;104:383–9.
Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–5.
Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115:391–8.
Baker MW, Grossman WJ, Laessig RH, Hoffman GL, Brokopp CD, Kurtycz DF, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol. 2009;124:8272–9.
•• Routes JM, Grossman WJ, Verbsky J, Laessig RH, Hoffman GL, Brokopp CD, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA. 2009;302:2465–70. Results of the first statewide NBS for SCID, which formed the basis for the addition of SCID to the RUSP by HHS.
Verschuren MC, Wolvers-Tettero IL, Breit TM, Noordzij J, van Wering ER, van Dongen JJ. Preferential rearrangements of the T cell receptor-delta-deleting elements in human T cells. J Immunol. 1997;158:1208–16.
Verbsky JW, Baker MW, Grossman WJ, Hintermeyer M, Dasu T, Bonacci B, et al. Newborn screening for severe combined immunodeficiency; the Wisconsin experience (2008–2011). J Clin Immunol. 2012;32:82–8.
Mauracher AA, Pagliarulo F, Faes L, Vavassori S, Gungor T, Bachmann LM, et al. Causes of low neonatal T-cell receptor excision circles: a systematic review. J Allergy Clin Immunol Pract. 2017;5:1457–60 e22.
Jyonouchi S, Jongco AM, Puck J, Sullivan KE. Immunodeficiencies associated with abnormal newborn screening for T cell and B cell lymphopenia. J Clin Immunol. 2017;37:363–74.
Morsheimer M, Brown Whitehorn TF, Heimall J, Sullivan KE. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am J Med Genet A. 2017;173:2366–72.
Barry JC, Crowley TB, Jyonouchi S, Heimall J, Zackai EH, Sullivan KE, et al. Identification of 22q11.2 deletion syndrome via newborn screening for severe combined immunodeficiency. J Clin Immunol. 2017;37:476–85.
Albin-Leeds S, Ochoa J, Mehta H, Vogel BH, Caggana M, Bonagura V et. al. Idiopathic T cell lymphopenia identified in New York State Newborn Screening. Clin Immunol. 2017;183:36–40.
Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371:434–46.
Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood. 2017;130:2718–27.
• Thakar MS, Hintermeyer MK, Gries MG, Routes JM, Verbsky JWA. Practical Approach to Newborn Screening for Severe Combined Immunodeficiency Using the T Cell Receptor Excision Circle Assay. Front Immunol. 2017;8:1470. This article provides the detailed protocol at Children’s Hospital of Wisconsin for the evaluation and treatment of infants with SCID identified by NBS until the time of HSCT.
Daguindau N, Decot V, Nzietchueng R, Ferrand C, Picard C, Latger-Cannard V, et al. Immune constitution monitoring after PBMC transplantation in complete DiGeorge syndrome: an eight-year follow-up. Clin Immunol. 2008;128:164–71.
Yu H, Zhang VW, Stray-Pedersen A, Hanson IC, Forbes LR, de la Morena MT, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol. 2016;138:1142–51 e2.
Acknowledgements
The authors are grateful for the support provided by the Jeffrey Modell Foundation and Children’s Hospital of Wisconsin Foundation in funding the initial statewide, newborn screening for SCID in WI and Emma Cook for her expert editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pediatric Allergy and Immunology
Rights and permissions
About this article

Cite this article
Routes, J., Verbsky, J. Newborn Screening for Severe Combined Immunodeficiency. Curr Allergy Asthma Rep 18, 34 (2018). https://doi.org/10.1007/s11882-018-0783-9
Published:
DOI: https://doi.org/10.1007/s11882-018-0783-9