
Abstract
Niraparib (Zejula®), a poly (ADP-ribose) polymerase (PARP) inhibitor, is approved for the maintenance treatment of recurrent, epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who are in complete or partial response to platinum-based chemotherapy. Approval was based on the results of the randomized, double-blind, placebo-controlled phase III NOVA trial. In NOVA, niraparib significantly prolonged progression-free survival (primary endpoint), chemotherapy-free interval and time to first subsequent therapy compared with placebo in patients with recurrent, platinum-sensitive, high grade serous ovarian, fallopian tube or primary peritoneal cancer. The beneficial effects of niraparib were consistent regardless of BRCA mutation or homologous recombination deficiency (HRD) status. Niraparib had a manageable tolerability profile, with the majority of grade 3 or 4 adverse events being haematologic abnormalities (e.g. thrombocytopenia, anaemia, neutropenia). Adverse events were generally well managed with dose interruption or modification of niraparib. Current evidence suggests that niraparib is an effective new option with a manageable tolerability profile for the maintenance treatment of recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or primary peritoneal cancer in adults, with or without BRCA1/2 mutation or HRD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Oral PARP inhibitor administered once daily | |
Effective regardless of BRCA mutation or homologous recombination deficiency status | |
Significantly prolongs PFS, chemotherapy-free interval and time to first subsequent therapy compared with placebo | |
Manageable tolerability profile |
ᅟ
1 Introduction
Ovarian cancer is the deadliest gynaecological cancer in the western world [1]. High grade serous ovarian cancer is the most common histologic subtype of ovarian cancer, with most patients (≈ 70%) being diagnosed at advanced stages [2]. It is thought that the majority of high grade serous cancers of the ovary, fallopian tube or primary peritoneum appear to arise from fimbriae of the fallopian tube [1, 2]. A platinum-based chemotherapy regimen after debulking surgery is currently the standard of care in ovarian cancer [3]. However, despite high initial response rates with platinum-based chemotherapy, many patients relapse and most then receive second-line and subsequent line chemotherapies based on the platinum sensitivity of the tumour [4, 5]. Attempts to improve these poor outcomes by using intensive therapy (e.g. high-dose sequential chemotherapy) or multiple agents as first-line therapy have largely failed [6].
More recently, targeted therapies for ovarian cancer have been developed in a maintenance setting, with the aim of prolonging remission and preventing disease progression in patients who have shown an initial response to platinum-based therapy [6]. Inhibition of poly (ADP-ribose) polymerase (PARP), an enzyme that is involved in single-stranded DNA break repair, has proven to be an effective treatment strategy in cancers that involve DNA repair mechanism defects [7, 8]. These genetic aberrations, which can predispose women to hereditary ovarian cancer, are caused by specific mutations in DNA repair genes, including BRCA 1 and 2, leading to cellular homologous recombination deficiency (HRD) [5]. Targeting PARP inhibition in the presence of HRD results in genetic instability and cell death in a process known as ‘synthetic lethality’ [7, 8]. Recently, a number of small molecule PARP inhibitors, including niraparib (Zejula®, the focus of this article), have emerged in the maintenance therapy setting in ovarian cancer.
Oral niraparib is approved for the maintenance monotherapy treatment of recurrent (in the USA [9]) or platinum-sensitive, relapsed, high grade serous (in the EU [10]) epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who are in complete or partial response (CR or PR) to platinum-based chemotherapy [9, 10]. Niraparib can be administered irrespective of BRCA mutation or HRD status. This article reviews the pharmacological properties, therapeutic efficacy and tolerability of niraparib in this indication.
2 Pharmacodynamic Properties of Niraparib
Niraparib is a potent and selective inhibitor of PARP-1 and -2, with its activity against these enzymes being 100-fold higher than against other PARP family members (i.e. PARP-3, v-PARP and Tankyrase-1) [11]. Preclinical data suggest that niraparib-induced cytotoxicity involves inhibition of PARP enzymatic activity resulting in increased formation of PARP-DNA complexes, leading to DNA damage, apoptosis and cell death [9, 10]. In vitro, the antitumour activity of niraparib was demonstrated across tumour cell lines with or without BRCA1/2 mutations [9, 10].
Niraparib also reduced tumour growth in xenograft mouse models with various tumour types, including HRD tumours (BRCA1/2 mutant [9, 12] or BRCA1/2 wild type [9, 10]), BRCA1/2 mutant [9, 10], as well as tumours that are BRCA wild type and without detectable HRD [10].
In vitro, niraparib inhibited dopamine, norepinephrine and serotonin transporters and may therefore have the potential to cause effects related to these transporters in patients receiving niraparib [9]. This pharmacological inhibition may alter pulse rate and blood pressure (BP) in patients who receive the recommended dose of niraparib (Sect. 5.1). In the phase III NOVA trial (Sect. 4), the mean greatest increase from baseline in pulse rate was 24.1 beats/min in the niraparib group versus 15.8 beats/min in the placebo group. The mean greatest increase from baseline in systolic BP in the respective groups was 24.5 versus 18.3 mmHg and that for diastolic BP was 16.5 versus 11.6 mmHg [9]. In a QTc substudy of the NOVA trial (n = 26), once daily dosing of niraparib 300 mg was not associated with changes > 20 ms in the mean QTc interval [13].
3 Pharmacokinetic Properties of Niraparib
Niraparib displayed dose-proportional systemic exposure (i.e. peak plasma concentration [Cmax] and area under the plasma concentration-time curve [AUC]) after single oral doses of 30–400 mg [9, 10]. Following an oral administration of niraparib 300 mg in the fasted state, the drug is rapidly absorbed and Cmax is reached within 3 h; the absolute bioavailability is ≈ 73% [9, 10]. The accumulation ratio was ≈ 2- [9, 10] to 3-fold [10] after multiple doses of niraparib (30–400 mg). Administration of niraparib 300 mg with a high-fat meal (≈ 50% of fat) did not affect the pharmacokinetics of niraparib [14]; as a result, niraparib can be taken with or without food [9, 10].
In vitro, niraparib was 83% plasma protein bound [9, 10]. In a population pharmacokinetic analysis in cancer patients, the large apparent volume of distribution of niraparib (1074 L) suggests that the drug is extensively distributed into the tissues [9, 10]. Niraparib is primarily metabolized by carboxylesterases to its major inactive metabolite M1, which subsequently undergoes glucuronidation [15].
The role of CYP450 enzymes in the metabolism of niraparib is negligible [15]. Niraparib is primarily eliminated via renal and hepatic routes [10]. Following a single dose of radiolabelled niraparib (300 mg) in patients with advanced cancer, 47.5% of the dose (range 33.4–60.2%) was recovered in the urine and 38.8% (28.3–47.0%) was recovered in the faeces over 21 days after administration [16]. The mean terminal half-life following a single 300 mg dose ranged from 48 to 51 h (≈ 2 days) [10] and the drug had an apparent total clearance of 16.2 L/h in patients with cancer [9, 10].
The pharmacokinetics of niraparib are not significantly affected by age (18–65 years), race, or bodyweight, and population pharmacokinetic studies show that dosage adjustments are not required in patients with mild or moderate renal impairment [9, 10]. Niraparib dosage adjustments are also not required in patients with mild [9, 10] or moderate [10] hepatic impairment. There are no data regarding the use of niraparib in patients with severe renal (including those with end-stage renal disease on haemodialysis) or hepatic impairment [9, 10]; thus, the safety of niraparib in these settings are unknown [9] and niraparib should be used with caution [10].
While no formal drug interaction studies have been conducted [9], in vitro studies suggest that niraparib and M1 do not inhibit any active substance-metabolising CYP enzymes [9, 10]. However, potential inhibition of CYP3A4 at the intestinal level has not been established with the relevant concentrations of niraparib; thus, caution is advised in the EU if niraparib is used in combination with drugs that are CYP3A4 metabolism-dependent, particularly those with a narrow therapeutic index (e.g. ciclosporin, tacrolimus, alfentanil, ergotamine, pimozide, quetiapine and halofantrine) [10].
In vitro, niraparib weakly induces CYP1A2 [9, 10] and caution is advised in the EU if niraparib is coadministered with drugs that are CYP1A2 metabolism-dependent, particularly those with a narrow therapeutic index (e.g. clozapine, theophylline and ropinirole) [10].
Niraparib weakly inhibits organic cation transporter 1 (OCT1), and inhibits BCRP, MATE1 and 2; thus, in the EU, caution is required when niraparib is coadministered with substrates of OCT1 (e.g. metformin), BCRP (e.g. irinotecan, rosuvastatin, simvastatin, atorvastatin and methotrexate) or MATE1 and 2 (e.g. metformin) [10].
4 Therapeutic Efficacy of Niraparib
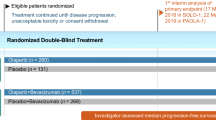
This section focuses on the efficacy of oral niraparib as a maintenance therapy for adults with platinum-sensitive, recurrent ovarian, fallopian tube, or primary peritoneal cancer, as evaluated in a randomized, double-blind, placebo-controlled phase III trial (NOVA) [17]. On the basis of the finding of an earlier dose-escalation phase I study in patients with advanced solid cancers [8], patients in NOVA received niraparib at a starting dosage of 300 mg once daily in continuous 28-day cycles [17].
NOVA included patients (aged ≥ 18 years) who were diagnosed with histologically confirmed high grade serous ovarian, fallopian tube or primary peritoneal cancer [17]. Eligible patients had received ≥ 2 platinum-based therapies, had platinum-sensitive disease after the penultimate platinum therapy (i.e. had achieved a CR or PR and disease progressed ≥ 6 months after the therapy), had responded (CR or PR) to the most recent platinum-based therapy and completed the last dose of such therapy ≤ 8 weeks before randomization. Patients were required to have Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0 or 1 and adequate haematologic, renal, and liver function. Patients who had previously received a PARP inhibitor were excluded [17].
Enrolled patients were assigned to one of two cohorts on the basis of their germline mutation status, gBRCA (n = 203) or non-gBRCA (n = 350) [17]. Patients were randomized in a 2:1 ratio to receive niraparib 300 mg or placebo once daily in continuous 28-day cycles. Treatment interruption and/or dose reductions were allowed at any time for toxicity of any grade that was considered intolerable by the patient. Briefly, treatment could be interrupted for up to 28 days because of hematologic toxicity; after the resolution of such toxicity, treatment could be restarted at a reduced dose of 200 mg according to protocol-specified criteria to manage adverse events and minimize drug discontinuation. Dose reductions were mandated for thrombocytopenia (i.e. grade ≥ 2 or recurrence of grade 1), and additional dose reduction down to 100 mg was permitted if needed. Treatment continued until disease progression [as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1], unacceptable toxicity, death, consent withdrawal or loss to follow-up. Randomization was stratified based on the time to progression after penultimate platinum-based therapy (6 to < 12 vs. ≥ 12 months), use of bevacizumab in conjunction with penultimate or last platinum therapy (yes vs. no) and the best response during the last platinum-based therapy (CR vs. PR) [17].
The niraparib and placebo groups in each cohort were balanced in terms of baseline clinical and demographic characteristics [17]. At baseline, most patients were diagnosed with stage III cancer (69–74%), and had an ECOG-PS of 0 (66–74%); 33–54% of patients previously received ≥ 3 chemotherapies, 24–27% had a history of prior bevacizumab use. In the gBRCA cohort, 62% of patients had BRCA1 mutation and 46% of patients in the non-gBRCA cohort had an HRD-positive tumour (as assessed by myChoice HRD test® [18]; HRD-positive subgroup) [17].
The primary endpoint was the duration of progression-free survival (PFS; assessed by a blinded, independent, central review of both clinical and radiographic progression) in the intent-to-treat (ITT) populations (all randomized patients) [17]. The primary endpoint was analysed independently in the gBRCA cohort and in the non-gBRCA cohort. A hierarchical testing procedure was applied in the non-gBRCA cohort; PFS was first analysed in the HRD-positive subgroup and if the subgroup showed statistical significance over placebo, PFS was subsequently analysed in the overall non-gBRCA cohort [17].
At the cutoff time for the efficacy analysis in NOVA (after disease progression or death occurred in 103 patients in the gBRCA cohort and 101 patients in the HRD-positive subgroup of the non-gBRCA cohort), 109 patients were receiving niraparib or placebo [17]. The most commonly administered niraparib dose after dose modification in NOVA was 200 mg [19]. A retrospective analysis of NOVA indicated that niraparib recipients with baseline platelet level < 150 × 109/L or those weighing < 77 kg required early dose modification and only 17% of these patients continued to receive the starting dosage of 300 mg/day after the first three cycles. According to the retrospective analysis, the efficacy of niraparib did not appear to be dose dependent [19]; however, it is not known if efficacy would not have been impacted in those patients who remained on higher doses, had they been reduced due to differences in exposure.
After a median follow-up of 16.9 months (database lock date June 20, 2016), niraparib significantly (p < 0.001) prolonged PFS compared with placebo in the three primary efficacy populations; the gBRCA cohort, the HRD-positive subgroup of the non-gBRCA cohort and in the overall non-gBRCA cohort (Table 1). This represented a prolongation in median duration of PFS of 15.5, 9.1 and 5.4 months versus placebo in the respective populations [17]. The PFS benefit of niraparib over placebo [i.e. hazard ratios (HRs) < 1] in all three ITT populations was consistently seen across all prespecified subgroup analyses, including age (18 to < 65 or ≥ 65 years), stratification factors (i.e. time to progression after penultimate platinum-based therapy, use of bevacizumab and the best response during the last platinum-based therapy) and cumulative number of previous chemotherapies (2 vs. > 2); however, significance (based on 95% CIs) was not reached in non-white race, possibly due to small sample size. Additionally, niraparib was associated with a greater estimated probability of PFS at 12, 18 and 24 month time points post randomization than placebo, with these effects observed regardless of BRCA mutation or HRD status (abstract [20]).
In terms of secondary endpoints, niraparib significantly prolonged chemotherapy-free interval (CFI) and time to first subsequent therapy (TFST), compared with placebo (Table 1) [17]. Although data for progression-free survival 2 (PFS2) were immature, a preliminary analysis indicated that niraparib significantly prolonged PFS2 compared with placebo in each cohort, but not in the HRD-positive subgroup of the non-gBRCA cohort (Table 1) [17].
According to an immature data set, overall survival (OS) in the entire population (HR 0.73) and the time to second subsequent therapy (TSST) in the gBRCA (HR 0.48) and the non-gBRCA (HR 0.74) cohorts favoured niraparib over placebo [21]. Mature data for these secondary endpoints are not yet available.
In prespecified exploratory analyses, niraparib provided consistent PFS benefit compared with placebo across subgroups of the non-gBRCA cohort that included patients with specific biomarkers; HRD-positive with somatic BRCA mutation (HR 0.27), HRD-positive with BRCA wild type (HR 0.38) and HRD-negative with BRCA wild type (HR 0.58) [17]. The degree of niraparib treatment benefit varied between the groups with different HRD status and BRCA mutation status; however, none of these biomarkers appeared to be sufficient to precisely predict magnitude of benefit to niraparib [17].
Niraparib maintenance treatment did not appear to adversely affect patient-reported outcomes, as assessed by Functional Assessment of Cancer Therapy–Ovarian Symptom Index (FOSI) and the European Quality of Life–5 Dimensions (EQ-5D-5L) questionnaires, with similar outcomes reported in the niraparib and placebo treatment groups [17].
5 Tolerability of Niraparib
Oral niraparib had a manageable tolerability profile in patients with recurrent ovarian, fallopian tube or primary peritoneal cancer in the NOVA trial (Sect. 4). The incidence of treatment-related adverse events (TRAEs) of any grade was 97.5% in the niraparib group and 70.9% in the placebo group [17]. TRAEs of any grade occurring with an incidence of > 25% in niraparib recipients were nausea (68.9 vs. 25.1% with placebo), anaemia (46.3 vs. 4.5%), thrombocytopenia (44.7 vs. 2.2%) and fatigue (37.3 vs. 20.7%) [13]. The incidence of grade 3 or 4 TRAEs in patients receiving niraparib was 65% (vs. 5% with placebo), with thrombocytopenia and anaemia being the most frequently reported. Serious TRAEs occurred in 16.9% of niraparib and 1.1% of placebo recipients, respectively, with the most common being thrombocytopenia (10.9 vs. 0%) and anaemia (3.8 vs. 0%) [13, 17].
In NOVA, among patients who received niraparib or placebo, 66.5 and 14.5% had dose reductions and 68.9 and 5.0% had treatment interruption because of treatment-emergent adverse events (TEAEs) [17]. TEAEs led to treatment discontinuation in 14.7 and 2.2% of niraparib and placebo recipients respectively; the most common TEAE leading to discontinuation in the respective groups was haematologic adverse events (9.3 vs. 0.6%) [17]. There were no treatment-related deaths during the study period; however, two treatment-related deaths (one patient in each treatment group) were reported during the follow-up period [due to myelodysplastic syndrome/acute myeloid leukemia (MDS/AML)] [17].
The tolerability profile of niraparib in elderly patients (aged ≥ 65 or ≥ 70 years) was similar to that observed in those aged < 65 or < 70 years (abstract [22]). Niraparib treatment in patients with low bodyweight however, may be associated with a higher incidence of grade ≥ 3 TEAEs, serious adverse events, and TEAEs leading to dose reduction or treatment discontinuation [19]. In a retrospective analysis of NOVA, the incidence of grade ≥ 3 TEAEs within 30 days of first dose was higher in niraparib recipients who weighed < 58 kg than in those who weighed ≥ 77 kg (58.6 vs. 36.2%). Dose reduction due to TEAEs in the respective groups was required by 50.6 versus 33.0% of patients and treatment discontinuation was required in 10.3 versus 1.1% of patients [19]. As a consequence, a lower dosage may be considered in patients with low bodyweight (Sect. 6).
5.1 Adverse Events of Special Interest
In NOVA, haematologic abnormalities made up the majority of grade 3 or 4 TEAEs seen in niraparib recipients, with the most common being thrombocytopenia (28.3 vs. 0.6% with placebo), anaemia (24.8 vs. 0%) and neutropenia (11.2 vs. 0.6%) [13]. Bleeding events concurrent with thrombocytopenia occurred in 13% of niraparib recipients; all events [including petechiae (5% incidence)] were grade 1 or 2 [10], except one case where a patient experienced grade 3 petechiae and hematoma (with pancytopenia) [10, 17]. Grade 3 or 4 thrombocytopenia occurred more frequently in niraparib recipients with lower baseline platelet level (< 180 × 109/L) [10]. Most haematologic adverse events in the niraparib group were observed in the first three cycles and the incidence of such adverse events decreased beyond cycle three, when the majority of affected patients had appropriate dose reductions [17]. Thrombocytopenia was transient and following dosage adjustments, patients stabilized on their individual dosage [17]. Haematologic adverse events (including pancytopenia) should be managed by treatment interruption, dosage modification or discontinuation of niraparib, depending upon the severity and persistence of these adverse events [9, 10]. Blood count monitoring weekly for the first month, followed by monthly monitoring for the next 10 months of treatment and periodically after this time is recommended to detect any clinically significant changes in haematologic parameters [9, 10].
The incidence of MDS/AML was similar in the niraparib and placebo groups (1.4 vs. 1.1%); niraparib should be discontinued if MDS/AML is confirmed [9, 10].
Although grade 3 or 4 hypertension was more common in niraparib than placebo recipients (8.2 vs. 2.2% [17]) in NOVA, < 1% of patients discontinued treatment because of hypertension [10]. In clinical trials in patients receiving niraparib, hypertension was generally well managed with standard antihypertensive treatment, with or without niraparib dosage adjustments [9, 10]. Blood pressure should be monitored regularly during treatment with niraparib [9, 10].
6 Dosage and Administration of Niraparib
Oral niraparib monotherapy is indicated for the maintenance treatment of recurrent (in the USA [9]) or platinum-sensitive relapsed high grade serous (in the EU [10]) epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who are in CR or PR to platinum-based chemotherapy [9, 10]. The approved dosage of oral niraparib is three capsules (3 × 100 mg) taken once daily; the capsules should be swallowed whole and can be taken with or without food [9, 10]. In the USA, niraparib should be initiated ≤ 8 weeks of last platinum-based therapy [9]. Niraparib should be continued until disease progression or unacceptable toxicity occurs [9, 10].
Dose interruption/modification or discontinuation of niraparib may be required for the management of adverse events [9, 10]. In addition, a lower starting dose of niraparib (200 mg) may be considered in patients weighing < 58 kg in the EU, given the greater incidence of grade 3 or 4 TEAEs observed in these patients (Sect. 5) [10]. Local prescribing information should be consulted for details regarding warning and precautions, dosage adjustments for adverse events and use of niraparib in special patient populations.
7 Current Status of Niraparib in the Management of Ovarian Cancer
In a recently updated National Comprehensive Cancer Network (NCCN) guideline, niraparib is recommended as a maintenance therapy for recurrent platinum-sensitive ovarian cancer as are other PARP inhibitors [3]. There are currently no clear preferences for one PARP inhibitor over any other [3]. Approval of niraparib is too recent to have been included in the European Society for Medical Oncology guideline for this indication [23, 24].
In the pivotal NOVA trial (Sect. 4), niraparib maintenance therapy significantly improved PFS (as well as other efficacy measures, such as CFI and TFST) versus placebo in platinum-sensitive patients with recurrent ovarian, fallopian tube, or primary peritoneal cancer. The PFS benefit of niraparib over placebo was evident across all predefined patient subgroups and across multiple tumour types with different BRCA mutation status and HRD status. In the recent National Institute for Health Care and Excellence appraisal, niraparib was recommended for use within the Cancer Drugs Fund as an option for the maintenance treatment of recurrent platinum-sensitive ovarian cancer in patients with or without BRCA1/2 mutation and those who have received ≥ 2 platinum-based chemotherapies [25]. However, cost effectiveness of niraparib could not be established with any certainty, because mature OS data are not yet available [25]. The mature OS data for niraparib are awaited with interest.
Niraparib had a manageable tolerability profile in NOVA (Sect. 5), with most of the grade 3 or 4 TEAEs reported being haematologic in nature, while hypertension was also identified as a safety signal [13]. The majority of these adverse events were observed in the first three treatment cycles and were generally manageable with dosage interruption/modifications without compromising efficacy. This may be due to variability in drug exposure between patients (Sect. 4). The adverse events associated with niraparib did not appear to affect patient-reported outcomes in niraparib recipients (Sect. 4), although longer-term data for niraparib (i.e. from clinical experience and real-world studies) to confirm the tolerability profile will be of paramount importance.
In conclusion, current evidence suggests that niraparib is an effective new option with a manageable tolerability profile for the maintenance treatment of recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or primary peritoneal cancer in adults, with or without BRCA1/2 mutation or HRD.
Data Selection Niraparib: 108 records identified
Duplicates removed | 15 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 36 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 32 |
Cited efficacy/tolerability articles | 6 |
Cited articles not efficacy/tolerability | 19 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were Niraparib, Zejula, JNJ-64091742, MK-4827, ovary, ovarian, cancer, carcinoma, tumor, tumour, neoplasm, fallopian, peritoneal. Records were limited to those in English language. Searches last updated 4 July 2018 | |
References
Erickson BK, Conner MG, Landen CN Jr. The role of the fallopian tube in the origin of ovarian cancer. Am J Obstet Gynecol. 2013;209(5):409–14.
Labidi-Galy SI, Papp E, Hallberg D, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun. 2017; https://doi.org/10.1038/s41467-017-00962-1.
National Comprehensive Cancer Network. Ovarian cancer including fallopian tube cancer and primary peritoneal cancer (NCCN clinical practice guidelines in oncology). 2018. http://www.nccn.org/. Accessed 10 May 2018.
Cortez AJ, Tudrej P, Kujawa KA, et al. Advances in ovarian cancer therapy. Cancer Chemother Pharmacol. 2018;81(1):17–38.
Jayson GC, Kohn EC, Kitchener HC, et al. Ovarian cancer. Lancet. 2014;384(9951):1376–88.
Khalique S, Hook JM, Ledermann JA. Maintenance therapy in ovarian cancer. Curr Opin Oncol. 2014;26(5):521–8.
Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i40–i4.
Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–92.
TESARO Inc. Zejula® (niraparib): US prescribing information 2017. http://www.fda.gov. Accessed 27 June 2018.
European Medicines Agency. Zejula (niraparib) 100 mg hard capsules: summary of product characteristics. 2018. http://www.ema.europa.eu/. Accessed 27 June 2018.
Jones P, Altamura S, Boueres J, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose) polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem. 2009;52(22):7170–85.
AlHilli MM, Becker MA, Weroha SJ, et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol Oncol. 2016;143(2):379–88.
European Medicines Agency. Zejula: European Public Assessment Report 2017. http://www.ema.europa.eu/. Accessed 27 Dec 2017.
Moore K, Zhang ZY, Agarwal S, et al. The effect of food on the pharmacokinetics of niraparib, a poly(ADP-ribose) polymerase (PARP) inhibitor, in patients with recurrent ovarian cancer. Cancer Chemother Pharmacol. 2018;81(3):497–503.
Zhang ZY, Kansra V, Van Andel L, et al. Characterization of absorption, metabolism, and elimination of niraparib, an investigational poly (adp-ribose) polymerase inhibitor, in cancer patients [abstract]. Clin Ther. 2017;39(8 Suppl 1):e7–8.
van Andel L, Zhang Z, Lu S, et al. Human mass balance study and metabolite profiling of 14C-niraparib, a novel poly(ADP-ribose) polymerase (PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer. Investig New Drugs. 2017;35(6):751–65.
Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Telli ML, Timms KM, Reid J, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–73.
Berek JS, Matulonis UA, Peen U, et al. Safety and dose modification for patients receiving niraparib. Ann Oncol. 2018; https://doi.org/10.1093/annonc/mdy181.
Matulonis UA, Herrstedt J, Tinker A, et al. Long-term benefit of niraparib treatment of recurrent ovarian cancer (OC) [abstract no. 5534]. J Clin Oncol. 2017;35(15 Suppl 1):5534.
Mahner S, Mirza MR, Moore K, et al. ENGOT-OV16/NOVA: results of secondary efficacy endpoints of niraparib maintenance therapy in ovarian cancer. Clin Adv Hematol Oncol. 2017;15(5 Suppl 5):2–3.
Fabbro M, Moore K, Dørum A, et al. Safety and efficacy of niraparib in elderly patients (pts) with recurrent ovarian cancer (OC) [abstract no. 934PD]. Ann Oncol. 2017;28(Suppl 5):v330–v54.
Ledermann JA, Raja FA, Fotopoulou C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi24–32.
Ledermann J, Sessa C, Colombo N on behalf of the ESMO guidelines committee. eUpdate – ovarian cancer treatment recommendations 2016. http://www.esmo.org. Accessed 19 Mar 2018.
National Institue for Health and Care Excellence. Final appraisal determination: niraparib for maintenance treatment of relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer. 2018. http://www.nice.org.uk. Accessed 5 June 2018.
Acknowledgments
During the peer review process, the manufacturer of niraparib was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflicts of Interest
Young-A Heo and Sean Duggan are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: M Markman, Eastern Regional Medical Center, Cancer Treatment Centers of America, Philadelphia, PA, USA; M Hall, Department of Medical Oncology, Mount Vernon Cancer Centre, Northwood, UK.
Rights and permissions
About this article

Cite this article
Heo, YA., Duggan, S.T. Niraparib: A Review in Ovarian Cancer. Targ Oncol 13, 533–539 (2018). https://doi.org/10.1007/s11523-018-0582-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-018-0582-1