
Abstract
Newborn screening (NBS) for severe combined immunodeficiency (SCID) utilizing the T-cell receptor excision circle assay is a sensitive and specific method to detect T-cell lymphopenia in early infancy. Starting in 2008, several programs have implemented SCID NBS with successful detection of SCID-affected infants; currently over two thirds of U.S. infants receive SCID NBS. Population-based, unbiased screening has established an incidence of SCID to be 1 in 58,000 births, and has revealed a distribution of SCID genotypes different from prior reports from specific SCID treatment centers. Detecting SCID-affected infants in the newborn period allows for timely implementation of protective measures and optimal definitive treatment prior to the onset of life-threatening infections. Infants with non-SCID T-cell lymphopenia also detected by NBS may have one of several recognized syndromes in which lymphocyte development may be impaired, as well as other conditions associated with secondary T-cell lymphopenia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe combined immunodeficiency (SCID) includes a group of genetically heterogeneous diseases characterized by impaired development of a diverse repertoire of functional T-lymphocytes combined with inability to produce specific antibodies, either due to impaired B-cell development or lack of T-cell help [1, 2]. SCID-affected infants are asymptomatic at birth and are protected in the first 2–4 months of life by transplacentally acquired maternal immunoglobulin G (IgG) antibodies. However, in the absence of diagnosis and immune system restoring treatment, infants with SCID develop recurrent, increasingly severe, and opportunistic infections leading to growth failure and early demise. SCID-affected infants can be rescued by establishment of a healthy immune system, usually by means of an allogeneic hematopoietic cell transplant derived from bone marrow or mobilized peripheral stem cells or umbilical cord blood. Experimental gene therapy has been successful for some infants with adenosine deaminase (ADA) gene defects or X-linked SCID (SCID-XL). ADA enzyme replacement for ADA-deficient SCID is also an effective treatment.
The best survival and health outcomes for SCID are achieved if hematopoietic cell transplantation (HCT) is performed early in infancy or before the development of uncontrollable, severe infections [3, 4•, 5, 6••]. As SCID-affected infants appear healthy at birth, only those with a recognized family history of SCID, fewer than 20 % of all cases, were able to have the diagnosis made early in the past [5, 7, 8•]. Early diagnosis and treatment for all infants, not just those with a positive family history, is possible only with population-based screening for SCID [9•].
There were initial attempts to screen infant dried blood spots for ADA-deficient SCID using a colorimetric ADA enzyme assay, but this was unsuccessful due to missed cases and false positives [10–12]. A real-time polymerase chain reaction (PCR) test was subsequently devised that amplifies DNA extracted from dried blood spots (DBS) to detect T-cell receptor excision circles (TRECs), a biomarker of naïve T cells [13]. Absence or low numbers of TRECs are found in infants who have an inadequate number of naïve T cells from any cause [14•]. Thus the test detects any of over a dozen genetic causes of SCID as well as conditions in which there is abnormal loss of T cells from the peripheral circulation. The TREC test was predicted by a cost analysis [15] and a Markov model [16] to be an effective strategy, both in terms of health and economic benefit, to save lives of infants with SCID under a range of assumptions about incidence and cost of early versus late treatment.
SCID caused by genetic defects that adversely affects T-cell generation or maturation prior to and including the formation of TRECs are expected to be identified by the TREC assay. As a secondary target, non-SCID immunodeficiencies in which there is a profound decrease in circulating naïve T cells may also be identified [14•]. Some of the non-SCID causes of low T cells include certain genetic syndromes (DiGeorge syndrome, trisomy 21 or Down syndrome, and others) as well as non-immune disorders such as congenital leukemia, vascular leakage, or chylous effusions that lead to increased T-cell loss until the underlying problem is corrected [17••]. Infants with very low T-cell counts from whatever cause are considered to have impaired immunity and may benefit from avoidance of live vaccines and environmentally infectious exposures, transfusion precautions, and in some cases administration of prophylactic antibiotics and immunoglobulin infusions [18, 19].
Biology of the TREC Test
The development of a diverse repertoire of T lymphocytes, each with its own T-cell receptor (TCR), is essential for recognition of foreign antigen presented bound to self-MHC molecules, leading to immune system activation and control of invading pathogens. The generation of T-cell receptors involves rearrangement and linear assembly of unique combinations of single segments of the TCR genes that encode alternate variable (V), diversity (D), and junctional (J) sequences. This VDJ recombination is carried out in a series of steps mediated by enzymes that induce double-strand breaks at specific sequences that flank each V, D, and J segment; upon successful cutting and rejoining of the DNA, unique VDJ T-cell receptors are generated, and T-cell maturation and selection continue, eventually resulting in a mature population of naïve T cells that are released from the thymus into peripheral blood [20]. The excised DNA fragments of the locus that are not destined to be incorporated into a recombinant TCR gene can be joined at their ends, forming a variety of circular DNA byproducts called T-cell receptor excision circles (TRECs) (Fig. 1). One particular circular species, the δrec-ψJα TREC, is produced late in maturation by 70 % of all T cells that express αβ TCRs [21]. The circles are stable, but are not replicated during mitosis, and therefore become diluted as T cells proliferate [21, 22]. TRECs can be detected and quantified using primers designed to amplify a segment spanning the joint of the circle. Thus, the number of TREC copies correlates with the production of naïve T cells by the thymus, and a normal TREC number signifies adequate autologous T-cell production.

Generation of the δRec-ψJα TREC, showing primers, black arrows, used to amplify and quantitate TREC junction fragment. Excision of the TCRD locus from the TCRA locus results in the excised fragment which circularizes to form the δRec-ψJα TREC, found in >70 % of αβ T-cell receptor expressing T cells. (schematic from [14•])
TRECs were initially used to monitor generation of new T cells in HIV-infected individuals who received effective antiretroviral treatment [21]. TREC copies in normal blood were highest in young infants, gradually decreasing with age, reflecting a successively lower contribution of thymic output of new T cells with TRECs versus peripheral T-cell expansion in older children and adults. The TREC assay was adapted to newborn screening by extracting genomic DNA from the dried blood spots (DBS) on filter paper already collected by newborn screening programs [13, 23]. Absence of TRECs identifies low or absent T-cell production from any cause. A PCR control consisting of primers amplifying a genomic DNA segment (typically from the β-actin or RNaseP gene) serves as control for DNA quality extracted from DBS and differentiates low TRECs due to genuinely low T cells from insufficient or poor quality DNA [14•]. With the development of the TREC assay, SCID became the first immune disorder for which newborn screening was possible, and at the same time became the first DNA-based test to be run as a high-throughput test for DBS samples from all newborns.
Although the TREC test is a DNA-based test, its measurement of TRECs as a DNA byproduct is not specific to particular gene mutations that cause SCID; rather, low TRECs will identify low naïve T cells from any cause. Mutations in a number of genes which play a role in T-cell development can cause SCID, and those mutations that affect successful VDJ recombination will manifest with low TRECs, a non-normal newborn screening test [14•]. (for genetic causes of SCID; see Picard et al., review in this series) It is important to remember that there are genetic forms of combined immunodeficiency (CID), sometimes previously grouped with SCID, in which mutations occur in genes that affect T cells at a developmental stage beyond the recombination of the TCR. Examples include ZAP-70 and MHC class II deficiency and also T-cell activation defects such as ORAI1, STIM1, LCK, IKK2, etc. (see Picard et al., review in this series). Although infants affected with these conditions may be phenotypically similar to SCID patients in their lack of T-cell function and susceptibility to opportunistic infections, TRECs are not expected to be low and the TREC screening test may be normal [24, 25].
Current Implementation
From a public health perspective, the characteristics of SCID that merit its inclusion among disorders in newborn screening (NBS) panels, as outlined by Wilson and Jungner [26], are its lack of recognizable physical features, the asymptomatic phase in early infancy, high disease burden for affected infants, effective available treatment, improved survival and outcome for infants detected early, and availability of a low-cost screening test. In the U.S., NBS programs are under the jurisdiction of the individual states and territories, each developing its own public health policies, budgets, and testing strategies for its citizens. Pilot SCID NBS programs were implemented in Wisconsin in 2008, and Massachusetts and selected Navajo Native American hospitals in 2009; in May, 2010, the national U.S. Department of Health and Human Services Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children recommended adding SCID to the uniform panel of NBS diseases based on an independent evidence-based review [27]. However, this recommendation was advisory in nature.
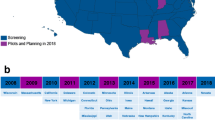
States with a large number of births, including California, New York, Florida, and Texas, as well as a number of smaller states, have added SCID screening to their NBS panels (Fig. 2). By the end of 2013, over 50 % of births in the U.S. were screened with a TREC test [28]. SCID NBS is now being conducted in 26 states, the District of Columbia and the Navajo Nation (spanning parts of Arizona and New Mexico), encompassing an estimated two thirds of births in 2014. In other nations, SCID NBS is occurring in Ontario, Canada, and Taiwan; in several additional countries in Europe and the Middle East steps toward the full implementation of SCID NBS are under way [29–31].

Annual U.S. births from 2008 to 2014, with proportions of births screened for SCID in states implementing NBS. Birth data taken from National Vital Statistics birth reports [28]. SCID screening data obtained from the Newborn Screening Translational Research Network
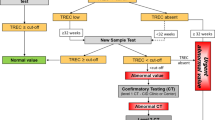
In the U.S., all public health laboratories that perform TREC testing have followed the general guidelines issued by the Clinical Laboratory Standards Institute [32••]. However, different programs have developed their own TREC cutoffs and rules for handling testing of ill and preterm infants, reflecting each program’s characteristics and population. Thus there is variability in the particulars of criteria for recalling infants for additional specimens, referral to specialists for follow-up, and immunological investigations undertaken after non-normal TREC results [33••]. Individual state programs have published SCID NBS technologies and findings so far [17••, 34•, 35, 36•], and a recent publication compared SCID newborn screening outcomes in 11 programs in the U.S. [33••]. The latter publication, which included over 3 million infants screened, noted that all programs readily detected infants with SCID, that no SCID cases were missed by NBS and then detected later, and that affected infants underwent immune restorative treatments in a timely fashion.
Findings and Impact in Clinical Setting
SCID is the primary target for TREC newborn screening. While the historical definition of SCID included low to absent T cells, growth failure, and severe and opportunistic infections, early detection of SCID enabled by newborn screening has required a new definition of SCID based on laboratory criteria in the absence of infectious complications. The Primary Immune Deficiency Treatment Consortium (PIDTC), comprising 33 centers in North America to study the treatment of rare and severe primary immunodeficiencies, now defines typical SCID as infants with <300 autologous T cells/μL blood, <10 % of control proliferation to the mitogen phytohemagglutinin A (PHA), frequently with detectable spontaneously engrafted maternal T cells, and supported by deleterious mutations in known SCID-associated genes [8•, 37••]. If the underlying gene mutations are not totally null, a diagnosis of leaky SCID is made [38, 39•]; infants with leaky SCID are defined as having 300–1,500 autologous T cells/μL (or more if there is an expansion of oligoclonal T cells), absence of maternal T-cell engraftment, and reduced proliferation to PHA of 10–50 %. Infants affected with SCID or leaky SCID may appear totally healthy for the first few months of life.
With wide implementation of SCID NBS in unbiased populations, accurate incidence data have become available; Kwan et al. reported 1 in 58,000 infants (95 % CI 1/46,000–80,000) to have SCID or leaky SCID (including Omenn syndrome), nearly twice the previous estimates based on population data [33••]. In countries where there has not been SCID NBS, fewer cases have been reported, in part because of greater reliance on family history or characteristic infections that are considered for the diagnosis [40–48]. Thus, the true burden of SCID worldwide is probably underestimated, with many infants succumbing to infectious diseases without a primary immunodeficiency being considered. Cost-benefit arguments and worldwide publicity continue to advocate widespread implementation of SCID NBS [49–52]. In settings where newborn screening for SCID is not available, SCID diagnosis relies on the occurrence of infections, which may be life-threatening: opportunistic pathogens such as Pneumocystis jiroveci, persistent and severe cytomegalovirus, adenovirus or other viral infections, oral thrush, invasive bacterial, mycobacterial and fungal infections, diarrhea, and failure to thrive. Further findings may include rashes, lack of tonsillar tissue and lymph nodes, absence of thymus on chest radiograph, and lymphopenia. Secondary causes of immune deficiency such as HIV must be excluded.
The distribution of gene mutations causing SCID detected by NBS has shown a larger proportion of autosomal recessive genes compared to SCID-XL IL2RG mutations than in pre-NBS series of cases reported by SCID transplant centers, possibly reflecting greater ascertainment of sporadic cases with no family history (Fig. 3) [33••, 53, 54]. As X-linked disorders with severe or lethal phenotypes maintain constant frequency due to replenishment in the gene pool by new mutations [55], the lower proportion of SCID-XL reflects an actual increase in the number of autosomal recessive SCID cases detected by unbiased screening. Kwan et al. also reported a larger proportion of cases with RAG1 and RAG2 defects, half of which were leaky [33••]; prior to newborn screening, the heterogeneous phenotypes of leaky RAG mutations meant that some patients were not diagnosed until later in childhood [39•, 56] and may have manifested autoimmunity or Omenn syndrome [57•, 58, 59]. Additionally, genes that were not known to be previously associated with SCID have been discovered with the advent of SCID NBS, e.g., TTC7A [60•, 61•, 62, 63]. Indeed, a higher proportion of SCID cases detected by NBS have had unknown gene defects, even after sequencing multiple typical SCID genes [33••]. These infants with typical SCID phenotypes represent opportunities for the discovery of new gene mutations or identification of previously unrecognized SCID-causing mutations. In facilities where high-throughput sequencing can be performed, T-lymphopenic infants identified by newborn screening can be investigated in a systematic way.

Distribution of SCID genotypes in the presence of newborn screening in California, with 1,980,133 infants screened in 4 years. SCID incidence was 1 per 53,000 births. An earlier study of 11 SCID NBS programs throughout the U.S., with 3,030,083 infants screened, found an incidence of 1 per 58,000 births with a similar distribution, and also detected single incidences of additional SCID genotypes CD3D, TTC7A, and chromosome 12p duplication [34•]
Immune System Restoring Therapies
The premise of SCID newborn screening is to identify affected infants such that their treatments can be optimized and tailored for the most favorable outcomes. Treatments for SCID include primarily HCT (see Wahlstrom et al., in this series), with enzyme replacement therapy (ERT) as an option for ADA-deficient SCID [6••, 8•, 64–69]. ADA-deficient SCID and γc-deficient SCID-XL can also be treated by experimental ex vivo addition of a correct cDNA to autologous hematopoietic stem cells, followed by reinfusion [70–74] (see Calero et al., review in this series). Current gene therapy trials with improved safety and efficiency may become approved as a standard care in the future [75, 76]. In addition, advances in genome editing technologies now under development may offer gene correction therapies without the use of viral vectors, reducing the risks of insertional mutagenesis and nonphysiologic regulation of gene expression [77, 78].
Since newborn screening for SCID has been implemented, infants have been transplanted at an earlier age when there are suitable donors, and importantly, before opportunistic bacterial infections take place because prophylactic antimicrobials and immunoglobulin replacement therapy have been started soon after birth. Viral infections in these immune-compromised infants are still problematic, and physicians need to remain vigilant to warn parents regarding exposure to respiratory viruses, potential for transmission of cytomegalovirus in breastmilk, and avoidance of live rotavirus vaccines [18, 79, 80].
Non-SCID Newborns with T-Cell Lymphopenia Detected by NBS
In addition to SCID and leaky SCID, secondary targets of TREC newborn screening include infants with T-cell lymphopenia (TCL) that does not meet the SCID definitions given above. These TCL conditions can be categorized into genetic syndromes with T-cell impairment (“Syndromes”), T-cell lymphopenia arising because of a non-immune illness (“Secondary”), certain preterm and low-birth-weight infants (“Preterm”), and infants with “variant SCID” or idiopathic T-lymphopenia (“Idiopathic TCL”). Screening programs have detected low TRECs in these non-SCID TCL infants at different rates that depend on programmatic selection of TREC and T-cell cutoffs. In the most common TCL category of Syndromes, DiGeorge/chromosome 22q11 deletion leads the list. This syndrome includes a failure of T-cell production due to thymic atrophy and/or intrinsic T- and B-cell defects related to the TBX1 gene in the commonly deleted region.
Among newborns with DiGeorge syndrome only a small proportion have sufficiently low T cells to be identified by SCID NBS. In complete DiGeorge syndrome, a very rare disorder in which there is aplasia of the thymus, TRECs and T cells are undetectable, and experimental thymus transplantation may be required. The number of partial DiGeorge cases detected by NBS depends on where each screening program has set its TREC and T-cell cutoffs. For example, in the California newborn screening program and several others that define significant TCL as <1,500 T cells/μL, only about 5 % of all infants with 22q deletion are expected to be identified by TREC screening [17••].
Down syndrome/trisomy 21 is also a common cause of low TRECs and low T cells, with other causes being CHARGE (heart defect, atresia choanae, retarded growth and development, genital abnormality, and ear abnormality) syndrome, Fryns syndrome, Nijmegen breakage syndrome, and ataxia telangiectasia [81].
Secondary causes of TCL include congenital heart disease (other than in association with chromosome 22q deletion) in which neonatal surgery or vascular leakage cause third-spacing of fluid and lymphocytes; gastrointestinal malformations, intestinal lymphangiectasia, and hydrops also cause increased T-cell loss, while neonatal leukemia is associated with low T cells due to leukemic cell infiltration of the bone marrow. The Preterm category of TCL refers to a small proportion of the infants born often at or before 30 weeks’ gestation and who weigh <1,500 gm at birth. If they survive their prematurity with its attendant complications, these infants generally recover T cells as they mature [17].
Idiopathic TCL describes infants with isolated low T cells without a recognized congenital syndrome who are not as severely immunologically impaired as infants with SCID and do not have hypomorphic SCID mutations characteristic of leaky SCID. T- or B-cell functions may be impaired and the condition may or may not resolve over time. This group of infants, whose existence was not recognized prior to SCID NBS, offer an opportunity to discover, and study genes not previously implicated in T-cell development. Furthermore, the natural history of these infants is of considerable interest. If a gene diagnosis associated with a known syndrome is discovered, the infant is moved to the appropriate category.
As a group, these infants with low T cells from any cause are advised to receive follow-up until their T-cell numbers improve, and some are advised to receive prophylactic antibiotics, immunoglobulin support, irradiated blood products, and avoid live vaccines. Further study will be required to know the benefit of immune interventions in these cases.
Conclusions
As of today, 26 states, District of Columbia, and the Navajo Nation in the U.S. are performing SCID screening with the TREC test, and SCID-affected infants are reliably being detected and promptly referred so as to receive immune-restoring treatments. Population-based screening has determined an unbiased, overall incidence of SCID to be 1 in 58,000 births, nearly double that of previous estimates. As newborn screening for SCID becomes more widespread, subpopulations with higher incidence due to founder mutations will be further elucidated [8•, 33••]. Early detection of SCID-affected infants provides new opportunities to investigate and define molecular etiologies and optimal treatment strategies for SCID infants. Large multi-center collaborations are needed to define and investigate the impact of each of many variables involved in HCT—donor selection, donor cell preparation, SCID genotype, conditioning regimen, GvHD prophylaxis—to determine optimal transplant strategies that can be tailored to specific SCID genotypes. New protocols may be required to take into account the small size and immaturity of the blood-brain barrier in SCID infants detected by NBS to minimize toxicity from treatments previously established for older individuals [82•].
The availability of newborn screening for SCID still requires vigilance from health care professionals to understand and interpret the screen results and to be aware of forms of immune deficiency that are not detected by abnormal TRECs. These include infants who for whatever reason are not screened or appropriately followed up; infants with defects in late T-cell development or function whose ability to make TRECs is preserved; and infants with primary deficiencies of B cells, granulocytes, or other cells that TREC testing did not reveal. Although screening for B-cell immunoglobulin gene rearrangement such as by measuring κ-chain excision circles (KRECs) has been suggested, data on the effectiveness in terms of specificity and sensitivity of this test as used on entire populations has not been reported to date. Protection from infection is paramount for all infants with SCID, T-cell lymphopenia, and indeed all primary immune defects. With further implementation of SCID NBS, we anticipate improved understanding of the underlying disorders and how best to treat them.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182–94.
van der Burg M, Gennery AR. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Pediatr. 2011;170:561–71.
Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002;99:872–8.
• Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. 2011;117:3243–6. A retrospective study of 60 SCID-affected infants demonstrating superior survival in those with SCID diagnosis in newborn period, compared to infants without previous family history.
Chan A, Scalchunes C, Boyle M, Puck JM. Early vs. delayed diagnosis of severe combined immunodeficiency: a family perspective survey. Clin Immunol. 2011;138:3–8.
•• Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014;371:434–46. A large retrospective study of transplantation outcomes of 240 SCID-affected infants over 10 years, that demonstrate excellent survival after hematopoietic cell transplants from all graft sources in infants with SCID identified before the onset of infections, as well as in asypmtomatic infants who had resolved infections.
Buckley RH, Schiff RI, Schiff SE, Markert ML, Williams LW, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr. 1997;130:378–87.
• Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. 2013;33:1156–64. A multi-center collaboration describing detailed baseline clinical and immunologic features of 50 SCID-affected infants, and initial outcomes following treatments.
• Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129:597–604 quiz 605-596. A review on early findings of several SCID newborn screening programs, including implementation and follow up..
Moore EC, Meuwissen HJ. Screening for ADA deficiency. J Pediatr. 1974;85:802–4.
Hirschhorn R. Adenosine deaminase deficiency. Immunodefic Rev. 1990;2:175–98.
Kalman L, Lindegren ML, Kobrynski L, Vogt R, Hannon H, et al. Mutations in genes required for T-cell development: IL7R, CD45, IL2RG, JAK3, RAG1, RAG2, ARTEMIS, and ADA and severe combined immunodeficiency: HuGE review. Genet Med. 2004;6:16–26.
Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115:391–8.
• Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: The winner is T-cell receptor excision circles. J Allergy Clin Immunol. 2012;129:607–16. A review on early efforts to develop a newborn screening test for SCID, the biology of the TREC test, its validation and application as a screening test to detect SCID and other T cell lymphopenic disorders.
McGhee SA, Stiehm ER, McCabe ER. Potential costs and benefits of newborn screening for severe combined immunodeficiency. J Pediatr. 2005;147:603–8.
Chan K, Davis J, Pai SY, Bonilla FA, Puck JM, et al. A Markov model to analyze cost-effectiveness of screening for severe combined immunodeficiency (SCID). Mol Genet Metab. 2011;104:383–9.
•• Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: Results of the first 2 years. J Allergy Clin Immunol. 2013;132:140–150 e147. Newborn screen outcomes from California, U.S., with nearly 1 million infants screened by TREC test. SCID-affected infants were detected, as well as other conditions with low T cells, all of whom were referred for prompt treatment.
Shearer WT, Fleisher TA, Buckley RH, Ballas Z, Ballow M, et al. Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J Allergy Clin Immunol. 2014;. doi:10.1016/j.jaci.2013.11.043.
Bakare N, Menschik D, Tiernan R, Hua W, Martin D. Severe combined immunodeficiency (SCID) and rotavirus vaccination: reports to the Vaccine Adverse Events Reporting System (VAERS). Vaccine. 2010;28:6609–12.
de Villartay JP. V(D)J recombination deficiencies. Adv Exp Med Biol. 2009;650:46–58.
Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–5.
Hazenberg MD, Otto SA, Cohen Stuart JW, Verschuren MC, Borleffs JC, et al. Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nat Med. 2000;6:1036–42.
Morinishi Y, Imai K, Nakagawa N, Sato H, Horiuchi K, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J Pediatr. 2009;155:829–33.
Grazioli S, Bennett M, Hildebrand KJ, Vallance H, Turvey SE, et al. Limitation of TREC-based newborn screening for ZAP70 Severe Combined Immunodeficiency. Clin Immunol. 2014;153:209–10.
Kuo CY, Chase J, Lloret MG, Stiehm ER, Moore T, et al. Newborn screening for severe combined immunodeficiency does not identify bare lymphocyte syndrome. J Allergy Clin Immunol. 2013;. doi:10.1016/j.jaci.2013.04.024.
Wilson JM, Jungner YG. Principles and practice of mass screening for disease. Bol Oficina Sanit Panam. 1968;65:281–393.
Lipstein EA, Knapp AA, Perrin JM. Evidence review: severe combined Immunodeficiency (SCID). Edited by Perrin JM; 2009:1–80.
Hamilton BE, Martin JA, Osterman MHK, Curtin SC. Births: preliminary data for 2013. National Vital Statistics Reports, vol. 63. Hyattsville: National Center for Health Statistics; 2014.
Adams SP, Rashid S, Premachandra T, Harvey K, Ifederu A, et al. Screening of neonatal UK dried blood spots using a duplex TREC screening assay. J Clin Immunol. 2014;34:323–30.
Audrain M, Thomas C, Mirallie S, Bourgeois N, Sebille V, et al. Evaluation of the T-cell receptor excision circle assay performances for severe combined immunodeficiency neonatal screening on Guthrie cards in a French single centre study. Clin Immunol. 2014;150:137–9.
Somech R, Lev A, Simon AJ, Korn D, Garty BZ, et al. Newborn screening for severe T and B cell immunodeficiency in Israel: a pilot study. Isr Med Assoc J. 2013;15:404–9.
•• Hannon WH, Abraham RS, Kobrynski L, Vogt RF Jr, Adair O, et al. Newborn blood spot screening for severe combined immunodeficiency by measurement of T-cell receptor excision circles; Approved Guideline. Wayne: Clinical and Laboratory Standards Institute; 2013 [Institute CaLS (Series Editor), vol 33.]. Comprehensive guidelines and standards for laboratories implementing SCID newborn screening using PCR detection of TRECs on dried blood spots.
•• Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312:729–38. This article describes the SCID newborn screening results from 11 screening programs in the U.S., including over 3 million infants screened, and establishes the first unbiased population estimate of SCID incidence.
• Verbsky JW, Baker MW, Grossman WJ, Hintermeyer M, Dasu T, et al. Newborn screening for severe combined immunodeficiency; the Wisconsin experience (2008-2011). J Clin Immunol. 2012;32:82–8. Newborn screening outcomes of 3 years from Wisconsin, U.S., with 200,000 infants screened.
Gerstel-Thompson JL, Wilkey JF, Baptiste JC, Navas JS, Pai SY, et al. High-throughput multiplexed T-cell-receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin Chem. 2010;56:1466–74.
• Vogel BH, Bonagura V, Weinberg GA, Ballow M, Isabelle J, et al. Newborn screening for SCID in New York State: experience from the first two years. J Clin Immunol. 2014;34:289–303. Newborn screening outcomes of 2 years from New York, U.S., with nearly 500,000 infants screened.
•• Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. 2014;133:1092–8. This paper describes the establishment of laboratory criteria to define SCID and leaky SCID in the era of newborn screening.
Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122:1082–6.
• Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol. 2011;141:73–82. A description of the variable presentations of infants with hypomorphic mutations in genes associated with SCID.
Yao CM, Han XH, Zhang YD, Zhang H, Jin YY, et al. Clinical characteristics and genetic profiles of 44 patients with severe combined immunodeficiency (SCID): report from Shanghai, China (2004-2011). J Clin Immunol. 2013;33:526–39.
Rozmus J, Junker A, Thibodeau ML, Grenier D, Turvey SE, et al. Severe combined immunodeficiency (SCID) in Canadian children: A National Surveillance Study. J Clin Immunol. 2013;. doi:10.1007/s10875-013-9952-8.
Suliaman F, Al-Ghonaium A, Harfi H. High incidence of severe combined immune deficiency in the Eastern Province of Saudi Arabia. Pediatr Asthma Allergy Immunol. 2006;19:14–8.
Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000;20:477–85.
Baumgart KW, Britton WJ, Kemp A, French M, Roberton D. The spectrum of primary immunodeficiency disorders in Australia. J Allergy Clin Immunol. 1997;100:415–23.
Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, et al. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 1993;123:564–72.
Ryser O, Morell A. Hitzig WH: Primary immunodeficiencies in Switzerland: first report of the national registry in adults and children. J Clin Immunol. 1988;8:479–85.
Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974-1979. J Clin Immunol. 1982;2:86–92.
Hayakawa H, Iwata T, Yata J, Kobayashi N. Primary immunodeficiency syndrome in Japan. I. Overview of a nationwide survey on primary immunodeficiency syndrome. J Clin Immunol. 1981;1:31–9.
Modell V, Knaus M, Modell F. An analysis and decision tool to measure cost benefit of newborn screening for severe combined immunodeficiency (SCID) and related T-cell lymphopenia. Immunol Res. 2014;60:145–52.
Etzioni A. World Primary Immunodeficiency Week: a call for newborn screening. Eur J Immunol. 2014;44:925–6.
Gaspar HB, Hammarstrom L, Mahlaoui N, Borte M, Borte S. The case for mandatory newborn screening for severe combined immunodeficiency (SCID). J Clin Immunol. 2014;34:393–7.
Grunebaum E. A drop of prevention is worth a liter of cure: the case for newborn screening for severe T cell immune deficiency in Israel. Isr Med Assoc J. 2013;15:445–6.
Griffith LM, Cowan MJ, Kohn DB, Notarangelo LD, Puck JM, et al. Allogeneic hematopoietic cell transplantation for primary immune deficiency diseases: current status and critical needs. J Allergy Clin Immunol. 2008;122:1087–96.
Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–55.
Haldane JB. The rate of spontaneous mutation of a human gene. 1935. J Genet. 2004;83:235–44.
Kutukculer N, Gulez N, Karaca NE, Aksu G, Berdeli A. Novel mutations and diverse clinical phenotypes in recombinase-activating gene 1 deficiency. Ital J Pediatr. 2012;38:8.
• Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133:1099–108. A systematic study of recombination activity and its correlation to phenotype and clinical presentation and immunologic manifestations.
Ij H, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol. 2014;133:1124–33.
Patiroglu T, Akar HH, Gilmour K, Ozdemir MA, Bibi S, et al. Atypical severe combined immunodeficiency caused by a novel homozygous mutation in Rag1 gene in a girl who presented with pyoderma gangrenosum: a case report and literature review. J Clin Immunol. 2014;34:792–5.
• Samuels ME, Majewski J, Alirezaie N, Fernandez I, Casals F, et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J Med Genet. 2013;50:324–9. Two whole exome sequencing studies that describe a new gene mutation identified to cause gastrointestinal atresias and immune deficiency in neonates.
• Chen R, Giliani S, Lanzi G, Mias GI, Lonardi S, et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol. 2013;132:656–664 e617. Two whole exome sequencing studies that describe a new gene mutation identified to cause gastrointestinal atresias and immune deficiency in neonates.
Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology. 2014;146:1028–39.
Agarwal NS, Northrop L, Anyane-Yeboa K, Aggarwal VS, Nagy PL, et al. Tetratricopeptide repeat domain 7A (TTC7A) mutation in a newborn with multiple intestinal atresia and combined immunodeficiency. J Clin Immunol. 2014;34:607–10.
Worth AJ, Booth C, Veys P. Stem cell transplantation for primary immune deficiency. Curr Opin Hematol. 2013;20:501–8.
Gaspar HB, Qasim W, Davies EG, Rao K, Amrolia PJ, et al. How I treat severe combined immunodeficiency. Blood. 2013;122:3749–58.
Hassan A, Booth C, Brightwell A, Allwood Z, Veys P, et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012;120:3615–24 quiz 3626.
Eapen M, Ahn KW, Orchard PJ, Cowan MJ, Davies SM, et al. Long-term survival and late deaths after hematopoietic cell transplantation for primary immunodeficiency diseases and inborn errors of metabolism. Biol Blood Marrow Transplant. 2012;18:1438–45.
Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126(602–610):e601–11.
Szabolcs P, Cavazzana-Calvo M, Fischer A, Veys P. Bone marrow transplantation for primary immunodeficiency diseases. Pediatr Clin North Am. 2010;57:207–37.
Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–58.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3:97ra79.
Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635–46.
Cavazzana-Calvo M, Fischer A, Hacein-Bey-Abina S, Aiuti A. Gene therapy for primary immunodeficiencies: part 1. Curr Opin Immunol. 2012;24:580–4.
Gaspar HB, Bjorkegren E, Parsley K, Gilmour KC, King D, et al. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14:505–13.
Qasim W, Gennery AR. Gene therapy for primary immunodeficiencies: current status and future prospects. Drugs. 2014;74:963–9.
Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Expert Opin Biol Ther. 2014;14:789–98.
Matsubara Y, Chiba T, Kashimada K, Morio T, Takada S, et al. Transcription activator-like effector nuclease-mediated transduction of exogenous gene into IL2RG locus. Sci Rep. 2014;4:5043.
Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–40.
Lanzieri TM, Dollard SC, Josephson CD, Schmid DS, Bialek SR. Breast milk-acquired cytomegalovirus infection and disease in VLBW and premature infants. Pediatrics. 2013;131:e1937–45.
Nijman J, de Vries LS, Koopman-Esseboom C, Uiterwaal CS, van Loon AM, et al. Postnatally acquired cytomegalovirus infection in preterm infants: a prospective study on risk factors and cranial ultrasound findings. Arch Dis Child Fetal Neonatal Ed. 2012;97:F259–63.
Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol. 2013;33:540–9.
• Long-Boyle J, Savic R, Yan S, Bartelink I, Musick L, et al. Population pharmacokinetics of busulfan in Pediatric and young adult patients undergoing hematopoietic cell transplant: a model-based dosing algorithm for personalized therapy and implementation into routine clinical use. Ther Drug Monit. 2014;. doi:10.1097/FTD.0000000000000131. This paper demonstrates the use of pharmacokinetic models to individualize busulfan chemotherapy dosing for infants to achieve targeted dosing and limit drug-related toxicity.
Acknowledgments
The authors would like to acknowledge Dr. Robert Currier, California Department of Public Health, and the many individuals who have contributed to SCID newborn screening. Funding was received from the National Institutes of Health (NIH) to JMP (R01 AI078248, AI105776), HCA International Foundation Traveling Scholarship (AK), Primary Immune Deficiency Treatment Consortium (NIH U54 A1082973), Immune Deficiency Foundation, and the Jeffrey Modell Foundation.
Disclosure
Antonia Kwan and Jennifer M. Puck declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Immunology.
Rights and permissions
About this article

Cite this article
Kwan, A., Puck, J.M. Newborn Screening for Severe Combined Immunodeficiency. Curr Pediatr Rep 3, 34–42 (2015). https://doi.org/10.1007/s40124-014-0068-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-014-0068-2