
Abstract
Oral niraparib, a highly-selective, potent poly(ADP-ribose) polymerase (PARP)-1 and PARP-2 inhibitor, is approved in the USA for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy. It is also under regulatory review in the EU for use in maintenance treatment in patients with platinum-sensitive, recurrent epithelial ovarian cancer who are in response to platinum-based chemotherapy. In the multinational, phase 3 NOVA trial in adult patients with platinum-sensitive, recurrent ovarian cancer, niraparib significantly prolonged median progression-free survival, irrespective of the presence or absence of a germline BRCA (gBRCA) mutation and irrespective of the presence or absence of homologous recombinant deficiency. Niraparib is also in development for use in other solid tumours, including breast and prostate cancer. This article summarizes the milestones in the development of niraparib leading to its first global approval for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Niraparib (Zejula™) is an oral, small molecule inhibitor of poly(ADP-ribose) polymerase (PARP) enzymes, including PARP-1 and PARP-2 [1, 2]. The drug is being developed by Tesaro Inc. for use in the treatment of various solid tumours [3]. The PARP family of enzymes (≥17 enzymes) are primarily involved in detecting single-stranded DNA breaks and triggering a cascade of events leading to the recruitment of DNA repair factors [1, 2]. Inhibition of PARPs is an effective strategy for treating cancers involving defects in DNA repair mechanisms that are caused by specific aberrations in DNA repair genes such as BRCA1 and BRCA2, mutations that predispose individuals to hereditary breast and ovarian cancer. In preclinical studies, PARP inhibition of tumour cells deficient in BRCA1 or BRCA2 (i.e. BRCA−/BRCA− cancer cells) resulted in accumulation of DNA damage and consequent cell death, with cell death requiring both processes (i.e. inhibition of base excision repair mediated by PARP inhibition and defects in DNA double-strand break repair mediated by the loss of BRCA1 and BRCA2) [1, 2].
Oral niraparib has been approved and launched in the USA for maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy [4]. Niraparib is also under regulatory review in the EU for use in maintenance treatment in patients with platinum-sensitive, recurrent epithelial ovarian cancer who are in response to platinum-based chemotherapy [5, 6]. Niraparib is also in development for use in other solid tumours, including breast and prostate cancer.
In the USA, the recommended regimen of niraparib is 300 mg once daily, with treatment continuing until disease progression or unacceptable adverse reaction [4].

Key milestones in the development of oral niraparib for the treatment of ovarian, fallopian tube or primary peritoneal cancer. Est estimated completion date
To manage adverse reactions, consider interruption of treatment, dose reductions or treatment discontinuation [4].
2 Scientific Summary
2.1 Pharmacodynamics
Niraparib is a potent, highly-selective PARP-1 and PARP-2 inhibitor (50% inhibitory concentrations of 3.8 and 2.1 nmol/L), with a 100-fold higher selectivity for these than for other PARP-family members (PARP-3, v-PARP and TANK-1) [7]. In cultured BRCA1 and BRCA2 deficient cancer cell lines, niraparib selectively inhibited cancer cell proliferation but not normal cell line proliferation, with niraparib-induced cytotoxicity in BRCA-deficient cells resulting from cell cycle arrest at the G2/M phase leading to apoptosis and mitotic catastrophe [1].
Preclinical studies [8,9,10] suggest that niraparib enhances the effects of radiation therapy in a p53-independent manner, based on in vitro studies [8,9,10] and murine xenograft models [9, 10]. In vitro, niraparib radiosensitized human breast, lung and/or prostate tumour cell lines, but not normal tissue-derived cell lines, in a p53-independent manner; an effect that appears to involve conversion of radiation-induced sublethal single stranded breaks into lethal double strand breaks [8,9,10]. In lung and breast cancer xenografts, including in a triple negative breast cancer xenograft model, niraparib enhanced the response to radiation therapy in a p53-independent manner using clinically relevant radiation-dose fractionation schedules [10]. Similarly, in a murine xenograft model of metastatic neuroblastoma, combining niraparib treatment with radiation therapy prolonged survival compared with either treatment alone, with mechanisms involved including augmentation of DNA repair, apoptotic cell death and, in mice treated with niraparib, down regulation of poly-ADP-ribose levels [9].
At recommended doses, niraparib has the potential to effect pulse rate and BP, which may be related to the pharmacological inhibition of the dopamine transporter, norepinephrine transporter and serotonin transporter [4]. In the NOVA trial in adult patients with platinum-sensitive, recurrent ovarian cancer (Sect. 2.3), mean greatest increases from baseline in pulse rate in the niraparib and placebo groups were 24.1 and 15.8 beats/min, with respective mean greatest increases in systolic BP of 24.5 and 18.3 mmHg and in diastolic BP of 16.5 and 11.6 mmHg. There were no large (>20 ms) changes in the mean corrected QT interval in patients treated with niraparib 300 mg once daily in a randomized, placebo-controlled trial in cancer patients (n = 546) [4].
2.2 Pharmacokinetics
Oral niraparib exhibits dose-proportional pharmacokinetics across the dose range of 30–400 mg [4]. Niraparib is rapidly absorbed with maximum plasma concentrations attained within 3 h. The mean absolute bioavailability of niraparib is ≈73%, with 83% of the drug bound to human plasma proteins. The average apparent volume of distribution (Vd) was 1220 (±1114) L; based on population pharmacokinetic analysis, the Vd of niraparib in cancer patients was 1074 L. Concomitant administration of niraparib with a high-fat meal had no clinically relevant effect on the pharmacokinetics of niraparib [4].
Niraparib is primarily metabolized by carboxylesterases (CEs) to form a major inactive metabolite (M1), which subsequently undergoes glucuronidation [4]. The mean elimination half-life of niraparib is 36 h following multiple daily 300 mg doses [4]. After a single radiolabeled 300 mg dose of niraparib, 47.5 and 38.8% of the administered dose was recovered within 21 days in the urine and faeces, respectively; of which, unchanged drug accounted for 11 and 19% of the administered dose [11].
There was no clinically relevant effect on the pharmacokinetics of niraparib based on age (aged 18–65 years), race or mild to moderate renal impairment [4]. The effect of severe renal impairment or end-stage renal disease undergoing haemodialysis on the pharmacokinetics of niraparib is unknown, as is the effect of moderate or severe hepatic impairment [4].
No formal drug interaction studies have been conducted with niraparib [4]. In vitro, niraparib and the M1 metabolite do not inhibit CYPA2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 enzymes or induce CYP3A4; niraparib weakly induces CYP1A2. In vivo, niraparib is a substrate for CEs and UGTs. Niraparib, but not M1, is a weak inhibitor of BCRP, with neither niraparib nor M1 inhibiting p-glycoprotein (P-gp), bile salt export pump (BSEP), OATP1B1, OATP1B3, OCT1, OAT1, OAT3 or OCT2. Niraparib is a substrate for P-gp and BCRP, but not BSEP, with M1 not a substrate for any of these. Niraparib and M1 are not a substrate of OATP1B1, OATP1B3, OCT1, OAT1, OAT3 or OCT2 [4].
Features and properties of niraparib
Alternative names | MK-4827 |
Class | Antineoplastics, Benzamides, Indazoles, Piperidines, Small molecules |
Mechanism of action | Poly(ADP-ribose) polymerase (PARP)inhibitor |
Route of administration | Oral |
Pharmacodynamics | Highly-selective, potent inhibitor of PARP-1 and -2, with antitumour activity in cells with mutations in the BRCA1/2 genes |
Pharmacokinetics | Dose-proportional pharmacokinetics; rapidly absorbed, with a mean half-life of 36 h |
Adverse events | |
Most frequent (incidence ≥10%) | Haematological abnormalities, palpitations, gastrointestinal events, mucositis/stomatitis, dry mouth, fatigue/asthenia, urinary tract infection, aminotransferase enzyme elevations, myalgia, back pain, arthralgia, headache, dizziness, dysgeusia, insomnia, anxiety, nasopharyngitis, dyspnoea, cough, rash, hypertension |
Occasional (incidence <1%) | Myelodysplastic syndrome/acute myeloid leukaemia |
ATC codes | |
WHO ATC code | LO1 |
EphMRA ATC code | L1 |
Chemical name | 2-[4-(Piperidin-3-yl)phenyl]-2H-indazole-7-carboxamide 4-methylbenzenesulfonate hydrate |
2.3 Therapeutic Trials
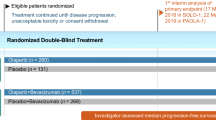
The efficacy of oral niraparib in adult patients with platinum-sensitive, recurrent, histologically-confirmed ovarian cancer was established in the pivotal, randomized, double-blind, multinational, phase 3 NOVA trial (NCT01847274) [12]. The trial enrolled two independent cohorts on the basis of the presence (n = 203) or absence (n = 350) of a germline BRCA mutation (the gBRCA and non-gBRCA cohorts). Patients received niraparib 300 mg or placebo once daily in 28-day cycles until disease progression, unacceptable toxicity, death, withdrawal of consent or loss to follow-up, whichever came first. Treatment could be interrupted for up to 28 days because of haematological toxicity, with dose reductions mandated for thrombocytopaenia (recurrence of grade 1 or occurrence of grade 2 or above). Across treatment groups, the median age of patients ranged from 57 to 63 years, approximately two-thirds of patients had an ECOG performance status of 0 and one-third an ECOG performance status of 1, and ≈50 and 33% of patients in the gBRCA and non-gBRCA cohorts had received ≥3 lines of chemotherapy. The primary endpoint was the duration of progression-free survival (PFS) in intent-to-treat analyses of the three predefined primary efficacy populations [the gBRCA cohort (n = 138 and 65 in the niraparib and placebo groups), a subgroup of patients with homologous recombinant deficiency (HRD) in the non-gBRCA cohort (n = 106 and 56), and the overall non-gBRCA cohort (n = 234 and 116)], using a predefined hierarchical testing procedure; if the primary endpoint was significant in the subgroup of patients with HRD tumours, then the overall non-gBRCA efficacy population was tested [12].
At a median follow-up of 16.9 months, median PFS was significantly (p < 0.001) prolonged in the niraparib groups compared with the placebo groups in the three predefined primary efficacy populations [12]. The risk of disease progression in niraparib recipients was reduced by 73% in the gBRCA cohort [median PFS 21.0 vs. 5.5 months in the placebo group; hazard ratio (HR) 0.27; 95% CI 0.17–0.41; p < 0.001], by 62% in the subgroup of patients in the non-gBRCA cohort with HRD-positivity (median PFS 12.9 vs. 3.8 months; HR 0.38; 95% CI 0.24–0.59; p < 0.001) and by 55% in the overall non-gBRCA cohort (median PFS 9.3 vs. 3.9 months; HR 0.45; 95% CI 0.34–0.61; p < 0.001). For all three primary efficacy populations, with the exception of patients who were non-white or of unknown race (potentially reflecting low patient numbers in this subgroup), niraparib significantly (i.e. the 95% CIs for the HR did not cross 1) prolonged median PFS compared with placebo in all prespecified subgroup analyses, including based on age, geographic region, time to disease progression prior to study enrolment, bevacizumab use, best overall response to platinum therapy, platinum in the last and penultimate therapies, total number of previous platinum regimens and the cumulative number of previous chemotherapy regimens [12].
Secondary endpoints also favoured niraparib treatment over placebo at this timepoint in the gBRCA and overall non-gBRCA cohorts [12]. Median chemotherapy-free intervals in the niraparib and placebo group in the gBRCA cohort were 22.8 and 9.4 months (HR 0.26; 95% CI 0.17–0.41; p < 0.001) and in the overall non-gBRCA cohort were 12.7 and 8.6 months (HR 0.50; 95% CI 0.37–0.67; p < 0.001). The median time to first subsequent treatment was also significantly delayed in the niraparib groups in the gBRCA cohort (median 21.0 vs. 8.4 months in the placebo group; HR 0.31; 95% CI 0.21–0.48; p < 0.001) and the overall non-gBRCA cohort (median 11.8 vs. 7.2 months; HR 0.55; 95% CI 0.41–0.72; p < 0.001). Data relating to the time from randomization until progression during receipt of the next anticancer therapy after termination of study treatment (i.e. PFS2) were not mature at the time of the database unlock; preliminary data indicate that PFS2 was prolonged in the gBRCA (p = 0.006 vs. placebo) and non-gBRCA cohorts (p = 0.03) [12].
Prespecified exploratory analyses in the HRD-positive non-gBRCA subgroup indicated that niraparib significantly (p ≤ 0.02 vs. placebo) prolonged median PFS in patients with wild-type BRCA and in those with a BRCA somatic mutation [12]. Niraparib treatment also prolonged PFS in patients with HRD-negative tumours in the non-gBRCA cohort (p = 0.02 vs. placebo) [12].
Niraparib treatment did not adversely impact health-related quality of life, with similar results for patient-reported outcomes in the niraparib and placebo groups [12].
Key clinical trials of niraparib
Drug(s) | Cancer indication | Phase | Status | Location(s) | Identifiers | Sponsors |
|---|---|---|---|---|---|---|
Niraparib vs. placebo | Ovarian | 3 | Completed | Multinational | NCT01847274 (NOVA); PR-30-5011-C | Tesaro Inc. |
Niraparib vs. placebo | Ovarian | 3 | Recruiting | Multinational | NCT02655016 (PRIMA); PR-30-5017-C | Tesaro Inc. |
Niraparib | Ovarian | 2 | Recruiting | Canada/USA | NCT02354586 (QUADRA); PR-30-5020-C | Tesaro Inc. |
Niraparib ± bevacizumab vs. bevacizumab | Ovarian | 1/2 | Recruiting | Denmark | NCT02354131 (AVANOVA); ENGOT-OV24-NSGO/AVANOVA | Nordic Society for Gynaecologic Oncology |
Niraparib + pembrolizumab | Breast or ovarian | 1/2 | Recruiting | USA | NCT02657889 (TOPACIO); 3000-PN162-01-001 | Tesaro Inc. |
Niraparib vs. physician’s choice | Breast | 3 | Recruiting | Multinational | NCT01905592 (BRAVO); PR-30-5010-C | Tesaro Inc |
Niraparib | Breast | 2 | Not yet open for recruiting | Netherlands | NCT02826512; M14ABC | Netherlands Cancer Institute |
Niraparib | Prostate | 2 | Recruiting | Multinational | NCT02854436 (Galahad); CR108208; 64091724PCR2001; 2016-002057-38 | Janssen Research and Development, LLC |
Niraparib | Endometrial | 2 | Not yet open for recruiting | Canada | NCT03016338; NEC | University Health Network, Toronto |
2.4 Adverse Events
Oral once-daily niraparib had a manageable safety profile in adults with platinum-sensitive, recurrent ovarian cancer in the pivotal NOVA trial [12]. Although at least 95% of patients experienced at least one treatment-emergent adverse event (TEAE; 100% in the niraparib group and 95.5% in the placebo group; n = 367 and 179), relatively few patients discontinued treatment because of these events (14.7 vs. 2.2%). In the niraparib group, TEAEs lead to treatment interruption in 68.9% of patients (vs. 5.0% in the placebo group) and dosage reductions in 66.5% (vs. 14.5%). The incidence of TEAES of at least grade 3 in the niraparib and placebo groups was 74.1 and 22.9%, most of which were haematological abnormalities. No patients died during the treatment period, with three deaths from myelodysplastic syndrome (MDS) or acute myeloid leukaemia (AML) occurring during the follow-up period; two of which were considered by investigators to be treatment-related (one in each group) [12].
TEAEs of any grade occurring in at least 15% of patients in the niraparib group were nausea (73.6 vs. 35.2% in the placebo group), thrombocytopaenia (61.3 vs. 5.6%), fatigue (59.4 vs. 41.3%), anaemia (50.1 vs. 6.7%), constipation (39.8 vs. 20.1%), vomiting (34.3 vs. 16.2%), neutropaenia (30.2 vs. 6.1%), headache (25.9 vs. 9.5%), decreased appetite (25.3 vs. 14.5%), insomnia (24.3 vs. 7.3%), abdominal pain (22.6 vs. 29.6%), dyspnoea (19.3 vs. 8.4%), hypertension (19.3 vs. 4.5%), diarrhoea (19.1 vs. 20.7%), dizziness (16.6 vs. 7.3%) and cough (15.0 vs. 4.5%) [12].
The most common (incidence ≥10% in either group) treatment-emergent haematological adverse events occurring in the niraparib and placebo groups were thrombocytopenia (61.3 vs. 5.6%), anaemia (50.1 vs. 6.7%) and neutropaenia (30.2 vs. 6.1%) [12]. Most of these events occurred within the first three treatment cycles and very few patients discontinued treatment because of these events (≤3.3% in the niraparib group and ≤0.06% in the placebo group). Most haematological adverse events were manageable with dose modifications and/or interruptions. In the niraparib group, common grade 3 or 4 haematological adverse events were thrombocytopaenia (33.8% of patients), anaemia (25.3%0 and neutropaenia (19.6%) [12]. In clinical studies, MDS/AML was reported in 0.9% of 751 niraparib-treated patients after up to 2 years’ treatment [4].
In the NOVA trial, grade 3 or 4 hypertension occurred in 9% of patients in the niraparib group and 2% of patients in the placebo group, with <1% of patients discontinuing treatment because of these events [4].
2.5 Ongoing Clinical Trials
A phase 3 trial is currently recruiting patients with advanced ovarian cancer following response to front-line platinum-based chemotherapy to evaluate niraparib maintenance therapy (NCT02655016; PRIMA) [13]. A phase 2 trial will evaluate niraparib treatment in patients with ovarian cancer who have received three or four previous chemotherapy regimens (NCT02354586; QUADRA; recruiting) [14]. The phase 3 BRAVO trial will evaluate niraparib treatment in HER2-negative, germline BRCA mutation-positive breast cancer (NCT01905592; recruiting) [15]; a phase 2 trial will evaluate niraparib in patients with advanced, BRCA1-like, HER2-negative breast cancer (NCT02826512; not yet open for recruiting). Niraparib treatment will also be investigated in phase 2 trials in patients with recurrent endometrial cancer (NCT03016338; not yet open for recruiting) and in men with metastatic castration-resistant prostate cancer (NCT 02854436; Galahad; recruiting).
Several planned or ongoing trials will evaluate niraparib combination therapy. A phase 1/2 trial will evaluate niraparib plus bevacizumab combination therapy in patients with HRD platinum-sensitive ovarian cancer (NCT02354131; AVANOVA; recruiting) [16, 17]. A phase 1/2 trial will evaluate niraparib in combination with pembrolizumab in patients with triple-negative breast cancer or ovarian cancer (NCT02657889; TOPACIO; recruiting) [18]. Phase 1 trials will evaluate niraparib in combination with enzalutamide in patients with metastatic, castrate-resistant prostate cancer (NCT02500901; ongoing trial) [19] and in combination with radium Ra 223 dichloride in patients with hormone-resistant prostate cancer (NCT03076203; not yet open for recruiting). Niraparib is also being investigated in a phase 1 trial in combination with temozolomide or irinotecan in patients with previously-treated, incurable Ewing’s sarcoma (NCT02044120; recruiting).
3 Current Status
Niraparib received its first global approval on the 27th of March 2017 for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy in the USA.
References
Jones P, Wilcoxen K, Rowley M, et al. Niraparib: a poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem. 2015;58(8):3302–14.
O’Sullivan Coyne G, Chen AP, Meehan R, et al. PARP inhibitors in reproductive system cancers: current use and developments. Drugs. 2017;77(2):113–30.
Tesaro. Tesaro announces expanded development program for niraparib focused on the treatment of front-line metastatic ovarian and lung cancer and metastatic breast cancer. 2017. http://www.tesarobio.com. Accessed 3 Apr 2017.
Tesaro. ZEJULA™ (Niraparib): US prescribing information. 2017. https://www.accessdata.fda.gov/. Accessed 29 Mar 2017.
European Medicines Agency. Applications for new human medicines under evaluation by the Committee for Medicinal Products for Human Use. 2016. http://www.ema.europa.eu. Accessed 3 Apr 2017.
Tesaro. Tesaro announces acceptance for review of niraparib marketing authorization by EMA. 2016. http://tesarobio.com. Accessed 3 Apr 2017.
Jones P, Altamura S, Boueres J, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem. 2009;52(22):7170–85.
Bridges KA, Toniatti C, Buser CA, et al. Niraparib (MK-4827), a novel poly(ADP-ribose) polymerase inhibitor, radiosensitizes human lung and breast cancer cells. Oncotarget. 2014;5(13):5076–86.
Mueller S, Bhargava S, Molinaro AM, et al. Poly (ADP-Ribose) polymerase inhibitor MK-4827 together with radiation as a novel therapy for metastatic neuroblastoma. Anticancer Res. 2013;33(3):755–62.
Wang L, Mason KA, Ang KK, et al. MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Invest New Drugs. 2012;30(6):2113–20.
van Andel L, Zhang Z, Lu S, et al. Human mass balance study and metabolite profiling of 14C-niraparib, a novel poly(ADP-Ribose) polymerase (PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer. Invest New Drugs. 2017. doi:10.1007/s10637-017-0451-2.
Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Gonzalez-Martin A, Backes FJ, Baumann KH, et al. A randomized, double-blind phase III trial of niraparib maintenance treatment in patients with HRD+ advanced ovarian cancer after response to front-line platinum-based chemotherapy [abstract no. TPS5606]. J Clin Oncol. 2016;34(Suppl.).
Moore KN, Rimel B, Agarwal S, et al. A Phase 2, open label study of niraparib in women with advanced, relapsed, highgrade serous epithelial ovarian, fallopian tube or primary peritoneal cancer after 3 previous chemotherapy regimens [abstract no. TPS5609]. J Clin Oncol. 2015;33(Suppl. 1).
Balmana J, Tryfonidis K, Audeh W, et al. A phase III, randomized, open label, multicenter, controlled trial of niraparib versus physician’s choice in previously-treated, HER2 negative, germline BRCA mutation-positive breast cancer patients. An EORTC-BIG intergroup study (BRAVO study) [abstract no. OT1-03-05]. Cancer Res. 2016;76(4 Suppl. 1).
Mirza MR, Mortensen CE, DePont Christensen R, et al. A phase I study of bevacizumab in combination with niraparib in patients with platinum-sensitive epithelial ovarian cancer: the ENGOT-OV24/AVANOVA1 trial [abstract no. 5555]. J Clin Oncol. 2016;34(Suppl.).
Mirza MR, Mortensen CE, Avall-Lundqvist E, et al. ENGOT-OV24-NSGO/AVANOVA: niraparib versus bevacizumab-niraparib combination versus bevacizumab and niraparib as sequential therapy in women with platinumsensitive epithelial ovarian, fallopian tube, or peritoneal cancer [abstract no. TPS5607]. J Clin Oncol. 2015;33(Suppl. 1).
Konstantinopoulos P, Moore KN, Sachdev JC, et al. Phase I/II study of niraparib plus pembrolizumab in patients with triple-negative breast cancer or recurrent ovarian cancer (KEYNOTE-162) [abstract no. TPS5599]. J Clin Oncol. 2016;34(Suppl.).
Flores JP, Mathew P. Combination therapy with enzalutamide and the poly (ADPribose) polymerase-1 (PARP1) inhibitor niraparib in castration-resistant prostate cancer (CRPC): HCRN GU 14-202 [abstract no. TPS5095]. J Clin Oncol. 2016;34(Suppl.).
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. L.J. Scott is a salaried employee of Adis, Springer SBM.
Author information
Authors and Affiliations
Corresponding author
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Scott, L.J. Niraparib: First Global Approval. Drugs 77, 1029–1034 (2017). https://doi.org/10.1007/s40265-017-0752-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0752-y