
Abstract
Because ischaemic stroke is one of the most common brain disorders, diverse effective therapies are urgently required. Recent studies reported a variety of azetidine-based scaffolds for the development of central nervous system-focused lead-like libraries. However, their mechanisms of action and in vivo functions remain unclear. Here, we investigated the potential mechanism and beneficial effects of 3-(naphthalen-2-yl(propoxy)methyl)azetidine hydrochloride (KHG26792), a novel azetidine derivative, on ischaemia/reperfusion (I/R) brain injury. We adapted a mouse brain ischaemia model induced by 2 h of middle cerebral artery occlusion followed by 24 h of reperfusion. We measured apoptotic cell death, inflammatory mediators, free radical generation, and anti-oxidative enzymes activities. We also measured the mitochondrial ATP level and Na+, K+-ATPase and cytochrome c oxidase activities. Using western blotting, we analysed the protein levels of inducible NOS, hypoxia-upregulated protein 1, PTEN-induced putative kinase, uncoupling protein 2, p-Akt, MMP-3, and full-length receptor for advanced glycation end-products (RAGE). KHG26792 significantly improved neurological deficits and brain oedema and suppressed I/R-induced apoptosis. KHG26792 significantly attenuated I/R-induced inflammation and oxidative stress by upregulating SOD and catalase activity, GSH, p-Akt, mitochondrial ATP, Na+, K+-ATPase, cytochrome c oxidase, and soluble RAGE and downregulating iNOS, HYOUP1, and MMP-3, suggesting a potential anti-inflammatory and antioxidant role of KHG26792. This is the first study to show that KHG26792 can protect mouse brains against I/R injury by inhibiting apoptotic damage, modulating inflammation, scavenging free radicals, ameliorating oxidative stress, and improving the energy metabolism of the brain, although the clinical relevance of our findings remains unknown.
Similar content being viewed by others
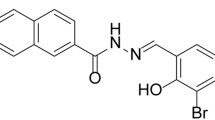

Avoid common mistakes on your manuscript.
Introduction
Ischaemic stroke is one of the most common brain disorders because the brain is particularly susceptible to cerebral ischaemic reperfusion injury due to its high demand for oxygen, high level of polyunsaturated fatty acids, and low activity of ROS-scavenging enzymes (Shi et al. 2013; Guo et al. 2014). Thus, diverse effective therapeutic approaches are urgently required to protect the brain from ischaemic brain injury (Urban et al. 2010). ROS production is dramatically increased during cerebral ischaemia/reperfusion (I/R), overwhelming endogenous antioxidant systems and leading to oxidative stress (Love 1999; Alexandrova et al. 2004; Allen and Bayraktutan 2009; Niatsetskaya et al. 2012). ROS cause tissue damage by inducing lipid peroxidation or protein nitrosylation of the cell membrane and initiating an inflammatory cascade involving cytokine release and inducible NOS (iNOS) activation (Allan and Rothwell 2001; Lo et al. 2003; Mehta et al. 2007). Inflammation and oxidative stress are considered major factors in the pathophysiology of I/R injury (Doyle et al. 2008; Broughton et al. 2009; de Vries et al. 2013). Hence, the use of agents that scavenge free radicals and suppress both inflammation and neural cell apoptosis might be one promising way to prevent or treat I/R-induced brain damage (Slemmer et al. 2008; Broughton et al. 2009; Wang et al. 2013).
Although much effort has been expended to identify agents capable of treating I/R brain injury, neuroprotective drugs that are fully effective against I/R-induced brain damage are not yet available because the overall pathophysiological process of brain ischaemia is complex and multifactorial. We are interested in the development of novel efficacious compounds able to control neuronal injury related to oxidative stress and inflammation. During the course of our studies, we recently reported the in vitro anti-inflammatory effects of 3-(naphthalen-2-yl(propoxy)methyl)azetidine hydrochloride (KHG26792), a novel azetidine derivative, on ATP- or hypoxia-induced inflammation in microglia (Kim et al. 2015a; Kim et al. in press). Previous reports have revealed a variety of azetidine-based scaffolds for the development of central nervous system (CNS)-focused lead-like libraries (Lowe et al. 2012). The beneficial functions of azetidine derivatives include memory-restorative effects on dysfunctions related to Alzheimer’s disease dementia and the attenuation of inflammation with reductions in TNF-α and NF-κB (Dalla et al. 2009; Gómez-Garre et al. 2009; Mnich et al. 2010). Recently, other studies reported biological activities of azetidine derivatives against α4β2 neuronal nicotinic receptors and serotonin, norepinephrine, and dopamine transporters (Han et al. 2014; Yun et al. 2014).
Nonetheless, the mechanism of action and in vivo function of KHG26792 remain largely unclear. In our present study, we characterized the potential mechanism and beneficial effects of KHG26792 on I/R brain injury. To do this, we investigated whether KHG26792 can exert a neuroprotective effect on middle cerebral artery occlusion (MCAO)-induced ischaemia in mouse brains by attenuating I/R-induced changes in inflammatory mediators, oxidative damage, and various signal pathways.
Methods
Materials
PBS and DMSO were obtained from Sigma-Aldrich (St. Louis, MO). Anti-caspase-3, anti-Bax, anti-iNOS, anti-hypoxia-upregulated protein 1 (HYOU1), anti-PTEN-induced putative kinase 1 (PINK1), anti-uncoupling protein 2 (UCP2), anti-Akt, anti-p-Akt, anti-MMP-3, and anti-full-length receptor for advanced glycation end-products (fl-RAGE), and anti-β-actin were purchased from Cell Signaling Technology (Beverly, MA). 3-(naphthalen-2-yl(propoxy)methyl)azetidine hydrochloride (KHG26792) was synthesized and purified as described previously (Han et al. 2012). All other reagents were of the highest purity among commercially available products.
Animals
Male C57BL/6 mice (Harlan Sprague Dawley, Indianapolis, IN) were individually housed in plastic cages in a pathogen-free facility and maintained under a standard 12/12-h light/dark cycle with food and water ad libitum. This study was reviewed and approved by the Institutional Animal Care and Use Committee of the Asian Institute for Life Sciences, Asian Medical Center, which abides by the Institute of Laboratory Animal Resources guidelines.
Induction of Cerebral I/R Injury and the Experimental Design
For the present study, mouse cerebral I/R injury was induced via 2 h of MCAO followed by 24 h of reperfusion according to the previously published methods (Zea Longa et al. 1989; Belayev et al. 1996; Liu et al. 2013; Ruan et al. 2013; Ahmed et al. 2014) with a slight modification. Briefly, mice were anaesthetized with 3.5% chloral hydrate in 0.9% NaCl and placed in dorsal recumbency. Through a midline incision of the neck, the external and internal carotid arteries were exposed. A nylon monofilament (15 mm in length and 0.15 mm in diameter) coated with poly-L-lysine was inserted into the external carotid artery and advanced to the internal carotid artery until a slight resistance was felt. On achieving occlusion, the filament was held in place with a ligature and the external incision was temporarily sutured. After 2 h of ischaemia, the mice were anaesthetised, the suture was opened, and the monofilament was withdrawn to induce a cycle of I/R injury. The wound area was treated with bupivacaine (0.5% v/v in PBS) and the animals were allowed to recover in heated isolation cages with appropriate chow/water provided ad libitum for 24 h. Then, the mice were decapitated and the brains were removed under visual inspection and placed on a piece of gauze moistened with ice-cold 0.9% saline. Since the most affected region is the midbrain in the MCAO model (Justin et al. 2014), brains were further dissected into the hippocampus to analyse the drug effect. Crude brain extracts were prepared using Dounce pestle fragmentation and sonication in HEPES buffer containing a mixture of protease inhibitors (1 μg mL−1 leupeptin, 1 μM dithiothreitol, 2 mM sodium orthovanadate, and 1 μM phenylmethylsulfonylfluoride), followed by centrifugation at 21,600×g for 10 min at 4 °C. The crude extracts were stored at −80 °C until use. The protein concentrations of the crude extracts were determined by the BCA Protein Assay (Pierce, Rockford, IL).
Mice were randomly divided into four groups (n = 6 per group): (1) sham group, (2) sham group treated with KHG26792, (3) I/R group, and (4) I/R group treated with KHG26792. KHG26792 (3 mg/kg) dissolved in DMSO was intraperitoneally injected 2 h before MCAO in groups 2 and 4. The dose of KHG26792 was chosen based on our pilot study as shown in supplementary Table 1. No significant differences in biomarkers of liver damage were observed at this dosage (data not shown). Equivalent amounts of DMSO were administered to the other groups. There were no differences in the mortality rate between both sham and sham + KHG26792 groups with no deaths in each group. Two of eight rats in I/R group and one of eight rats in I/R + KHG26792 group died, respectively. Therefore, we selected 6 animals from each group randomly based on the number of animals remained in I/R group to be included in this study.
Evaluation of the Neurological Deficit, Infarction Volume and Brain Oedema
Neurological deficits were scored on a five-point scale as described elsewhere (Zea Longa et al. 1989) with a slight modification: 0, normal function; 1, contralateral forelimb flexion when the animal is lifted by the tail; 2, torso turning to the ipsilateral side when the animal is held by the tail with moderate forelimb weakness; 3, spontaneous movement in all directions; contralateral circling only if the animal is pulled by the tail; 4, spontaneous contralateral circling; 5, death. A neurologic examination of all animals of the four groups was blindly performed by a single experimenter.
Infarct volume was determined by the methods as described by others with a slight modification (Ruan et al. 2013). Briefly, the brains were removed and frozen immediately. Then, brains were cut into 2 mm thick slices using a rodent brain matrix slicer. The brain slices were stained with 2% 2,3,5-triphenyltetrazolium chloride at 37 °C for 20 min in dark and then fixed with 10% formalin. The infarct volumes were analyzed and normalized to the volume of the contralateral (non-occluded) hemisphere using a computer image analysis system (Image J 1.46R, NIH, USA). Application of the volume algorithm and statistical analysis was performed by two other investigators, blinded to experimental conditions.
Measurement of brain water content as an indicator of brain oedema was performed as described elsewhere (Ruan et al. 2013). Briefly, brains were blotted to remove residual absorbent moisture and dissected through the interhemispheric fissure into right and left hemispheres. The wet weight was determined with a resolution of 0.1 mg. The hemispheres was dried for 24 h at 100 °C in a drying oven and the dry weight was measured. Brain water content was calculated using the following formula: (wet weight–dry weight)/wet weight) × 100, as previously used as an index of brain oedema (Vakili et al. 2005).
TUNEL Staining
The hippocampus was removed and fixed in 30% sucrose in 10% formaldehyde-saline solution for 72 h. Micrometer paraffin-embedded sections of the hippocampus were processed for a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining in the per-infarct area in accordance with the manufacturer’s specifications with a slight modification, as described elsewhere (Choi et al. 2015; Jo et al. 2016). The DeadEnd Fluorometric TUNEL system (Promega, Madison, WI) was used for nick-end labelling with terminal deoxynucleotidyl transferase. TUNEL-positive cells with green fluorescent staining were detected by fluorescent microscopy. For cell counts, TUNEL-positive cells were counted manually in three different fields of each of three coverslips by three individuals blinded to the experiment. Results are expressed as the percentage of positive cells/total numbers of cells (65 cells per condition), with the KHG26792-treated groups assigned a value of 100%.
Western Blotting
For western blot analysis, total proteins of brain tissues were mixed with SDS-PAGE loading buffer, boiled for 5 min, and analysed by 10% SDS-PAGE. Resolved proteins were transferred onto nitrocellulose membranes, detected by enhanced chemiluminescence according to the manufacturer’s instructions (Amersham, Buckinghamshire, UK), and analysed using a Molecular Imager ChemiDoc XRS system (Bio-Rad, Hercules, CA) as described elsewhere (Eom et al. 2015). Densitometry was performed using ImageJ software (NIH; Bethesda, MD). The following antibodies were used for western blotting: anti-caspase-3, anti-Bax, anti-iNOS, anti-HYOU1, anti-PINK, anti-UCP2, anti-Akt, anti-p-Akt, anti-MMP3, anti-fl-RAGE. β-actin was used to confirm equal protein loading.
Elisa
Levels of inflammatory mediators and cytokines in the crude extracts of the brain tissues were measured using commercially available ELISA kits for TNF-α, IL-1β, MMP-2, intercellular adhesion molecule-1 (ICAM-1), monocyte chemoattractant protein-1 (MCP-1) (R&D Systems, Minneapolis, MN), and keratinocyte-derived chemokine (KC) (Invitrogen, Carlsbad, CA). All tests were carried out in accordance with the manufacturers’ instructions and as described elsewhere (Doverhag et al. 2010; Ju et al. 2015; Liu et al. 2014; Kim et al. 2016).
Measurement of MDA, HNE, ROS, and GSH Levels
The malondialdehyde (MDA) level was measured as an index of lipid peroxidation using the thiobarbituric acid method as previously described (Kim et al. 2009; Uddin et al. 2015). Briefly, samples were treated as described above and suspended in a reaction mixture containing 8.1% SDS (100 μL), 20% acetic acid (pH 3.5, 750 μL), 0.8% thiobarbituric acid (750 μL), and distilled water (300 μL). After being boiled for 1 h at 95 °C, samples were centrifuged at 4000×g for 10 min and the absorbance of the supernatant was measured at 532 nm. In addition, the 4-hydroxy-2-nonenal (HNE) level was measured as an indicator of lipid peroxidation using an HNE assay kit (Oxis-Research, Portland, OR) as described elsewhere (Kim et al. 2015b). The sample was mixed with N-methyl-2-phenylindole in acetonitrile and methanesulfonic acid. After centrifugation, the absorbance of the supernatant was determined at 586 nm. The HNE concentrations in the experimental samples were determined from the standard curve obtained with the HNE standards provided in the assay kit.
A microfluorescence assay using 2′,7′-dichlorofluorescin diacetate (DCF-DA) was used to monitor the production of ROS as described previously (Kim et al. 2015a; Uddin et al. 2015). ROS concentrations were assessed using the oxidant-sensitive probe DCF-DA according to the manufacturer’s protocol. The fluorescence intensity of the DCF product was measured by using a SpectraMax GEMINI XS fluorescence spectrophotometer (Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 485 nm and an emission wavelength of 538 nm. All experiments were performed in the dark.
The GSH level was measured as described elsewhere with minor modifications (Lombardi et al. 2002). Briefly, 100 μL of 5′,5′-dithio-bis(2-nitrobenzoic acid) (6 mM), 15 μL of protein-free extracts, 875 μL of NADPH (0.3 mM), and 10 μL of GSH reductase (10 U·mL−1) were mixed, and the absorbance change was measured at 412 nm with a spectrophotometer.
Measurement of Catalase and SOD Activities
A catalase (CAT) activity assay was performed by determining the H2O2 decomposition rate at 240 nm, according to a method described elsewhere (Aebi 1984). SOD activity was measured by monitoring inhibition of the ferricytochrome c reduction reaction by xanthine/xanthine oxidase (Kim et al. 2009). The reaction mixture contained 10 μM cytochrome c, 50 μM xanthine, and sufficient xanthine oxidase to produce a cytochrome c reduction rate of 0.025 absorbance units per minute at 550 nm. The assay was performed in 3 mL of 50 mM potassium phosphate buffer (pH 7.8), containing 0.1 mM EDTA in a cuvette at 25 °C.
Isolation of Mitochondria
Isolation of mitochondria was carried out according to the procedure described by Clark and Nicklas (1970). Brain tissues were gently homogenized in 10 mM HEPES (pH 7.5) containing 0.25 M sucrose using a loose-fit Teflon pestle. The homogenate was centrifuged at 2000×g for 3 min at 4 °C, and the supernatant obtained was further centrifuged at 12000×g for 8 min. The precipitate was resuspended in the isotonic sucrose medium. Then, the suspension was centrifuged at 12000×g for 10 min, and the mitochondrial pellet was resuspended in sucrose 0.25 M and centrifuged at 12000×g for 10 min. Finally, the mitochondrial pellets were suspended in a minimal amount of 10 mM HEPES (pH 7.5) containing 0.25 M sucrose and stored at −80 °C until the start of the analysis. All of the above procedures were performed at 4 °C.
Measurement of ATP, Na+, K+-ATPase, Cytochrome c Oxidase, and Soluble RAGE (sRAGE)
ATP concentration was measured in accordance with the guidelines of an ATP ELISA kit (Abcam, Cambridge, MA) by measuring the absorbance at 570 nm. The ATP concentration of each sample was determined from a standard curve and normalized to total protein content.
Na+, K+-ATPase was assayed using 50 mM Tris/HCl, pH 7.4 containing 5 mM MgCl2, 80 mM NaCl, 20 mM KCl, and 3 mM ATP. The reaction was initiated by adding 25 μL of 10% homogenate to the samples and incubating them at 37 °C for 1 h. Released inorganic phosphate was measured by the method of Chan et al. (1986). The absorbance intensity at 640 nm was detected against a control that was carried out under the same conditions containing all of the reagents except the crude extracts. The standard curve was plotted using potassium dihydrogen phosphate. The specific activity of the enzyme was expressed as nanomoles of inorganic phosphorus released per minute per microgram of protein.
Cytochrome c oxidase activity in the mitochondrial fractions was assessed using an activity assay kit (BioChain Institute, Hayward, CA). The activity of cytochrome c oxidase to oxidize ferrocytochrome c to ferricytochrome c was measured using spectrophotometry. The absorbance of ferricytochrome c was measured as a loss of absorbance at 550 nm and normalized to total protein content.
Total levels of soluble RAGE (sRAGE) were quantified using a commercially available ELISA kit (R&D Systems) according to the manufacturer’s instructions. Plasma samples were obtained from trunk blood. Blood samples were rapidly centrifuged for 10 min at 500×g and stored at −80 °C until the analysis was performed.
Data Analyses
Statistical data are expressed as the mean ± SD. Individual differences between the groups were analysed using one-way ANOVA. Student’s t-test was used to analyse differences between two groups, and a threshold of P < 0.01 was used to define statistical significance.
Results
Effects of KHG26792 on Neurological Assessment, Infarction Volume and Water Content in I/R Brain Injury
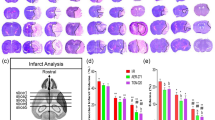
To investigate the neuroprotective effects of KHG26792, the neurological deficit scores and infarct volume after cerebral I/R were measured. The neurological deficit scores and infarct volume of the different groups after 24-h reperfusion are shown in Fig. 1. The neurological deficit score and infarct volume in the I/R + KHG26792 group was significantly lower than that of the I/R group (Fig. 1a, b), suggesting that KHG26792 improved the I/R-induced neurological deficit in mouse brains. However, we can’t exclude the possibility that the neurological score changes may be due to hemorrhage at least in part. Further studies such as laser-Doppler measurement might be necessary to support for exclusion of data based on the in cerebral blood flow following MCAO as described by Morris et al. (2016). Brain water content as an indicator of brain oedema was also measured. After 24 h of reperfusion, the brain water content in the I/R group was markedly higher than that of the sham group (Fig. 1c). However, KHG26792 treatment significantly reduced the water content in the I/R + KHG26792 group.

Effects of KHG26792 on neurological deficit scores and water content in I/R brain injury. Neurological deficit scores a, infarction volume b and brain water content c were measured in mouse brains. Data are presented as means ± SD. The asterisk indicates a significant difference between the I/R group and the I/R group pretreated with KHG26792 (P < 0.01)
Anti-Apoptotic Properties of KHG26792 in I/R Brain Injury
Next, we examined the anti-apoptotic properties of KHG26792. The protein levels of caspase-3 and Bax as measured by western blot are shown in Fig. 2a. The protein levels of β-actin remained relatively constant in the four groups. The protein levels of caspase-3 and Bax were not significantly different between the sham-operated and KHG26792-treated sham groups. On the other hand, significant increases in caspase-3 and Bax protein levels were detected in the I/R group exposed to 2-h MCAO followed by 24-h reperfusion (Fig. 2b, c). However, caspase-3 and Bax protein levels were significantly reduced in the I/R + KHG26792 group (Fig. 2b, c). Our results are consistent with those of previous studies, in which agents that protect neuronal cells from caspase-3 activation ameliorated ischaemic brain injury (Doverhag et al. 2010; Hou et al. 2014).

Anti-apoptotic properties of KHG26792 in I/R brain injury. a Western blot results showing changes in caspase-3 and Bax expression in the I/R group when the animals were pretreated with KHG26792. (b and c) Bar graphs showing the quantified levels of caspase-3/β-actin and Bax/β-actin calculated using densitometry and expressed as the percentage of the I/R group. d Treatment with KHG26792 significantly reduced the number of TUNEL-positive cells. Data are presented as means ± SD. The asterisk indicates a significant difference between the I/R group and the I/R group treated with KHG26792 (P < 0.01)
Apoptosis was also evaluated using a TUNEL assay. Consistent with the caspase-3 and Bax data, the number of TUNEL-positive cells significantly increased in the I/R group, whereas TUNEL staining was almost absent in the sham and KHG26792-treated sham groups (Fig. 2d). Compared with the I/R group, only weak TUNEL labelling was detected in the I/R + KHG26792 group (Fig. 2d). These results suggest that apoptosis after ischaemia was significantly inhibited by KHG26792 treatment.
Effects of KHG26792 on the Levels of Inflammatory Cytokines and ICAM-1 in I/R Brain Injury
We have previously reported the anti-inflammatory and anti-oxidative effects of KHG26792 on ATP- or hypoxia-induced inflammation in BV-2 cells (Kim et al. 2015a; Kim et al. in press). Here, we investigated the effects of KHG26792 on ischaemia-induced changes in the levels of the proinflammatory cytokines TNF-α, IL-1β, and MCP-1 in mouse brains. As expected, ischaemia significantly increased the levels of TNF-α, IL-1β, and MCP-1 by 3.2-fold, 4.4-fold, and 25.1-fold, respectively, compared with the sham group (Fig. 3a, b and c). Remarkably, KHG26792 treatment markedly attenuated proinflammatory cytokine levels compared with the I/R group (Fig. 3a, b and c).

Effects of KHG26792 on TNF-α a, IL-1β b, MCP-1 c, MIP-2 d, KC e, and ICAM-1 f levels in I/R brain injury. Data are presented as the means ± SD and expressed as the percentage of the I/R group. The asterisk indicates a significant difference between the I/R group and the I/R group pretreated with KHG26792 (P < 0.01)
As shown in Fig. 3d, e and f, we further determined the levels of macrophage inflammatory protein-2 (MIP-2), murine homologue KC, and ICAM-1. MIP-2, KC, and ICAM-1 levels were increased 2.1-fold, 3.4-fold, and 4.1-fold in I/R, respectively. In contrast, KHG26792 treatment significantly reduced the levels of these inflammatory markers, as indicated by a comparison of the I/R + KHG26792 and I/R groups. No significant changes in the levels of these markers were observed between the sham and sham + KHG26792 groups. Together, these data suggest that the protective effects of KHG26792 are partly mediated by regulation of inflammatory cytokine release in I/R brain injury.
Effects of KHG26792 on MDA, HNE, ROS, and GSH Levels and SOD and CAT Activity in I/R Brain Injury
To examine the inhibitory effects of KHG26792 on ischaemia-induced lipid peroxidation, we measured the levels of the lipid peroxidation markers MDA and HNE. As shown in Fig. 4a, b, the levels of MDA and HNE in the I/R group were 2.2-fold and 3.4-fold higher, respectively, than those of the sham group. However, significant reductions in MDA and HNE levels were observed in the I/R + KHG26792 group compared with the I/R group (Fig. 4a, b). In addition, we also measured the effects of KHG26792 on the levels of ROS, an indicator of oxidative stress. ROS levels were approximately 3.2-fold higher in the I/R group than in the sham group, indicating that ROS generation was accelerated (Fig. 4c). However, KHG26792 administration significantly reduced the ischaemia-induced increase in ROS levels (Fig. 4c), suggesting a potential antioxidant role of KHG26792.

Effects of KHG26792 on MDA a, HNE b, ROS c, GSH d, SOD activity e, and CAT activity f in I/R brain injury. Data are presented as means ± SD and expressed as the percentage of the I/R group (a–c) or the percentage of the control (d–f). The asterisk indicates a significant difference between the I/R group and the I/R group pretreated with KHG26792 (P < 0.01)
GSH, a central component in the antioxidant defence of cells, acts both by detoxifying ROS directly and by serving as a substrate for various peroxidases (Conrad and Sato 2012; Ma et al. 2012). GSH content was lower in animals in the I/R group, whereas KHG26792 treatment significantly attenuated I/R-induced depletion of GSH (Fig. 4d). In addition to GSH, antioxidant enzymes such as SOD and CAT play important roles in cellular defence against lipid peroxidation and ROS production (Kunwar et al. 2009). Accordingly, we also investigated the effects of KHG26792 on the activities of these enzymes. There was a significant decrease in SOD (Fig. 4e) and CAT (Fig. 4f) activities in the I/R group. In contrast, the mice in the I/R + KHG26792 group exhibited significantly higher levels of SOD and CAT than those in the I/R group. Because significant decreases in MDA, HNE, and ROS levels with concomitant increases in SOD and CAT activities and the GSH level were seen in the I/R + KHG26792 group compared with the I/R group (Fig. 4), we suggest that KHG26792 protects the brain against I/R injury at least partly through regulation of the oxidation–reduction system, specifically by increasing antioxidant capacity.
Effects of KHG26792 on the Total Brain ATP Level, Mitochondrial ATP Level, Na+, K+-ATPase Activity, and Cytochrome c Oxidase Activity in I/R Brain Injury
The ATP content of brain mitochondria was quantified to examine the effects of KHG26792 on brain energy metabolism in cerebral I/R mice. MCAO reduced the brain ATP level compared with the sham group, whereas KHG26792 treatment significantly attenuated the ATP level compared with the I/R group (Fig. 5a). Because mitochondria can become dysfunctional and adversely affect ATP bioavailability after an I/R insult, ATP production in isolated mitochondria was also determined. As shown in Fig. 5b, cerebral I/R dramatically reduced the mitochondrial ATP level in the I/R group compared with the sham control group. However, KHG26792 administration significantly increased the mitochondrial ATP level compared with the I/R group (Fig. 5b), suggesting that KHG26792 protects against I/R-induced tissue energy dysfunction. In addition, we further examined the ability of KHG26792 to improve mitochondrial dysfunction following I/R using analysis of mitochondrial cytochrome c oxidase activity. Consistent with the changes in ATP levels, our results showed that I/R reduced mitochondrial cytochrome c oxidase activity, indicating the dysfunction of the mitochondrial electron transfer chain under I/R (Fig. 5c). Once again, KHG26792 treatment significantly recovered cytochrome c oxidase activity in the I/R + KHG26792 group compared with the I/R group (Fig. 5c).

Effects of KHG26792 on the total brain ATP level a, mitochondrial ATP level b, cytochrome c oxidase activity c, and Na+, K+-ATPase activity d in I/R brain injury. Data are presented as means ± SD and expressed as the percentage of the control. The asterisk indicates a significant difference between the I/R group and the I/R group pretreated with KHG26792 (P < 0.01)
Next, we determined the impact of KHG26792 on the Na+, K+-ATPase level in ischaemic and KHG26792 treatment groups because changes in Na+, K+-ATPase can affect the neuronal injury apoptosis cascade by destroying K+ homeostasis (Wang et al. 2003). I/R induction reduced the Na+, K+-ATPase level compared with the sham group (Fig. 5d). In contrast, KHG26792 treatment significantly increased the Na+, K+-ATPase level in the I/R + KHG26792 group compared with the I/R group.
Effects of KHG26792 on the Protein Levels of iNOS, HYOU1, PINK, and UCP2 in I/R Brain Injury
Western blotting analysis showed that I/R markedly increased the protein levels of iNOS, HYOU1, PINK, and UCP2 (Fig. 6a), in line with previous reports (Chen et al. 2006; Liu et al. 2013; Ruan et al. 2013; Chen et al. 2015). In contrast, KHG26792 treatment effectively attenuated the levels of iNOS and HYOU1 protein compared with the I/R group without KHG26792 (Fig. 6a, b, c and d). Taken together, iNOS and HYOU1 downregulation (Fig. 6b, c) was accompanied by decreased oxidative stress (Fig. 4). However, KHG26792 treatment did not show any significant effects on the I/R-induced protein levels of PINK and UCP2 (Fig. 6d, e). These results suggest that the suppression of iNOS and HYOU1 signalling, but not PINK and UCP2 signalling, may be involved in the protective effect of KHG26792 on I/R-induced injury in mouse brain, although the precise mechanism of KHG26792 activity under ischaemic conditions remains unclear. There were no significant differences in the levels of any of the proteins tested between the sham and sham + KHG26792 groups.

Effects of KHG26792 on the protein levels of iNOS, HYOU1, PINK, and UCP2 in I/R brain injury. a Equal amounts of crude extracts were immunoblotted using primary antibodies against each protein. (b–e) Bar graphs showing the quantified protein levels calculated using densitometry and the ratio of the protein intensity to β-actin intensity was expressed as the percentage of the I/R group. Data are presented as means ± SD. The asterisk indicates a significant difference between the I/R group and the I/R group pretreated with KHG26792 (P < 0.01)
Effects of KHG26792 on the Protein Expression of P-Akt and MMP-3 in I/R Brain Injury
As shown in Fig. 7a, the western blotting results revealed no significant changes in the protein level of Akt among the groups (Fig. 7a, b). However, compared with the sham group, I/R dramatically decreased the protein levels of p-Akt, but increased those of MMP-3 in the I/R group (Fig. 7a, b and c). Interesting, KHG26792 treatment almost completely restored the levels of both p-Akt and MMP-3 in the I/R + KHG26792 group, so that they were not significantly different from their corresponding values in the sham group (Fig. 7a, b and c). Therefore, these results strongly support the possible involvement of Akt and MMP-3 signalling, as well as iNOS and HYOU1 signalling, in the neuroprotective effects of KHG26792 on brain I/R injury.

Effects of KHG26792 on the protein expression of p-Akt and MMP-3 in I/R brain injury. a Western blot results showing changes in p-Akt and MMP-3 expression in the I/R group when the animals were pretreated with KHG26792 (a). (b, c) Bar graphs showing the quantified levels of p-Akt/Akt and MMP-3/β-actin calculated using densitometry and expressed as the percentage of the control group (b) or the I/R group (c). Data are presented as means ± SD. The asterisk indicates a significant difference between the I/R group and the I/R group treated with KHG26792 (P < 0.01)
Effects of KHG26792 on Fl-RAGE Protein Expression and sRAGE Activity in I/R Brain Injury
The western blot results showed that the protein levels of fl-RAGE after I/R were almost identical among the groups (Fig. 8a, b). Actually, fl-RAGE proteins were hardly detected in any of the groups. Interestingly, after 24 h of reperfusion, significant decreases in sRAGE plasma levels were found in the I/R group compared with the sham group, and treatment with KHG26792 significantly restored the decreased sRAGE circulating levels caused by I/R (Fig. 8c).

Effects of KHG26792 on the protein expression of fl-RAGE and the activity of sRAGE in I/R brain injury. a Western blot results showing changes in fl-RAGE expression in the I/R group when the animals were pretreated with KHG26792. b Bar graphs showing the quantified levels of fl-RAGE/β-actin calculated using densitometry and expressed as the percentage of the control group. c KHG26792 significantly attenuated the inhibition of sRAGE in I/R brain injury. Data are presented as means ± SD. The asterisk indicates a significant difference between the I/R group and the I/R group treated with KHG26792 (P < 0.01)
Discussion
Our current study is the first to investigate the anti-oxidative properties of KHG26792 in an ischaemia animal model. Our results showed that KHG26792 significantly reduced neurological deficits and brain oedema in mouse brains exposed to 2 h of MCAO followed by 24 h of reperfusion. Many studies have reported that hypoxia–ischaemia-induced apoptosis in the brain promotes cell death through caspase-3 and Bax activation (Doverhag et al. 2010; Yao et al. 2012; Hou et al. 2014). In the present study, we showed that caspase-3 and Bax protein levels were significantly higher in the I/R group, in agreement with the TUNEL assay results, suggesting apoptotic processes after I/R injury (Fig. 2). Interestingly, KHG26792 significantly decreased caspase-3 and Bax expression in mouse brains. These results indicate that KHG26792 promotes cell survival by inhibiting I/R-induced expression of caspase-3 and Bax, suggesting that it might act as an effective regulator of the apoptotic process. Our results further support previous studies in which agents that protect neuronal cells from caspase-3 activation ameliorated ischaemic brain injury (Doverhag et al. 2010; Hou et al. 2014).
Furthermore, our current analysis indicated increased levels of the inflammatory mediators TNF-α, IL-1β, MIP-2, KC, ICAM-1, and MCP-1 in the I/R group (Fig. 3), indicating the role of inflammatory events in ischaemia and further confirming the successful induction of cerebral ischaemia in mouse brains, as previously reported (Ramanathan et al. 2012; Orsu et al. 2013). Our results are therefore consistent with those of previous studies in which I/R increased the release of proinflammatory cytokines and inflammatory mediators in the brain (Shen et al. 2008; Justin et al. 2014). Oxidative stress and inflammation are the main factors for cell destruction and apoptosis in I/R-induced brain injury (Wilms et al. 2005; Doyle et al. 2008; Broughton et al. 2009). In addition, important roles for oxidative stress in neuroinflammation regulation after stroke during hypoxic processes have been reported (Nakanishi and Wu 2009; Guo et al. 2014). Therefore, agents that can scavenge free radical generation and reduce inflammation are expected to have neuroprotective effects. ROS levels are increased in the brains of experimental animal models of agent-induced hypoxia (Nakanishi and Wu 2009; Rathnasamy et al. 2011; Guo et al. 2014; Hou et al. 2014; Guan et al. 2015). Many studies also reported that ROS functions as a main factor in the pathophysiology of ischaemic stroke and cerebral injury by promoting oxidative damage of membrane lipids and proteins (Love 1999; White et al. 2000). In this study, we measured the effects of KHG26792 on the oxidative stress caused by I/R to examine the protective role of KHG26792 during I/R brain injury. Consistent with previous reports (Li et al. 2014; Guan et al. 2015), we found that MDA, HNE, and ROS levels in the I/R group were higher than those in the sham group (Fig. 4a, b and c). However, KHG26792 significantly reduced the oxidative stress observed in the I/R group, suggesting a potential antioxidant role of KHG26792.
SOD, CAT, and GSH play important roles in protecting the CNS against oxidative stress and inflammation (Kim et al. 2013; Zhang et al. 2015). Our present data indicated that SOD and CAT activities and GSH levels were reduced in the I/R group, with KHG26792 administration significantly recovering SOD and CAT activities as well as the GSH level compared with the I/R group (Fig. 4). Accordingly, these results showed an anti-oxidative effect of KHG26792 on both enzymatic and non-enzymatic antioxidant systems. Taken together, the results shown in Fig. 4 suggest that the antioxidant action of KHG26792 underlies its neuroprotective capacity in I/R brains. Therefore, KHG26792 may be a valuable and novel agent for studying the relationship between oxidation and disease pathophysiology.
iNOS plays a key role in inflammation, and cytokine signal transduction can be involved in the regulation of the iNOS gene (Cardenas et al. 2000; Shahani and Sawa 2011; Yao et al. 2013). In I/R brain injury, an elevated iNOS level increases the production of peroxynitrite in the presence of superoxide radicals, which is accompanied by depleted GSH levels (Iadecola et al. 1995; Samdani et al. 1997; Moro et al. 2004; Tang et al. 2012; Liu et al. 2013). This may explain the decreased GSH levels observed in the present study (Fig. 4d). On the other hand, KHG26792 treatment significantly attenuated the reduction in ROS generation by increasing antioxidants levels (Fig. 4) and downregulating iNOS (Fig. 6). Recently, we reported that KHG26792 exhibits anti-inflammatory and antioxidant activities as a consequence of iNOS downregulation in BV-2 cells (Kim et al. 2015a; Kim et al. in press), which is consistent with the anti-oxidative and anti-inflammatory properties of KHG26792 observed in the present study after iNOS downregulation (Fig. 6).
Interestingly, we found that the protein expression of HYOU1, an oxygen-regulated protein, was increased in I/R brain injury, with KHG26792 attenuating the I/R-induced increase (Fig. 6b). HYOU1 is induced by hypoxia/ischaemia (Sato et al. 2001; Ruan et al. 2013). Accordingly, we suggest that the reduction in HYOU1 by KHG26792 may at least partly contribute to the neuroprotective effect of KHG26792, although the mechanism regulating the relationship between HYOU1 and KHG26792 is currently unclear.
Mitochondria are mainly responsible for the production of cellular energy in the form of ATP. During reperfusion, excessive ROS produced by mitochondria can damage electron transport complexes, suppressing mitochondrial energy metabolism and increasing apoptosis (Krajewski et al. 1999; Blomgren et al. 2003; Niatsetskaya et al. 2012). Moreover, ROS induced by MCAO destroys the functional energy metabolism of mitochondria and reduces ATP levels, facilitating neuronal cell apoptosis after cerebral ischaemia (Wang et al. 2013; Sun et al. 2014; Justin et al. 2014). Figure 5 shows that total brain ATP levels as well as brain mitochondria ATP levels were lower in the I/R group than in the sham group. We found that KHG26792 can rescue I/R-induced ATP generation in the I/R mouse brain. Therefore, KHG26792 improved ATP levels to protect the brain against I/R brain injury by increasing antioxidant enzyme activities and suppressing ROS generation (Fig. 4).
We further evaluated whether the neuroprotective properties of KHG26792 involve a recovery of cytochrome c oxidase activity, part of the electron transfer chain complex, because caspase-3 activation requires cytochrome c release after mitochondrial dysfunction (Li et al. 1997). We found that cytochrome c oxidase activity was significantly reduced in the I/R group, supporting the evidence indicating electron respiratory chain disruption after I/R brain injury (Fig. 5). Because the reduced ATP level impairs the ion pumps in the neuronal membrane, the effect of KHG26792 was also reflected in the Na+, K+-ATPase activity. Na+, K+-ATPase plays an important role in the active transport of ions across the cell membrane. This transport system consumes approximately half of the ATP content of the brain to maintain the electrochemical gradient (Kaplan 2002). Consistent with previous results, about a 50% reduction in Na+, K+-ATPase activity was observed after I/R brain injury (Fig. 5d). This change in Na+, K+-ATPase may mainly activate neuronal apoptosis by changing the K+ homeostasis (Wang et al. 2003). A functional Na+, K+-ATPase consists of a catalytic α subunit and a regulatory β subunit, multiple α isoforms are found in mammals, and the regulative functions are also reflected in the expressive relationship among α isoforms in the CNS (Sweadner 1989; Isaksen and Lykke-Hartmann 2016). Therefore, further studies for the precise mechanisms of the effect of KHG26792 on the expressive relationship among Na+, K+-ATPase α isoforms in ischemia remain to be elucidated.
Next, we examined the effects of KHG26792 on PINK1 and UCP2 in I/R brain injury because previous studies reported that PINK1 is a mitochondrial serine/threonine-protein kinase that protects against mitochondrial dysfunction (Plun-Favreau et al. 2007; Pridgeon et al. 2007). UCP2 is another inner mitochondrial membrane protein that is implicated in reduced production of ROS (Arsenijevic et al. 2000). Moreover, increased UCP2 expression was observed in global ischaemia-induced injury (Chen et al. 2006). However, KHG26792 showed no effects on either PINK1 or UCP2 (Fig. 6d, e).
To further investigate the mechanism by which KHG26792 protects against I/R brain injury, we determined the effects of KHG26792 on the levels of p-Akt and MMP-3 protein. Akt regulates the survival response to oxidative stress-induced neuronal apoptosis and its phosphorylation is necessary for its neuroprotective activity by suppressing apoptosis in the CNS (Zhao et al. 2005; Manning and Cantley 2007; Zhang et al. 2007). MMP-3, a key endopeptidase involved in inflammatory reactions, is also increased after cerebral ischaemia in rats, mice, nonhuman primates, and humans, resulting in the proteolytic breakdown of the blood–brain barrier (Pfefferkorn and Rosenberg 2003; Cheng et al. 2011). Our present results support the notion that inhibition of p-Akt and MMP-3 could be important factors exacerbating brain damage after I/R injury. On the other hand, KHG26792 treatment efficiently increased p-Akt and reduced MMP-3 (Fig. 7). Therefore, it is possible that the effects of KHG26792 on reduced ROS generation and iNOS down-regulation may be closely associated with amelioration of MMP-3 and the Akt pathway. Because we only used cerebral homogenates in the present study, further studies including knockdown experiments would help to validate our findings.
Many studies have shown that RAGE, as well as promoting ROS production, functions as a damage-sensing molecule and is increased in pathological conditions such as cerebral ischaemia (Muhammad et al. 2008; Hassid et al. 2009; Kalea et al. 2009). Proteolysis of the full-length membrane-bound RAGE by MMPs generates soluble forms of RAGE (sRAGE), suggesting the potential use of sRAGE as a blood biomarker of stroke severity (Zhang et al. 2008; Greco et al. 2012, 2014). In addition, exogenous sRAGE treatment has been used to antagonize I/R brain injury (Tang et al. 2013). In the present study, we showed that sRAGE levels were reduced after MCAO followed by reperfusion, in line with previous findings indicating decreased sRAGE levels after either transient or permanent MCAO (Greco et al. 2012, 2014). In contrast, there were no significant changes in the protein expression of fl-RAGE in any of the animal groups (Fig. 8). More interestingly, KHG26792 treatment clearly attenuated the I/R-induced reduction in sRAGE, further indicating the mechanism for the neuroprotective effects of KHG26792 in I/R brain injury.
In conclusion, our present report is the first to show that KHG26792 can protect the mouse brain against ischaemic injury by inhibiting apoptotic damage, modulating inflammation, scavenging free radicals, ameliorating oxidative stress, and improving the energy metabolism of the brain. These beneficial effects are the possible pharmacological basis of the protective activity of KHG26792, although we cannot completely exclude other possible mechanisms by which KHG26792 might attenuate I/R-induced brain injury. Further supporting data are needed to clarify how KHG26792 protects against cerebral ischaemia and to understand the neuroprotective effects of this novel compound.
Abbreviations
- Bax:
-
Bcl-2-associated X protein
- CAT:
-
catalase
- CNS:
-
central nervous system
- DCF-DA:
-
2′,7′-dichlorofluorescin diacetate
- fl-RAGE:
-
full-length receptor for advanced glycation end-products
- HNE:
-
4-hydroxy-2-nonenal
- HYOU1:
-
hypoxia-upregulated protein 1
- ICAM-1:
-
intercellular adhesion molecule-1
- iNOS:
-
inducible nitric oxide synthase
- I/R:
-
ischaemia/reperfusion
- KC:
-
keratinocyte-derived chemokine
- MCAO:
-
middle cerebral artery occlusion
- MCP-1:
-
monocyte chemoattractant protein-1
- MDA:
-
malondialdehyde
- PINK1:
-
PTEN-induced putative kinase 1
- sRAGE:
-
soluble receptor for advanced glycation end-products
- UCP2:
-
uncoupling protein 2
References
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Ahmed MAE, El Morsy EM, Ahmed AAE (2014) Pomegranate extract protects against cerebral ischemia/reperfusion injury and preserves brain DNA integrity in rats. Life Sci 110:61–69
Alexandrova M, Bochev P, Markova V, Bechev B, Popova M, Danovska M, Simeonova V (2004) Dynamics of free radical processes in acute ischemic stroke: influence on neurological status and outcome. J Clin Neurosci 11:501–506
Allan SM, Rothwell NJ (2001) Cytokines and acute neurodegeneration. Nat Rev Neurosci 2:734–744
Allen CL, Bayraktutan U (2009) Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 4:461–470
Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26:435–439
Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD (1996) Middle cerebral artery occlusion in the rat by intraluminal suture. Neurol Pathol Eval Improved Model Stroke 27:1616–1622
Blomgren K, Zhu C, Hallin U, Hagberg H (2003) Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Biochem Biophys Res Commun 304:551–559
Broughton BR, Reutens DC, Sobey CG (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40:e331–e339
Cardenas A, Moro MA, Hurtado O, Leza JC, Lorenzo P, Castrillo A, Bodelón OG, Boscá L, Lizasoain I (2000) Implication of glutamate in the expression of inducible nitric oxide synthase after oxygen and glucose deprivation in rat forebrain slices. J Neurochem 74:2041–2048
Chan KM, Delfert D, Junger KD (1986) A direct colorimetric assay for Ca+-stimulated ATPase activity. Anal Biochem 157:375–380
Chen SD, Wu HY, Yang DI, Lee SY, Shaw FZ, Lin TK, Liou CW, Chuang YC (2006) Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem Biophys Res Commun 351:198–203
Chen SD, Lin TK, Yang DI, Lee SY, Shaw FZ, Liou CW, Chuang YC (2015) Roles of PTEN-induced putative kinase 1 and dynamin-related protein 1 in transient global ischemia-induced hippocampal neuronal injury. Biochem Biophys Res Commun 460:397–403
Cheng Z, He W, Zhou X, Lv Q, Xu X, Yang S, Zhao C, Guo L (2011) Cordycepin protects against cerebral ischemia/reperfusion injury in vivo and in vitro. Eur J Pharmacol 664:20–28
Choi SH, Park BK, Lee KW, Chang J, Lee Y, Kwon HJ (2015) Effect of respiratory syncytial virus on the growth of hepatocellular carcinoma cell-lines. BMB Rep 48:565–570
Clark JB, Nicklas WJ (1970) The metabolism of rat brain mitochondria. Prep Charact J Biol Chem 245:4724–4731
Conrad M, Sato H (2012) The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (−):cystine supplier and beyond. Amino Acids 42:231–246
Dalla Y, Singh N, Jaggi AS, Singh D, Ghulati P (2009) Potential of ezetimibe in memory deficits associated with dementia of Alzheimer’s type in mice. Indian J Pharm 41:262–267
de Vries DK, Kortekaas KA, Tsikas D, Wijermars LG, van Noorden CJ, Suchy MT, Cobbaert CM, Klautz RJ, Schaapherder AF, Lindeman JH (2013) Oxidative damage in clinical ischemia/reperfusion injury: a reappraisal. Antioxid Redox Signal 19:535–545
Doverhag C, Hedtjärn M, Poirier F, Mallard C, Hagberg H, Karlsson A, Sävman K (2010) Galectin-3 contributes to neonatal hypoxic–ischemic brain injury. Neurobiol Dis 38:36–46
Doyle KP, Simon RP, Stenzel-Poore MP (2008) Mechanisms of ischemic brain damage. Neuropharmacology 55:310–318
Eom SA, Kim DW, Shin MJ, Ahn EH, Chung SY, Sohn EJ, Jo HS, Jeon SJ, Kim DS, Kwon HY, Cho SW, Han KH, Park J, Eum WS, Choi SY (2015) Protective effects of PEP-1-catalase on stress-induced cellular toxicity and MPTP-induced Parkinson's disease. BMB Rep 48:395–400
Gómez-Garre D, Muñoz-Pacheco P, González-Rubio ML, Aragoncillo P, Granados R, Fernández-Cruz A (2009) Ezetimibe reduces plaque inflammation in a rabbit model of atherosclerosis and inhibits monocyte migration in addition to its lipid-lowering effect. Br J Pharmacol 156:1218–1227
Greco R, Amantea D, Mangione AS, Petrelli F, Gentile R, Nappi G, Blandini F, Corasaniti MT, Tassorelli C (2012) Modulation of RAGE isoforms expression in the brain and plasma of rats exposed to transient focal cerebral ischemia. Neurochem Res 37:1508–1516
Greco R, Tassorelli C, Mangione AS, Levandis G, Certo M, Nappi G, Bagetta G, Blandini F, Amantea D (2014) Neuroprotection by the PARP inhibitor PJ34 modulates cerebral and circulating RAGE levels in rats exposed to focal brain ischemia. Eur J Pharmacol 744:91–97
Guan D, Su Y, Li Y, Wu C, Meng Y, Peng X, Bagetta G, Blandini F, Amantea D (2015) Tetramethylpyrazine inhibits CoCl2-induced neurotoxicity through enhancement of Nrf2/GCLc/GSH and suppression of HIF1α/NOX2/ROS pathways. J Neurochem 134:551–565
Guo K, Mou X, Huang J, Xiong N, Li H (2014) Trans-caryophyllene suppresses hypoxia-induced neuroinflammatory responses by inhibiting NF-κB activation in microglia. J Mol Neurosci 54:41–48
Han Y, Han M, Shin D, Song C, Hahn HG (2012) Exploration of novel 3-substituted azetidine derivatives as triple reuptake inhibitors. J Med Chem 55:8188–8192
Han M, Song C, Jeong N, Hahn HG (2014) Exploration of 3-aminoazetidines as triple reuptake inhibitors by bioisosteric modification of 3-aoxyazetidine. ACS Med Chem Lett 5:999–1004
Hassid BG, Nair MN, Ducruet AF, Otten ML, Komotar RJ, Pinsky DJ, Schmidt AM, Yan SF, Connolly ES (2009) Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J Clin Neurosci 16:302–306
Hou CW, Chen YL, Chuang SH, Wang JS, Jeng KC (2014) Protective effect of a sesamin derivative, 3-bis (3-methoxybenzyl) butane-1, 4-diol on ischemic and hypoxic neuronal injury. J Biomed Sci 21:15
Iadecola C, Zhang F, Xu S, Casey R, Ross ME (1995) Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 15:378–384
Isaksen TJ, Lykke-Hartmann K (2016) Insights into the pathology of the α2-Na(+)/K(+)-ATPase in neurological disorders; lessons from animal models. Front Physiol 7:161
Jo HS, Yeo HJ, Cha HJ, Kim SJ, Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, Eum WS, Choi SY (2016) Transduced tat-DJ-1 protein inhibits cytokines-induced pancreatic RINm5F cell death. BMB Rep 49:297–302
Ju SM, Youn GS, Cho YS, Choi SY, Park J (2015) Celastrol ameliorates cytokine toxicity and pro-inflammatory immune responses by suppressing NF-κB activation in RINm5F beta cells. BMB Rep 48:172–177
Justin A, Sathishkumar M, Sudheer A, Shanthakumari S, Ramanathan M (2014) Non-hypotensive dose of telmisartan and nimodipine produced synergistic neuroprotective effect in cerebral ischemic model by attenuating brain cytokine levels. Pharmacol Biochem Behav 122:61–73
Kalea AZ, Schmidt AM, Hudson BI (2009) RAGE: a novel biological and genetic marker for vascular disease. Clin Sci 116:621–637
Kaplan JH (2002) Biochemistry of Na+, K+-ATPase. Annu Rev Biochem 71:511–535
Kim DW, Jeong HJ, Kang HW, Shin MJ, Sohn EJ, Kim MJ, Ahn EH, An JJ, Jang SH, Yoo KY, Won MH, Kang TC, Hwang IK, Kwon OS, Cho SW, Park J, Eum WS, Choi SY (2009) Transduced human PEP-1-catalase fusion protein attenuates ischemic neuronal damage. Free Radic Biol Med 47:941–952
Kim EA, Choi J, Han AR, Choi SY, Hahn HG, Cho SW (2013) Anti-oxidative and anti-inflammatory effects of 2-cyclopropylimino-3-methyl-1,3-thiazoline hydrochloride on glutamate-induced neurotoxicity in rat brain. Neurotoxicology 38:106–114
Kim EA, Cho CH, Kim J, Hahn HG, Choi SY, Yang SJ, Cho SW (2015a) The azetidine derivative, KHG26792 protects against ATP-induced activation of NFAT and MAPK pathways through P2X7 receptor in microglia. Neurotoxicology 51:198–206
Kim SM, Hwang IK, Yoo DY, Eum WS, Kim DW, Shin MJ, Ahn EH, Jo HS, Ryu EJ, Yong JI, Cho SW, Kwon OS, Lee KW, Cho YS, Han KH, Park J, Choi SY (2015b) Tat-antioxidant 1 protects against stress-induced hippocampal HT-22 cells death and attenuate ischaemic insult in animal model. J Cell Mol Med 19:1333–1345
Kim SJ, Cha JY, Kang HS, Lee JH, Lee JY, Park JH, Bae JH, Song DK, Im SS (2016) Corosolic acid ameliorates acute inflammation through inhibition of IRAK-1 phosphorylation in macrophages. BMB Rep 49:276–281
Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC (1999) Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci U S A 96:5752–5757
Kunwar A, Sandur SK, Krishna M, Priyadarsini KI (2009) Curcumin mediates time and concentration dependent regulation of redox homeostasis leading to cytotoxicity in macrophage cells. Eur J Pharmacol 611:8–16
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489
Li H, Wang Y, Feng D, Liu Y, Xu M, Gao A, Tian F, Zhang L, Cui Y, Wang Z, Chen G (2014) Alterations in the time course of expression of the Nox Family in the brain in a rat experimental cerebral ischemia and reperfusion model: effects of melatonin. J Pineal Res 57:110–119
Liu H, Wei X, Chen L, Liu X, Li S, Liu X, Zhang X (2013) Tetramethylpyrazine analogue CXC195 protects against cerebral ischemia/reperfusion injury in the rat by an antioxidant action via inhibition of NADPH oxidase and iNOS expression. Pharmacology 92:198–206
Liu Y, Zhao T, Yang Z, Li Q (2014) CX3CR1 RNAi inhibits hypoxia-induced microglia activation via p38MAPK/PKC pathway. Int J Exp Pathol 95:153–157
Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4:399–415
Lombardi G, Varsaldi F, Miglio G, Papini MG, Battaglia A, Canonico PL (2002) Cabergoline prevents necrotic neuronal death in an in vitro model of oxidative stress. Eur J Pharmacol 457:95–99
Love S (1999) Oxidative stress in brain ischemia. Brain Pathol 9:119–131
Lowe JT, Lee MD, Akella LB, Davoine E, Donckele EJ, Durak L, Duvall JR, Gerard B, Holson EB, Joliton A, Kesavan S, Lemercier BC, Liu H, Marié JC, Mulrooney CA, Muncipinto G, Welzel-O'Shea M, Panko LM, Rowley A, Suh BC, Thomas M, Wagner FF, Wei J, Foley MA, Marcaurelle LA (2012) Synthesis and profiling of a diverse collection of azetidine-based scaffolds for the development of CNS-focused lead-like libraries. J Organomet Chem 77:7187–7211
Ma S, Liu H, Jiao H, Wang L, Chen L, Liang J, Zhao M, Zhang X (2012) Neuroprotective effect of ginkgolide K on glutamate-induced cytotoxicity in PC 12 cells via inhibition of ROS generation and Ca2+ influx. Neurotoxicology 33:59–69
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274
Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66
Mnich SJ, Hiebsch RR, Huff RM, Muthian S (2010) Anti-inflammatory properties of CB1-receptor antagonist involves beta2 adrenoceptors. J Pharmacol Exp Ther 333:445–453
Moro MA, Cardenas A, Hurtado O, Leza JC, Lizasoain I (2004) Role of nitric oxide after brain ischaemia. Cell Calcium 36:265–275
Morris GP, Wright AL, Tan RP, Gladbach A, Ittner LM, Vissel B (2016) A comparative study of variables influencing ischemic injury in the longa and Koizumi methods of intraluminal filament middle cerebral artery occlusion in mice. PLoS One 11:e0148503
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M (2008) The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 28:12023–12031
Nakanishi H, Wu Z (2009) Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav Brain Res 201:1–7
Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS (2012) The oxygen free radicals originating frommitochondrial complex I contribute to oxidative brain injury following hypoxia–ischemia in neonatal mice. J Neurosci 32:3235–3244
Orsu P, Murthy BVSN, Akula A (2013) Cerebroprotective potential of resveratrol through anti-oxidant and anti-inflammatory mechanisms in rats. J Neural Transm 120:1217–1223
Pfefferkorn T, Rosenberg GA (2003) Closure of the blood–brain barrier by matrix metalloproteinase inhibition reduces rtPAmediated mortality in cerebral ischemia with delayed reperfusion. Stroke 34:2025–2030
Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J (2007) The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol 9:1243–1252
Pridgeon JW, Olzmann JA, Chin LS, Li L (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 5:e172
Ramanathan M, Babu CS, Justin A, Shanthakumari S (2012) Elucidation of neuroprotective role of endogenous GABA and energy metabolites middle cerebral artery occluded model in rats. Indian J Exp Biol 50:391–377
Rathnasamy G, Ling EA, Kaur C (2011) Iron and iron regulatory proteins in amoeboid microglial cells are linked to oligodendrocyte death in hypoxic neonatal rat periventricular white matter through production of proinflammatory cytokines and reactive oxygen/nitrogen species. J Neurosci 31:17982–17995
Ruan L, Huang HS, Jin WX, Chen HM, Li XJ, Gong QJ (2013) Tetrandrine attenuated cerebral ischemia/reperfusion injury and induced differential proteomic changes in a MCAO mice model using 2-D DIGE. Neurochem Res 38:1871–1879
Samdani AF, Dawson TM, Dawson VL (1997) Nitric oxide synthase in models of focal ischemia. Stroke 28:1283–1288
Sato M, Sugano N, Ohzono K, Nomura S, Kitamura Y, Tsukamoto Y, Ogawa S (2001) Apoptosis and expression of stress protein (ORP150, HO1) during development of ischaemic osteonecrosis in the rat. J Bone Joint Surg (Br) 83:751–759
Shahani N, Sawa A (2011) Nitric oxide signaling and nitrosative stress in neurons: role for S-nitrosylation. Antioxid Redox Signal 14:1493–1504
Shen YC, Wang YH, Chou YC, Liou KT, Yen JC, Wang WY, Liao JF (2008) Dimemorfan protects rats against ischemic stroke through activation of sigma-1 receptor-mediated mechanisms by decreasing glutamate accumulation. J Neurochem 104:558–572
Shi H, Sheng B, Zhang F, Wu C, Zhang R, Zhu J, Xu K, Kuang Y, Jameson SC, Lin Z, Wang Y, Chen J, Jain MK, Atkins GB (2013) Kruppel-like factor 2 protects against ischemic stroke by regulating endothelial blood brain barrier function. Am J Physiol Heart Circ Physiol 304:H796–H805
Slemmer JE, Shacka JJ, Sweeney MI, Weber JT (2008) Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem 15:404–414
Sun J, Li YZ, Ding YH, Wang J, Geng J, Yang H, Ren J, Tang JY, Gao J (2014) Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro and cerebral ischemia/reperfusion injury in vivo. Brain Res 1589:126–139
Sweadner KJ (1989) Isozymes of the Na+/K+−ATPase. Biochim Biophys Acta 988:185–220
Tang Q, Han R, Xia H, Shen J, Luo Q, Li J (2012) Neuroprotective effects of tanshinone IIA and/or tetramethylpyrazine in cerebral ischemic injury in vivo and in vitro. Brain Res 1488:81–91
Tang SC, Wang YC, Li YI, Lin HC, Manzanero S, Hsieh YH, Phipps S, Hu CJ, Chiou HY, Huang YS, Yang WS, Mattson MP, Arumugam TV, Jeng JS (2013) Functional role of soluble receptor for advanced glycation end products in stroke. Arterioscler Thromb Vasc Biol 33:585–594
Uddin GM, Kim CY, Chung D, Kim KA, Jung SH (2015) One-step isolation of sappanol and brazilin from Caesalpinia sappan and their effects on oxidative stress-induced retinal death. BMB Rep 48:289–294
Urban PP, Wolf T, Uebele M, Marx JJ, Vogt T, Stoeter P, Bauermann T, Weibrich C, Vucurevic GD, Schneider A, Wissel J (2010) Occurrence and clinical predictors of spasticity after ischemic stroke. Stroke 41:2016–2020
Vakili A, Kataoka H, Plesnila N (2005) Role of arginine vasopressin V1 and V2 receptors for brain damage after transient focal cerebral ischemia. J Cereb Blood Flow Metab 25:1012–1019
Wang XQ, Xiao AY, Sheline C, Hyrc K, Yang A, Goldberg MP, Choi DW, Yu SP (2003) Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J Cell Sci 116:2099–2110
Wang HB, Li YX, Hao YJ, Wang TF, Lei Z, Wu Y, Zhao QP, Ang H, Ma L, Liu J, Zhao CJ, Jiang YX, Wang YR, Dai XY, Zhang WN, Sun T, Yu JQ (2013) Neuroprotective effects of LBP on brain ischemic reperfusion neurodegeneration. Eur Rev Med Pharmacol Sci 17:2760–2765
White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 179:1–33
Wilms H, Rosenstiel P, Unger T, Deuschl G, Lucius R (2005) Neuroprotection with angiotensin receptor antagonists: a review of the evidence and potential mechanisms. Am J Cardiovasc Drugs 5:245–253
Yao RQ, Qi DS, Yu HL, Liu J, Yang LH, Wu XX (2012) Quercetin attenuates cell apoptosis in focal cerebral ischemia rat brain via activation of BDNFTrkB-PI3K/Akt signaling pathway. Neurochem Res 37:2777–2786
Yao L, Kan EM, Lu J, Hao A, Dheen ST, Kaur C, Ling EA (2013) Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J Neuroinflammation 10:23
Yun J, Han M, Song C, Cheon SH, Choi K, Hahn HG (2014) Synthesis and biological evaluation of 3-phenethylazetidine derivatives as triple reuptake inhibitors. Bioorg Med Chem Lett 24:3234–3237
Zea Longa E, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Zhang Y, Park TS, Gidday JM (2007) Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Aktdependent survivin activation. Am J Physiol Heart Circ Physiol 292:H2573–H2581
Zhang L, Bukulin M, Kojro E, Roth A, Metz VV, Fahrenholz F, Nawroth PP, Bierhaus A, Postina R (2008) Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem 283:35507–35516
Zhang Q, Yuana L, Zhang Q, Gao Y, Liu G, Xiu M, Wei X, Wang Z, Liu D (2015) Resveratrol attenuates hypoxia-induced neurotoxicity through inhibiting microglial activation. Int Immunopharmacol 28:578–587
Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK (2005) Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci 25:9794–9806
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A09056947).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Electronic supplementary material
Supplementary Table 1
(DOCX 24 kb)
Rights and permissions
About this article

Cite this article
Kim, EA., Na, JM., Kim, J. et al. Neuroprotective Effect of 3-(Naphthalen-2-Yl(Propoxy)Methyl)Azetidine Hydrochloride on Brain Ischaemia/Reperfusion Injury. J Neuroimmune Pharmacol 12, 447–461 (2017). https://doi.org/10.1007/s11481-017-9733-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-017-9733-x