
Opinion statement
Aging is the most potent of carcinogens, especially for the bone marrow stem cell clonal disorders called myelodysplastic syndromes (MDS). Age-associated changes in the microenvironment or the soil of the bone marrow (BM) as well as in the cell or the seed provide a growth advantage for clonal myeloid cells. Slowly accumulating senescent cells which can no longer divide because they have reached the end of their proliferative life cycle, but which continue to produce metabolic debris, overwhelm the natural autophagy mechanisms resulting in pro-inflammatory changes in the BM soil. In addition, the seed or stem cells acquire passenger mutations with each round of proliferation resulting from DNA copying errors. Some mutations commonly associated with MDS can be found in older, otherwise healthy individuals; however, when combined with other passenger mutations or in the setting of a noxious soil, the result could be a proliferative advantage for one stem cell over others, leading to its clonal expansion and development of the clinical syndrome. When considering therapeutic options for MDS patients, the important considerations are related to both the common co-morbidities of an elderly population along with the heterogeneous passenger mutations and the inflammatory changes in the soil. At present, allogeneic stem cell transplant is the only potentially curative option in MDS. Palliative strategies are directed at improving the quality of life and prolonging survival. Only three drugs are FDA approved, two being the hypomethylating agents azacytidine and decitabine while the third is lenalidomide which is restricted to lower risk MDS patients with deletion 5q. Promising future therapies are directed at reversing the pro-inflammatory changes in the microenvironment (luspatercept) or targeting specific mutations isocitrate dehydrogenase (IDH)1, IDH2, p53, EZH2. More durable responses are to be expected when the seed and soil are targeted simultaneously through a combination of drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of clonal myeloid disorders characterized by impaired hematopoiesis resulting in cytopenias and dysmyelopoiesis. MDS patients are at an increased risk of evolution to acute myeloid leukemia (AML). Owing to the fact that MDS are very dissimilar in their genetic characteristics and molecular pathogenesis, the natural history is also highly variable with survival ranging from few months to several years [1]. Accordingly, treatment options vary from supportive measures to hematopoietic stem cell transplant (HSCT). An accurate assessment of prognosis is mandatory to offer the most appropriate treatment option.
In the last decade, significant efforts have been made to understand the complexity of the disease biology, leading to the identification of recurrently mutated genes with well-defined clinical, prognostic, and therapeutic implications [2,3,4]. However, this has not been reflected in an improvement of the treatment options. Currently, there are only three approved drugs and none is curative. HSCT is the only potentially curative procedure but with limited applicability in patients with a median age of 76 years and age-related comorbidities [5,6,7,8].
In this review, we will discuss the therapeutic options of MDS patients using a risk-adapted algorithm and describe our recommendations for the best management of these patients. Finally, a review of the most encouraging novel agents will be provided.
Risk stratification
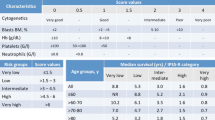
An individual risk-adapted treatment approach relying on the patient’s age, comorbidities, risk of death, and leukemic transformation is mandatory in MDS. Currently, the most commonly used tool for risk stratification is the International Prognostic Scoring System Revised (IPSS-R) [9]. This score is based on the number and severity of cytopenias, bone marrow blasts percentage, and specific cytogenetic abnormalities. Although IPSS-R provides a more accurate risk stratification than the former IPSS [10], it continues to have limitations: it is complex, it lacks molecular information, and its reproducibility is inconsistent with special concern related to the intermediate-risk group [11, 12]. The IPSS-R stratifies patients into five groups. However, in daily practice, the two lowest groups (very low and low) are considered lower-risk MDS, while the two highest groups (high and very high) are referred to as higher-risk MDS. The intermediate group can present very heterogeneous outcomes; some patients present an indolent disease, and others rapidly evolve to AML. Furthermore, lower- or intermediate-risk patients, who failed in first-line therapy, should be handled as higher-risk.
More recently, recurrent somatic mutations have been reported to influence the prognosis of MDS. Incorporating them into the risk assessment, especially in the lower- and intermediate-risk groups, may provide more accurate risk stratification. Among the mutated genes, TP53, ASXL1, NRAS, SFRSF2, and EZH2 are consistently associated with poor outcomes, whereas SF3B1 is the only related to a better survival [2,3,4, 13•, 14]. A molecular prognostic index (IPSS-Rm) including more than 3000 patients is under development. A recent study from our group focused on erythropoiesis has shown that presence or absence of quantifiable terminal erythroid differentiation (TED) is an independent prognostic factor for survival in MDS patients. Patients in whom TED was quantifiable (TED-positive) or not (TED-negative) showed significantly different overall survival (OS). Importantly, TED status further refined the prognostic power in all IPSS-R risk groups [15].
Treatment approach for lower-risk MDS patients
The erythroid lineage is the most frequently affected in MDS. Consequently, anemia is present in 85% MDS patients at diagnosis or during the course of the disease, with half of the patients with hemoglobin (Hb) levels < 10 g/dL [9]. Very little is known about the molecular and cellular basis of erythroid differentiation in MDS patients. The defects can affect early or late stages of differentiation. Our recent work showed that the majority of MDS patients had abnormal terminal erythroid differentiation [15]. Compared to normal individuals, MDS patients had a smaller number of cells in proerythroblast and early basophilic erythroblast stages, but significantly high number of cells in polychromatic stages with a reduced number of cells in orthochromatic stages [15]. Life expectancy of lower-risk patients is expected to be long [OS > 30 months]; therefore, the goal is to alleviate the anemia in order to improve their quality of life. Treatment of the anemia in lower-risk MDS patients depends on the presence of the deletion 5q [del(5q)]. A treatment algorithm is described in Fig. 1.

Treatment algorithm for myelodysplastic syndromes. G-CSF granulocyte colony-stimulating factor, HMAs hypomethylating agents, TPO ag thrombopoietin receptor analogs, IST immunosuppressive therapy, ESAs erythropoiesis-stimulating agents, RS ring sideroblasts. a If initially is managed as lower- risk but fails to respond move to higher risk management strategies. b IPSS-R intermediate patients may be managed as lower- or higher-risk depending on additional prognostic factors such as age, performance status, serum ferritin levels and serum LDH levels.
First-line treatment of anemia in MDS without del(5q)
Erythropoiesis-stimulating Agents
The off-label use of erythropoiesis-stimulating agents (ESAs) is the first choice of supportive care for anemia in low risk patients. High doses of ESA, epoetin or darbepoetin alpha, yield 60% of erythroid responses within 8–12 weeks with a median duration of response lasting up to 2 years [16]. It is recommended to administer ESAs regularly to maintain Hb levels from 10 to 12 g/dL. The main predictors of response include endogenous erythropoietin (EPO) of < 500 U/L and low number of blood transfusions (≤ 2 U/month) [17, 18]. The efficacy of ESAs can be improved with synergistic activity of granulocyte colony-stimulating factor (G-CSF); hence, a trial with GCS-F should be considered in patients unresponsive to ESAs after 4–8 weeks, especially in those with ≥ 15% ring sideroblasts [19]. Treatment should be abandoned if there is no improvement after 8–16 weeks.
Second-line treatments for anemia in MDS without del(5q)
Lower-risk MDS unresponsive or refractory to ESAs remains a challenge because of the intrinsically unfavorable prognosis and the discouraging treatment options, most patients remaining transfusion-dependent.
Hypomethylating Agents
At present, four prospective studies of azacitidine in low risk transfusion-dependent non-del(5q) MDS unresponsive or refractory to ESAs have been published with reported rates of transfusion independence (TI) ranging from 15 to 30% with a median duration of < 12 months [20,21,22,23]. In the Spanish study, there was no difference in overall survival between the azacitidine and BSC arm [23]. Similar results have been reported with decitabine [24]. Hypomethylating agents (HMAs) are approved for lower-risk non-del(5q) MDS in the USA and Japan but not in Europe.
Lenalidomide
Lenalidomide has been shown to induce erythroid differentiation in lower-risk non-del(5q) MDS ineligible or refractory to ESAs resulting in 26% of durable erythroid responses with best responses (42%) in patients with serum EPO levels < 100 U/L [25, 26•]. Recently, the combination of lenalidomide plus ESAs has proved to be a promising option with durable and significantly higher rates of erythroid responses without added toxicity [27•].
The question of the optimal order of lenalidomide and HMAs use after ESAs failure remains to be addressed. In a retrospective analysis, rates of erythroid improvement were significantly higher when lenalidomide was used as first-line rather than second-line treatment (38% vs. 12%), with no differences when azacitidine was used before or after lenalidomide [28]. Because of better responses with lenalidomide than azacitidine (26% vs. 17%), it is our practice to start with lenalidomide ESAs and switch to HMAs if there is no response.
First-line treatment of anemia in MDS with deletion 5q
Lenalidomide
Lenalidomide is an immunomodulating agent approved for lower-risk MDS with del(5q) with or without additional cytogenetic abnormalities and transfusion-dependent anemia. TI is observed in almost 70% of the subjects and 50–70% experience cytogenetic responses with a median response duration of 24 months [29, 30]. Obtaining TI was associated with a significantly reduced risk of AML progression and death [31]. A debate persists about the timing of lenalidomide initiation in anemic del(5q) patients: American guidelines advocate for lenalidomide as first-line, while European recommend it after a trial of ESAs [32•, 33,34,35,36]. However, ESAs are less effective in del(5q) as compared to non-del(5q) MDS, and lenalidomide can trigger deeper responses with cytogenetic remissions, potentially modifying the natural history of the disease [37]. Furthermore, there is evidence that lenalidomide is also effective in non-transfusion dependent del(5q) patients [38]. Accordingly, the Spanish study SINTRA-REV (NCT01243476) is assessing if early treatment with lenalidomide delays transfusion dependence in del(5q) MDS. In view of the above, we recommend starting lenalidomide as first-line. Treatment is maintained until loss of response, with no data available regarding drug interruption after sustained complete responses (CR).
Special consideration should be taken with del(5q) patients and TP53 mutations (18%) due to its elevated risk of AML evolution. These patients should be considered for a more aggressive approach such as HMAs, HSCT or clinical trials [39].
Second-line treatments for anemia in MDS with del(5q)
Despite the effectiveness of lenalidomide in del(5q) MDS, 30% patients remain unresponsive. Moreover, for responders, the median duration of response is approximately 2 years [29]. Treatment with HMAs may have some potential benefits in these patients [40]. However, HSCT or clinical trials should be considered when possible.
RBC transfusion and iron chelation
Up to 40% of all MDS patients fail treatments or cannot be considered for active interventions and will remain transfusion-dependent. Transfusion should be based on symptoms and comorbidities rather than Hb levels. However, maintaining a Hb level between 8 and 10 g/dL is recommended [32•, 33,34,35,36]. Regular RBC transfusions are associated with worse prognosis, in part reflecting a more aggressive disease but also due to severe anemia and toxicities, mainly organ damage due to iron overload [41]. Iron chelation with deferasirox may be helpful in selected cases; however, the effect of chelation on outcomes is still controversial and can be associated with adverse effects [42]. A randomized trial of deferasirox vs. placebo in transfusion-dependent lower-risk MDS with ferritin levels > 1000 ng/mL is about to be published (NCT00940602). At present, chelation is recommended in patients with regular transfusions (≥ 2 units/month) and/or serum ferritin concentration ≥ 1000 ng/mL, and a life expectancy of at least 1 year or candidate to HSCT [43].
Treatment of neutropenia and thrombocytopenia
Limited effective options exist for cytopenias beyond anemia in lower-risk patients. Neutropenia often does not respond to G-CSF and some concerns exist about promoting clonal expansion. Its use has not been shown to reduce episodes of febrile neutropenia or to improve survival [44]. Therefore, G-CSF or antibiotics should be recommended in selected patients with severe neutropenia and recurrent infections. Platelet transfusions may improve bleeding temporarily in severe thrombocytopenia, but are ineffective in the long term. Thrombopoiesis-stimulating agents have been used in lower-risk MDS with chronic severe thrombocytopenia (this will be discussed below). Interestingly, HMAs appear to give platelet and neutrophils responses in 30% of lower-risk patients, in addition to erythroid responses [23].
Watchful waiting strategy
Some patients with MDS present with borderline cytopenias and are asymptomatic at diagnosis. In these cases, observation is recommended with regular follow-up until progression. This approach is supported by several studies that showed comparable survival in stable lower-risk MDS and the general population [45]. There are no proven advantages of and early intervention in this group of MDS.
Emerging agents
The new drugs in lower risk MDS are directed at ameliorating cytopenias to improve the quality of life. Hereafter, we describe the most relevant emerging options. For more information, the reader is referred to Table 1.
Luspatercept
Luspatercept (ACE-536) is a recombinant fusion protein that promotes late-stage erythroid differentiation by blocking transforming growth factor beta (TGFβ) superfamily inhibitors of erythropoiesis, especially GDF11. Luspatercept has been effective for the treatment of anemia in lower-risk MDS, especially in those with ring sideroblasts [46••]. Based on these results, a phase 3 trial has been conducted to assess the safety and efficacy of luspatercept in lower-risk MDS syndromes with anemia and ≥ 15% ring sideroblasts and/or SF3B1 mutation (NCT02631070). Early results indicate that the trial met both its primary and secondary end points.
Sotatercept
Sotatercept (ACE-011), another novel molecule similar to luspatercept, has been well tolerated and effective for the treatment of anemia in patients with lower-risk MDS who previously failed ESA [47].
TPO receptor analogs
Two thrombopoietin (TPO) analogs, already approved for the treatment of immune thrombocytopenic purpura and aplastic anemia, have shown efficacy in thrombocytopenic lower-risk MDS. Romiplostim is a peptide TPO mimetic administered subcutaneously, whereas eltrombopag is an oral non-peptide TPO receptor that interacts with the transmembrane domain of the TPO receptors, resulting in megakaryocyte and platelet development. About 30% of patients have experienced improvement in platelet counts and decreased bleeding events [48, 49]. Initial concerns of AML risk were not confirmed [49]. However, as TPO stimulates early hematopoietic progenitor cells, an increase in the number of blasts can be expected. Thus, its use is limited to lower-risk MDS. Both TPO analogs have been used in combination with HMAs and lenalidomide with limited results, especially with romiplostim, and further studies are required [50].
ARRY-614
Activation of p38 MAPK has recently been implicated in the pathophysiology of MDS, and dysregulation of Tie2 has been correlated with poor prognosis. ARRY-614, an oral dual inhibitor of p38 MAPK and Tie2, was associated with clinical responses in a group of heavily pretreated lower-risk patients [51].
Treatment approach higher-risk MDS patients
Therapeutic strategies in higher-risk MDS are directed at improving survival (OS < 30 months and AML transformation in 30% of the patients). Initial approach depends on whether or not the patient is eligible for HSCT. A treatment algorithm is described in Fig. 1.
Allogenic HSCT
Allo-transplant represents the only potentially curative option but with limited applicability (≈ 10% of patients), because of patient’s age and the significant morbidity and mortality of the procedure. Two studies based on Markov model analysis concluded that early transplantation improves the life expectancy of higher-risk MDS, whereas the opposite results were seen in lower-risk MDS [52, 53••]. Transplant with HLA-matched unrelated donor produces comparable results to HLA-matched sibling transplant. Alternative sources, such as cord blood and HLA-haploidentical related donors, may be a valid option for patients without matched donors [54]. Recently, a phase 3 trial compared myeloablative conditioning regimen (MAC) with reduced-intensity conditioning (RIC). The latter resulted in lower treatment-related mortality but higher relapse rates compare to MAC conditioning [55]. These support the use of MAC in younger and fitter patients. Based on the best results of HSCT in CR, a pretransplant bridging therapy is appropriate, especially in those receiving RIC regimens or when > 10% blasts are present, and when transplant is delayed (i.e., donor availability). At present, HMAs are preferred for pretransplant cytoreduction instead of AML-like chemotherapy because of similar outcomes with both strategies but markedly lower toxicity with HMAs [56]. Another important issue is to identify which patients benefit most from transplant. In this regard, it has been reported that the survival of patients with monosomy 7 is very poor after HSTC [57], whereas azacitidine seems to be effective [58]. However, whether HSCT offers a survival advantage compared to HMAs remains a question to be elucidated. Two prospective trials are expected to shed light on this topic (NCT01404741 and BMT CTN 1102).
Hypomethylating agents
For non-transplant candidates, HMAs are the first choice, either azacitidine or decitabine. These cytosine analogs promote decreased methylation through DNA methyltransferase inhibition and subsequent gene expression. Azacitidine may be preferred compared to decitabine because it has demonstrated to be superior to conventional care approaches (OS 24.5 vs. 15 months), while with decitabine no clear survival improvement was shown [5, 59]. To date, there is no trial that has compared both HMAs directly.
The optimal schedule of azacitidine is 75 mg/m2/day for 7 days every 28 days, whereas for decitabine, it is 20 mg/m2 for 5 days every 28 days. Alternative schemes, avoiding the weekend, have been studied with similar response rates in lower-risk, but there is no data available in higher-risk MDS [60]. Responses to HMAs are slow; a minimum of six cycles of azacitidine or four of decitabine is recommended before assessing responses as long as there is no disease progression. HMAs do not eradicate the clonal stem cells; thus, relapse is unavoidable. Outcomes after failure or no response to HMAs are poor with median survival being less than 6 months [61]. There is no second-line approved treatment; however, if the patient is eligible, HSCT is recommended. Whenever possible, inclusion in clinical trials with investigational agents is mandatory.
Emerging therapies
Upcoming novel strategies targeting associated somatic mutations and specific altered molecular pathways in high risk patients are being explored. The most relevant therapies are described below. All new drugs under development in MDS are listed in Table 1.
DNA hypomethylation
Building on the success of HMAs in MDS, next-generation HMAs are under investigation. Guadecitabine (SGI-110), a hypomethylating dinucleotide of decitabine and deoxyguanosine resistant to degradation by cytidine deaminase resulting in prolonged drug exposure, showed 31% responses in relapsed or refractory MDS [62]. The oral formulation of azacitidine (CC-486) enables long-term, lower-doses schedules that may enhance therapeutic activity by increasing exposure to malignant cells.
Spliceosome modulation
Mutations in the genes encoding the RNA splicing factors (SF3B1, U2AF1, SRSF2) represent, overall, the most common mutations in MDS patients. A phase 1 study with an oral modulator of the SF3B1 complex (H3B-8800) which potently targets spliceosome-mutant cells is underway (NCT02841540) [63].
Immune checkpoint inhibitors
Abnormal activation of the innate immune system and its associated inflammation are involved not only in the pathogenesis but also in AML evolution. Immune checkpoints like CTLA-4, PD-1, and PDL-1 are involved in the maintenance of self-tolerance to prevent autoimmunity, thus preventing organ damage [64]. In high risk MDS, checkpoint receptors are upregulated favoring immune evasion. Nivolumab (anti-PD-1), pembrolizumab (anti-PD-1), ipilimumab (anti-CTL4), and atezolizumab (anti-PDL-1) have limited activity as single agents, though combination therapies, with HMAs or with other checkpoints inhibitors, have shown promising results in high risk MDS and AML [65].
Cell signaling pathway inhibitors
Rigosertib is a small molecule that acts as a RAS mimetic and binds to the Ras binding domain of RAS, PI3K, and RalGDS, resulting in the inhibition of the RAS-RAF-MEK pathway. Data from a recently published first phase 3 trial of a new drug for MDS after HMA failure showed that survival was significantly longer in very-high IPSS-R patients treated in the rigosertib arm compared with the BSC group (7.6 vs. 3.2 months). By contrast, no difference in survival between groups was evident among the patients with a lower-risk IPSS-R [66]. According to these results, a phase 3 trial with rigosertib in very high IPSS-R patients is underway (NCT02562443).
IDH inhibitors
Up to 5% of MDS patients have mutations in isocitrate dehydrogenase (IDH)1 or IDH2. Results with ivosidenib and enasidenib, IDH2 and IDH1 inhibitors, respectively, have been encouraging in MDS with overall responses rates of 50% [67••].
Anti-apoptotic protein inhibitors
The BCL-2 protein tends to be overexpressed in AML and high risk MDS where it has been implicated in chemotherapy resistance and poor outcomes. Venetoclax, a potent oral inhibitor of BCL-2, has shown promising results in AML in combination with cytarabine [68]. An ongoing phase 1 study is evaluating the efficacy of venetoclax alone and in combination with azacitidine in higher-risk MDS after HMA failure (NCT02966782).
Perspective
It is an undeniable fact that, so far, all approved treatments for MDS have arrived at the bedside by serendipity. The first FDA-approved strategy is the use of HMAs in MDS, azacytidine arriving in the market in 2004 followed shortly by decitabine. HMAs were not initially developed for MDS. Azacytidine was given to many types of cancer patients, but the best responses were seen in MDS. As for lenalidomide approved for del(5q) type of MDS, the story is even more strange. We had performed detailed cell kinetic studies in MDS patients in 1990s using intravenous infusions of the thymidine analogs bromo and iodo-deoxyuridine (BrdU and IUdR). We showed that the MDS clonal cells were highly proliferative with rapid transition through S-phase. This hyper-proliferation accounted for hyper-cellular bone marrow, but the question of cytopenia remained unanswered [69].
Apoptosis as a distinct process of programmed cellular suicide was described around this time. It would make sense if the so-called ineffective hematopoiesis of MDS was the result of excessive apoptosis in the clonal cells. We looked in the BM aspirates of MDS patients by the standard techniques of detecting apoptosis, which at the time was DNA laddering. We found none. Having already demonstrated the discordance between proliferative rates of BM aspirates and biopsies, the former being three times less than the latter, we were convinced that the real action was likely happening in the biopsy compartment. With this idea, we developed an in situ end labeling (ISEL) method to detect damaged, fragmented DNA in BM biopsies and were able to show that anywhere from 30 to 70% cells in BM biopsies were actively undergoing apoptosis. The paradox of cytopenia and a hyper-cellular marrow was explained by the presence of a hyper-proliferative clone which produced cells that died of premature apoptosis [70].
A study of the BM microenvironment revealed the presence of excessive amounts of tumor necrosis factor alpha (TNFa) and transforming growth factor beta (TGFb). If these cytokines were accelerating the apoptotic program, then their suppression should lead to decreased cell death. No anti-TGFb agents were available, but thalidomide had just been approved for a form of leprosy in the USA. Its mechanism of action was thought to include an anti-TNF component along with being anti-angiogenic and immune-modulatory [71]. We initiated a protocol of thalidomide in MDS patients, treated 83 cases with all types of MDS, and reported a 20% incidence of complete transfusion independence [72]. The company marketing thalidomide, Celgene, had already developed an analog, lenalidomide, which, to everyone’s surprise, showed spectacular responses in a subset of lower risk transfusion-dependent MDS patients who had a del(5q) abnormality [29]. This is the indication for which lenalidomide is approved in MDS. While we suspect many possible modes of action of this agent, we still cannot fully explain the precise basis of the positive effect seen in del(5q) as well as a third of the lower risk MDS patients without del(5q).
What are the next therapeutic frontiers? After more than a decade of stillborn clinical trials, luspatercept seems poised for approval for lower-risk patients with ring sideroblasts. If approved, this drug will be another agent for MDS, which arrives at the bedside by serendipity. It was originally developed for bone issues, but when given to healthy volunteers and some patients with multiple myeloma, it was shown to increase the hemoglobin. It was then tried in anemia patients with MDS and thalassemia and showed the best responses in those with ring sideroblasts. The precise mechanism of its effect remains a mystery.
In this review, we referred to our recent observations regarding TED issues. We showed that erythroid cells in patients with ring sideroblasts reach the penultimate step of TED before becoming arrested. It seems that luspatercept is somehow releasing a block in differentiation [15]. By extension, any MDS patient whose erythroid cells reach this stage should be sensitive to luspatercept. A response prediction test could be developed for luspatercept based on TED profiles in MDS patients regardless of the presence or absence of ring sideroblasts.
Finally, a drug targeting p53 mutation and silencing, APR246, was tried in ~ 250 patients with solid tumors and showed no responses but when tried in liquid tumors, MDS patents with the mutation appeared to benefit. Results are awaited from an ongoing study of this agent in combination with HMA.
Summary
To conclude, biologic studies of freshly obtained MDS cells need to be pursued for future drug development programs. Since most patients with MDS are older, it is impractical to develop potentially toxic therapies like cellular therapies and stem cell transplants. Rather, more attention should be paid to an earlier diagnosis of MDS by screening older individuals with routine mutational profiles to detect clonal hematopoiesis. Finally, more attention should be given to developing therapeutic strategies to target minimal initial disease, much like the minimal residual disease.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361:1872–85.
Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506.
Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27.
Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32.
Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–803.
List A, Kurtin S, Roe DJ, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352:549–57.
De Witte T, Hermans J, Vossen J, et al. Haematopoietic stem cell transplantation for patients with myelodysplastic syndromes and secondary acute myeloid leukaemias: a report on behalf of the Chronic Leukaemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Br J Haematol. 2000;110:620–30.
Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88.
Pfeilstöcker M, Tuechler H, Sanz G, et al. Time-dependent changes in mortality and transformation risk in MDS. Blood. 2016;128:902–10.
Kawabata H, Tohyama K, Matsuda A, et al. Validation of the revised International Prognostic Scoring System in patients with myelodysplastic syndrome in Japan: results from a prospective multicenter registry. Int J Hematol. 2017;106:375–84.
• Bejar R, Papaemmanuil E, Haferlach T, et al. Somatic mutations in MDS patients are associated with clinical features and predict prognosis independent of the IPSS-R: analysis of combined datasets from the International Working Group for Prognosis in MDS Molecular Committee. Blood. 2015;126:907 This study validates the prognostic value of mutations in several MDS-associated genes. Furthermore, mutations in several genes retain their prognostic significance after adjustment for IPSS-R risk groups refining risk prognostication. This study will serve as the template with which to build a molecular risk model for MDS.
Gangat N, Mudireddy M, Lasho TL, et al. Mutations and prognosis in myelodysplastic syndromes: karyotype-adjusted analysis of targeted sequencing in 300 consecutive cases and development of a genetic risk model. Am J Hematol. 2018;93:691–69.
Ali AM, Huang Y, Pinheiro RF, et al. Severely impaired terminal erythroid differentiation as an independent prognostic marker in myelodysplastic syndromes. Blood Adv. 2018;2:1393–402.
Moyo V, Lefebvre P, Duh MS, et al. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol. 2008;87(7):527–36.
Buckstein R, Balleari E, Wells R, et al. ITACA: a new validated international erythropoietic stimulating agent-response score that further refines the predictive power of previous scoring systems. Am J Hematol. 2017;92(10):1037–46.
Hellström-Lindberg E, Gulbrandsen N, Lindberg G, et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol. 2003;120(6):1037–46.
Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111:574–82.
Filı C, Malagola M, Follo MY, et al. Prospective phase II study on 5-days azacitidine for treatment of symptomatic and/or erythropoietin unresponsive patients with low/INT-1-risk myelodysplastic syndromes. Clin Cancer Res. 2013;19:3297–308.
Tobiasson M, Dybedahl I, Holm MS, et al. Limited clinical efficacy of azacitidine in transfusion-dependent, growth factor-resistant, low- and Int-1 risk MDS: results from the nordic NMDSG08 phase II trial. Blood Cancer J. 2014;4:e189.
Thépot S, Abdelali RB, Chevret S, et al. A randomized phase II trial of azacitidine þ/ epoetin-b in lower risk myelodysplastic syndromes resistant to erythropoietic stimulating agents. Haematologica. 2016;101:918–25.
Sanchez-Garcia J, Falantes J, Medina Perez A, et al. Prospective randomized trial of 5 days azacitidine versus supportive care in patients with lower-risk myelodysplastic syndromes without 5q deletion and transfusion-dependent anemia. Leuk Lymphoma. 2018;59(5):1095–110.
Garcia-Manero G, Jabbour E, Borthakur G, et al. Randomized open-label phase II study of decitabine in patients with low- or intermediate-risk myelodysplastic syndromes. J Clin Oncol. (2013);31:2548–53.
Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111(1):86–93.
• Santini V, Almeida A, Giagounidis A, et al. Randomized phase III study of lenalidomide versus placebo in RBC transfusion-dependent patients with lower-risk non-del(5q) myelodysplastic syndromes and ineligible for or refractory to erythropoiesis-stimulating agents. J Clin Oncol. 2016;34(25):2988–96 This study supports the clinical benefits of lenalidomide in lower-risk non-del(5q) MDS.
• List AF, Sun Z, Verma A, et al. Combined treatment with lenalidomide (LEN) and epoetin alfa (EA) is superior to lenalidomide alone in patients with erythropoietin (Epo)-refractory, lower risk (LR) non-deletion 5q [del(5q)] myelodysplastic syndrome (MDS): results of the E2905 Intergroup Study-an ECOG-ACRIN Cancer Research Group Study, Grant CA180820, and the National Cancer Institute of the National Institutes of Health. Blood. 2016;128(22):223 This study shows that combination treatment with lenalidomide and epoetin alfa results in higher response rates without added toxicity. Also describes CD45 isoform as a possible biomarker of response.
Zeidan AM, Al Ali NH, Padron E, et al. Lenalidomide treatment for lower risk nondeletion 5q myelodysplastic syndromes patients yields higher response rates when used before azacitidine. Clin Lymph Myeloma Leuk. 2015;15(11):705–10.
List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–65.
Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765–76.
Komrokji RS, List AF. Short- and long-term benefits of lenalidomide treatment in patients with lower-risk del(5q) myelodysplastic syndromes. Ann Oncol. 2016;27(1):62–8.
• Greenberg PL, Stone RM, Al-Kali A, et al. Myelodysplastic syndromes, version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw. 2017;15:60–87 Newest version of the American MDS clinical practice guidelines.
Grupo Español de Síndromes Mielodisplásicos (GESMD); Sociedad Española de Hematología y Hemoterapia (SEHH): Guías españolas de diagnóstico y tratamiento de los síndromes mielodisplásicos y la leucemia mielomonocítica crónica. http://gesmd.es/pdfs/guias_smd/Haematologia_Guias_SMD.pdf.
Giagounidis A. Current treatment algorithm for the management of lower-risk MDS. Hematology Am Soc Hematol Educ Program 2017;(1):453–59.
Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943–64.
Fenaux P, Haase D, Sanz GF, et al. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25 Suppl 3:iii57–69.
Kelaidi C, Park S, Brechignac S, et al. Treatment of myelodysplastic syndromes with 5q deletion before the lenalidomide era; the GFM experience with EPO and thalidomide. Leuk Res. 2008;32:1049–53.
Oliva EN, Lauseker M, Aloe Spiriti MA, et al. Early lenalidomide treatment for low and intermediate-1 International Prognostic Scoring System risk myelodysplastic syndromes with del(5q) before transfusion dependence. Cancer Med. 2015;4(12):1789–97.
Jadersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol. 2011;29(15):1971–9.
Prebet T, Cluzeau T, Park S, et al. Outcome of patients treated for myelodysplastic syndromes with 5q deletion after failure of lenalidomide therapy. Oncotarget. 2017;8(47):81926–35.
Castelli R, Schiavon R, Deliliers GL, et al. The impact of anaemia, transfusion dependency, comorbidities and polypharmacy in elderly patients with low-risk myelodysplastic syndromes. Med Oncol. 2018;35(3):33.
Cappellini MD, Porter J, El-Beshlawy A, et al. EPIC Study Investigators. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica. 2010;95(4):557–66.
Bennett JM. MDS Foundation’s Working Group on Transfusional Iron Overload. Consensus statement on iron overload in myelodysplastic syndromes. Am J Hematol. 2008;83(11):858–61.
Sloand EM, Yong AS, Ramkissoon S, et al. Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. Proc Natl Acad Sci U S A. 2006;103(39):14483–8.
Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25(23):3503–10.
•• Platzbecker U, Germin U, Götze KS, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338–47 This study shows the efficacy of luspatercept in anemic patients with lower-risk MDS, especially in those who have ≥ 15% ring sideroblasts and/or SF3B1 mutation.
Komrokji R, Garcia-Manero G, Ades L, et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol. 2018;5(2):e63–72.
Giagounidis A, Mufti GJ, Fenaux P, et al. Results of a randomized, double-blind study of romiplostim versus placebo in patients with low/intermediate-1-risk myelodysplastic syndrome and thrombocytopenia. Cancer. 2014;120(12):1838–46.
Oliva EN, Alati C, Santini V, et al. Eltrombopag versus placebo for low-risk myelodysplastic syndromes with thrombocytopenia (EQoL-MDS): phase 1 results of a single-blind, randomised, controlled, phase 2 superiority trial. Lancet Haematol. 2017;4(3):e127–36.
Kantarjian HM, Giles FJ, Greenberg PL, et al. Phase 2 study of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving azacitidine therapy. Blood. 2010;116:3163–70.
Garcia-Manero G, Khoury HJ, Jabbour E, et al. A phase I study of oral ARRY-614, a p38 MAPK/Tie2 dual inhibitor, in patients with low or intermediate-1 risk myelodysplastic syndromes. Clin Cancer Res. 2015;21(5):985–94.
Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104(2):579–85.
•• Della Porta MG, Jackson CH, Alessandrino EP, et al. Decision analysis of allogeneic hematopoietic stem cell transplantation for patients with myelodysplastic syndrome stratified according to the revised International Prognostic Scoring System. Leukemia. 2017;31:2449–57 This study provides the optimal timing and the benefit of cytorreduction before transplant in lower, intermediate, and higher-risk MDS.
Majhail NS, Brunstein CG, Shanley R, et al. Reduced-intensity hematopoietic cell transplantation in older patients with AML/MDS: umbilical cord blood is a feasible option for patients without HLA-matched sibling donors. Bone Marrow Transplant. 2012;47:494–8.
Scott BL, Pasquini MC, Logan BR, et al. Myeloablative versus reduced-intensity hematopoietic cell transplantation for acute myeloid leukemia and myelodysplastic syndromes. J Clin Oncol. 2017;35(11):1154–61.
Gerds AT, Gooley TA, Estey EH, et al. Pretransplantation therapy with azacitidine vs induction chemotherapy and posttransplantation outcome in patients with MDS. Biol Blood Marrow Transplant. 2012;18(8):1211–8.
Van Gelder M, Schetelig J, Volin L, et al. Monosomal karyotype predicts poor outcome for MDS/sAML patients with chromosome 7 abnormalities after allogeneic stem cell transplantation for MDS/sAML. A study of the MDS subcommittee of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Blood. 2009;114:293.
Díez-Campelo M, Lorenzo JI, Itzykson R, et al. Azacitidine improves outcome in higher-risk MDS patients with chromosome 7 abnormalities: a retrospective comparison of GESMD and GFM registries. Br J Haematol. 2018;181(3):350–9.
Lubbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29(15):1987–96.
Lyons RM, Cosgriff TM, Modi SS, et al. Hematologic response to three alternative dosing schedules of azacitidine in patients with myelodysplastic syndromes. J Clin Oncol. 2009;27(11):1850–6.
Prébet T, Gore SD, Thépot S, et al. Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br J Haematol. 2012;157(6):764–6.
Issa JJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16(9):1099–110.
Seiler M, Yoshimi A, Darman R, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24(4):497–504.
Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23.
Daver N, Boddu P, Garcia-Manero G, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia. 2018;32(5):1094–105.
Garcia-Manero G, Fenaux P, Al-Kali A, et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17:496–508.
•• Stein EM, Fathi AT, DiNardo CD, et al. Enasidenib (AG-221), a potent oral inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) enzyme, induces hematologic responses in patients with myelodysplastic syndromes (MDS). Blood. 2016;128:343 The new oral molecule enasidenib induces responses in more than one half of MDS with IDH2 mutation, which included those patients who failed to HMAs agents.
Wei A, Strickland SA, Roboz GJ, et al. Safety and efficacy of venetoclax plus low-dose cytarabine in treatment-naive patients aged ≥ 65 years with acute myeloid leukemia. Blood. 2016;128:102.
Raza A, Yousuf N, Bohkari SA, et al. In situ cell cycle kinetics in bone marrow biopsies following sequential infusions of IUdR/BrdU in patients with hematopoietic malignancies. Leuk Res. 1992;16(3):299–6.
Parcharidou A1, Raza A, Economopoulos T, et al. Extensive apoptosis of bone marrow cells as evaluated by the in situ end-labelling (ISEL) technique may be the basis for ineffective haematopoiesis in patients with myelodysplastic syndromes. Eur J Haematol. 1999;62(1):19–26.
Mundle SD, Ali A, Cartlidge JD, et al. Evidence for involvement of tumor necrosis factor-alpha in apoptotic death of bone marrow cells in myelodysplastic syndromes. Am J Hematol. 1999;60(1):36–47
Azra Raza, Peter Meyer, Diya Dutt et al. Thalidomide produces transfusion independence in long-standing refractory anemias of patients with myelodysplastic syndromes. Blood 2001 98:958–65
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Julia Montoro declares that she has no conflict of interest.
Aslihan Yerlikaya declares that she has no conflict of interest.
Abdullah Ali declares that he has no conflict of interest.
Azra Raza has received research funding through grants from Celgene, Kura Oncology, Janssen, and Syros Pharmaceuticals and has received honoraria from Novartis for Educational lectures on MDS.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Leukemia
Rights and permissions
About this article

Cite this article
Montoro, J., Yerlikaya, A., Ali, A. et al. Improving Treatment for Myelodysplastic Syndromes Patients. Curr. Treat. Options in Oncol. 19, 66 (2018). https://doi.org/10.1007/s11864-018-0583-4
Published:
DOI: https://doi.org/10.1007/s11864-018-0583-4