
Abstract
Purpose
When studying redox conditions in soils with manganese (Mn) and iron (Fe) oxide-coated redox bars, we observed the formation of Fe oxides along the Mn oxide coating and assumed sorption of other elements from soil solution to oxide surface. The objective of this study was to investigate the formation of Fe oxides along Mn redox bars and to analyze element sorption from soil solution to either Mn or Fe oxide along redox bar coatings.
Materials and methods
We protruded Mn redox bars into solutions with defined Fe2+ concentrations and removed the bars at distinct time intervals. The Mn oxide coating and potential Fe oxides were extracted using dithionite-citrate-bicarbonate (DCB). To investigate in situ element sorption behavior, we used previously field-installed redox bars, protruding these Mn redox bars into acidified hydroxylamine hydrochloride (AAH) to selectively extract Mn oxide and afterwards into DCB to dissolve the remaining Fe oxide coating. This two-step extraction procedure enabled the differentiation of elements bonded to either Mn or Fe oxide. Additionally, we analyzed the redox bar coatings at a very small scale (<1 mm2) via energy-dispersive x-ray spectroscopy (EDX).
Results and discussion
Iron oxides precipitated along the Mn oxide coating at low concentrations of 0.05 mg Fe2+ L−1, but did not trigger a color change. Although a change in color did occur instantaneously at 500 mg Fe2+ L−1, it is expected that Fe2+ concentrations are significantly lower under field conditions because ferrous Fe auto-oxidized within the artificial setup. Whereas Mn oxide sorbed cationic elements from the soil solution in the order Cu > Pb > Zn, Fe oxide preferentially sorbed oxyanions with As > P > Mo > V, respectively. “Field”-Fe oxides precipitating along the Mn redox bars sorbed elevated levels of As and P compared with the action of synthesized “lab”-Fe oxides along Fe redox bars, a finding which we attribute to short-range-ordered Fe phases with elevated sorption capacity.
Conclusions
Besides providing information regarding the monitoring of soil redox status, the developed sequential two-step extraction procedure enables the differentiation of the selective sorption of elements in the soil solution to the coating of Mn and Fe redox bars. The collection of Fe oxides formed naturally along the Mn redox bar coatings further enables the investigation of temporally and spatially diverse Fe oxide-forming processes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The characterization of soil redox status is of great importance because different pedogenetic responses are triggered under oxidizing or reducing soil conditions. In soil environments in which oxygen (O2) is scarce (e.g., due to groundwater or perched water tables), manganese (MnIII,IV) and iron (FeIII) oxides are important soil constituents because both minerals participate in reversible electron transfer reactions and are able to take up electrons released by the microbially mediated oxidation of reduced carbon (C) occurring in the soil organic matter pool (Ottow 2011). Whereas the transformation of MnIII,IV oxide to Mn2+ takes place at redox potentials (EH) as high as 500 mV at pH 5 (Gotoh and Patrick 1972), critical EH values for FeIII reduction have been recorded at 300 mV (Gotoh and Patrick 1974). Overall, a lowering of EH will result in enhanced Mn and Fe reduction rates (Atta et al. 1996) and furthermore, to the potential mobilization of trace elements bonded to the oxide surface.
One tool developed to test whether a soil is in a reduced state is the Indicator of Reduction in Soils (IRIS) (Jenkinson and Franzmeier 2006), in which synthesized Fe oxides consisting of ferrihydrite and goethite are coated on polyvinyl chloride (PVC) bars and installed in the soil for a defined period. In the absence of O2, the coating reductively dissolves, with the depletion patterns visually assessed to delineate soil-reducing conditions. Dorau and Mansfeldt (2015) adapted this method, coating PVC bars with synthesized Mn oxide consisting of birnessite (hereafter referred to as Mn and Fe redox bars or redox bars in general). Field monitoring of soil redox status was performed at a study site characterized by elevated concentrations of Fe and arsenic (As) in the groundwater, originating from the weathering of fossil bog Fe (Mansfeldt and Overesch 2013). The manganese redox bars proved their better suitability for the identification of reducing soil conditions, with oxide removal two to five times greater than that achieved using Fe redox bars (Dorau et al. 2015). Furthermore, sections of the Mn redox bar surfaces differed remarkably in color from the original dark brown Mn oxide coating. Chemical extraction using dithionite-citrate-bicarbonate (DCB) (Mehra and Jackson 1960) combined with elemental analysis of a previously installed bar revealed precipitated Fe to be the cause for this change in color. The presence of Fe2+ in soil solution mediates a non-enzymatic redox reaction that favors the dissolution of the Mn oxide coating. According to the reaction MnO2 + 2Fe2+ + 4H2O → Mn2+ + 2Fe(OH)3 + 2H+ (1) Fe2+ acts as a reductant that is oxidized to Fe3+ and hydrolyzes along the Mn oxide coating. In turn, Mn2+ is released from the surface, with in situ formed Fe oxides remaining as a durable coating (Dorau and Mansfeldt 2015). Along with the precipitated Fe oxides, we also hypothesized that As and other elements would be detectable in the extracts. Thus far, no study has investigated the sorption behavior of distinct elements to either the Mn or Fe oxide coating on redox bars.
In aerobic soil environments, MnIII,IV and FeIII (hydr)oxides are known to sorb and minimize the solubility of a variety of elements, for example, As (Manning and Goldberg 1997; Nesbitt et al. 1998), chromium (Cr) (Ajouyed et al. 2010; Bradl 2004; Rai et al. 1989; Zachara et al. 1987), copper (Cu) (Abd-Elfattah and Wada 1981), molybdenum (Mo) (Hooda 2010), nickel (Ni) (Arai 2008), phosphorus (P) (Peretyazhko and Sposito 2005), lead (Pb) (Abd-Elfattah and Wada 1981; Hooda 2010), vanadium (V) (Hooda 2010), and zinc (Zn) (Roberts et al. 2002; Scheinost et al. 2002). Variations in the mineral structure of birnessite (a common Mn oxide in soils), as well as ferrihydrite and goethite (prominent Fe oxides in soils), cause bonding mechanisms to differ between less reversible inner-sphere surface complexes and rather weakly bonded outer sphere surface complexes, with soil pH altering the element-specific pH adsorption edge (Bradl 2004). The linkage between the liberation of distinct trace metals into the soil solution under weakly reducing (MnIII,IV reduction; EH 400 to 200 mV at pH 7) and moderately reducing soil conditions (FeIII reduction; EH 200 to −100 mV at pH 7) was recently shown in microcosm experiments (Hindersmann and Mansfeldt 2014). These findings also highlighted that Mn and Fe oxides selectively sorb distinct trace metals from soil solution. However, it should be noted that (i) the EH controls the speciation of some trace elements via electron transfer reactions (e.g., As) (Smedley and Kinniburgh 2002; Mansfeldt and Overesch 2013), (ii) dissolved organic matter (DOM) is able to sequester certain trace metals (e.g., Pb) (Adriano 2001), and (iii) sulfide (S2−) in soil solution forms sparingly soluble trace metal-sulfide phases (e.g., CdS, PbS, and ZnS) (Morse and Luther 1999). Hence, various soil processes and properties alter the mobilization of trace metals and a universal concept is impossible to apply. Moreover, information regarding the in situ soil-surface chemistry of Mn and Fe oxides is difficult to obtain because major restrictions prevail when separating pure oxides from bulk soil. Rennert et al. (2013) developed a device with which to collect freshly precipitated Fe oxides without producing physical or chemical artifacts during sampling. Issues related to soil-mineral formation, e.g., redox-induced Fe oxide aging (Thompson et al. 2006) or interactions between Fe oxides and DOM, can be studied by applying microscopic and spectroscopic techniques (Rennert et al. 2013).
The objectives of this article were (i) to improve the understanding of Fe precipitation and the formation of Fe oxide along Mn redox bars, (ii) to investigate the sorption of various elements from the soil solution to either Mn or Fe oxide along the redox bar coating, using a selective chemical extraction procedure, and (iii) to analyze the Mn and Fe oxide coatings at a very small scale via energy-dispersive x-ray spectroscopy (EDX).
2 Materials and methods
2.1 Study site
The monitoring site is in the district of Recklinghausen, North Rhine-Westphalia, Germany (51° 48′ 59″ N, 7° 12′ 59″ E). Water table (WT) depths across the study site range from ponding surface water to 1 m below ground at small scale, reflecting the action of glaciofluvial processes that produced a micro-relief (47.6 to 49.0 m asl) segregating elevated dry sites from flooded sites in lower terrain.
2.2 Field monitoring
The manufacturing process for the Mn redox bars is presented in detail in Dorau and Mansfeldt (2015) and that for the Fe redox bars in Jenkinson and Franzmeier (2006) and Castenson and Rabenhorst (2006). Three Mn and three Fe redox bars 60 cm in length and with the lower 50 cm coated, were installed in a flooded plot in lower terrain where water was periodically ponding. At this site, Fe oxide formation was favorable because Fe2+ was abundant in the soil solution (plot C in Dorau and Mansfeldt 2015); we also assumed that the potential sorption of distinct elements to the oxide coating would be detectable by the selected analytical methods. We removed the redox bars on a monthly basis from March to July 2013. Environmental monitoring comprised measurement of WT depths (PDLR 70, EcoTech, Bonn, Germany); soil temperature (temperature lance with RS 485 interface, UIT, Dresden, Germany); and oxide removal along redox bars. A detailed description of soil properties and site-specific characteristics is presented in Mansfeldt et al. (2012).
2.3 Elemental analysis
Disturbed soil samples were taken from an excavation pit at the designated plot from 0 to 25 cm and from 25 to 50 cm depth. Soil sampling was not carried out genetically based on soil horizons but followed the analytical procedure of elements bonded to the upper (0 to 25 cm) and lower (25 to 50 cm) sections of redox bar coatings. The obtained soil samples were oven dried at 40 °C, sieved <2 mm, and homogenized, with subsamples then pulverized in a mixer mill (MM400, Retsch, Haan, Germany) to determine total element concentrations. Pseudo-total concentrations of As, Cr, Cu, Mo, Ni, Pb, V, and Zn were measured via inductively coupled plasma mass spectrometry (ICP-MS) (XSeries 2, Thermo Scientific, Waltham, USA), and those of Fe, Mn, and P via optical emission spectroscopy (ICP-OES) (ULTIMA 2, Horiba Scientific, Unterhaching, Germany) in an inverse aqua regia digestion solution (2 mL concentrated HCl and 6 mL concentrated HNO3, 200 mg sample weight).
2.4 Fe precipitation along Mn redox bars
As outlined in Dorau and Mansfeldt (2015), the presence of Fe2+ in the soil solution is responsible for the non-enzymatic reductive dissolution of the Mn oxide coating. To prove the reaction kinetics of this abiotic process and to investigate possible color changes, we protruded three Mn redox bars of 8 cm length into 20-mL solutions containing 0.05, 5, and 500 mg Fe2+ L−1, removing one bar from each solution after 1, 15, and 30 days. The concentration of 5 mg Fe2+ L−1 approximates the upper limit of divalent Fe found in the soil solution at the study site (Mansfeldt and Overesch 2013), while the 30-day limit is equal to the installation time of redox bars during monitoring. Additionally, we performed a run with 100 mg Fe2+ L−1 at shorter intervals of 1, 2, 4, 8, 16, and 24 h. After removing the redox bars from the solutions and prior to their dry storage, we rinsed the bars with deionized water to remove any adhering solution containing Fe2+. As the pH of the FeCl2 solution varied depending on the Fe2+ concentration, all solutions were standardized to the pH conditions found at the study site (pH 4.5), with sodium hydroxide (0.1 M NaOH) and nitric acid (0.1 M HNO3) used to increase and decrease solutions to the desired pH, respectively. After removing the redox bars, the pH of the solution was measured again. To establish whether Fe precipitated along the Mn oxide coating, we protruded the bars into 50 mL vials and performed a DCB extraction (Mehra and Jackson 1960). This extracting agent simultaneously dissolves both Mn and Fe oxides in soils. Afterwards the solution was analyzed for total Mn and Fe concentrations via flame atomic absorption spectroscopy (F-AAS) (iCE 3000 series, Thermo Scientific, Waltham, USA). The experiment was performed in duplicate repetition.
2.5 Sorption characteristics of redox bars for various elements
We performed a two-step extraction procedure to differentiate elements sorbed to either Mn or Fe oxide along the redox bar surface. In this analysis, we first cut two Mn and two Fe redox bars (the 50-cm coated surface) per month into 8.33-cm PVC sections. We investigated the elemental composition of the redox bar coating for both the topsoil (0 to 25 cm; upper three sections) and subsoil (25 to 50 cm; lower three sections), along with soil characterization. For this purpose, redox bar sections were protruded for 10 min into 50-mL vials 10 cm in height (2.5 cm Ø) and containing 25 mL of extracting agent; the complete surface of each section was in contact with the solution. To investigate elements bonded to Mn oxide, we used acidified hydroxylamine hydrochloride solution (AAH; 0.1 M HONH2·HCl in 0.01 M HNO3, pH 2), which selectively extracts Mn oxides in soils (Chao 1972) and dissolves the Mn oxide coating within minutes (Dorau and Mansfeldt 2015). The residual Fe oxide coating and elements bonded to it were extracted using DCB (Mehra and Jackson 1960). Manganese redox bars were thus sequentially treated with AAH and DCB, and Fe redox bars only with DCB. Total concentrations of As, Cr, Cu, Mo, Ni, Pb, V, and Zn were determined via ICP-MS, and those of Fe, Mn, and P via ICP-OES; these values were then converted to extractable element contents. To characterize the sorption behavior of elements bonded to either Mn or Fe oxide, we calculated the quotient between the mean AAH-extractable (assumed to bond to Mn oxide) and DCB-extractable element contents (assumed to bond to Fe oxide) exclusively for Mn redox bars. The final dataset also included two replicates each for the top and bottom of the Mn redox bars for the 5-month period (n = 20).
2.6 EDX analysis of redox bars
We used a scanning electron microscope (SEM) (NEON 40, Zeiss, Jena, Germany) at 15 keV accelerating voltage equipped with an EDX detector (Dry Cool, Oxford instruments, Abingdon, UK) to analyze the element contents of redox bar coatings at a very small scale (∼1 mm2). This approach was employed in order to address a scaling problem, because chemical extraction integrated the elemental content of 16,493 mm2, equal to 25 cm of coated PVC surface, according to the employed methodological setup. In contrast, for SEM investigations, smaller sample dimensions are required and thus the bars were prepared in the following way: Each previously field-installed Mn and Fe redox bar served as an example, and was first cut lengthwise into 1-cm sections and then crosswise to obtain plastic chips ∼4 mm in height. We also analyzed an uncoated PVC chip as a reference in order to exclude any elements incorporated into the plastic material. For SEM investigations, sample surfaces must be electrically conductive and hence the plastic chips were coated with a 15-nm-thick layer of C prior to analysis (K950X, EMITECH, Ashford, UK).
3 Results and discussion
3.1 Fe precipitation along Mn redox bars
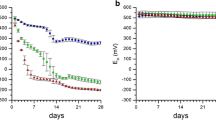
Figure 1 shows the DCB-extractable Fe contents recorded along Mn redox bars protruded into solutions of different Fe2+ concentration (a) and the corresponding pH values of these solutions (b) at distinct time intervals, and Fig. 2 illustrates the corresponding color of the Mn oxide coatings after removing the Mn redox bars from the Fe2+-containing solutions. A steady increase in DCB-extractable Fe content was observed in the experiment with the 100 mg Fe2+ L−1 solution, which reached a steady state and stagnated after 24 h of reaction. These findings indicate the fast reaction kinetics of Fe2+ (reductant) with the Mn oxide coating (oxidant), and are coherent with data published by Golden et al. (1986). The latter authors reported increased Mn2+ concentrations in the solution phase resulting from the reduction of MnIII,IV oxides in the presence of Fe2+, also proposing that Fe2+ oxidized to Fe3+ in turn hydrolyzes in a further step to form FeIII oxides according to Eq. (1). Presumably, the pseudomorphic Fe precipitates are composed of ferrihydrite or feroxyhite, which progressively transform into goethite (Golden et al. 1986). As the oxidation of ferrous Fe is a proton-producing process, pH values at 100 and 500 mg Fe2+ L−1 fell rapidly from pH 4.5 at the beginning of the experiment to pH 3.1 and 2.9, respectively, at the end of the experiment (Fig. 1b). The release of H+ due to the adsorption of Fe2+ and Fe3+ is also partly responsible for the pH drop. No decrease and only an insignificant pH decline occurred at 0.05 and 5 mg Fe2+ L−1, respectively. Although according to Le Chatelier’s principle, low pH values in a system tend to push the reaction to the left side of Eq. (1), with the formation of Fe oxides along Mn redox bars thus more favorable at sites with neutral soil, this was not the case in the present study. Elevated concentrations of Fe2+ in a solution resulted in enhanced Fe oxide formation along the Mn oxide coating, with 23, 63, 2000, and 3100 μg DCB-extractable Fe recorded at 0.05, 5, 100, and 500 mg Fe2+ L−1, respectively (Fig. 1a). At 5 mg Fe2+ L−1, which approximates concentrations found in the soil solution at the study site (Mansfeldt and Overesch 2013), oxidation and hydrolysis of Fe along the Mn oxide coating obviously occurred (63 μg Fe could be extracted by DCB) but without favoring a change in coating color (Fig. 2). Indeed, concentrations of at least 500 mg Fe2+ L−1 were necessary to obtain a significant change in the color of the original dark brown Mn oxide coating to a light orange Fe oxide coating (Fig. 2). After a protruding time of 30 days in the 500 mg Fe2+ L−1 solution, only a minor Mn content of 73 μg remained along the 8-cm-long redox bar, a figure which is significantly lower than the average of 740 μg Mn present directly after bar manufacture. Due to the non-uniform application procedure, Mn contents ranged from 500 to 2200 μg Mn. Assuming 500 μg Mn to be the upper and 2200 μg Mn to be the lower limit, Fe2+ contents of 1020 to 4470 μg Fe2+ (according to Eq. (1)) would have been required to completely dissolve the Mn oxide coating, which corresponds to 51 to 224 mg Fe2+ L−1. According to the stoichiometric calculation, a change of color should have occurred below 500 mg Fe2+ L−1; indeed, field observations highlighted that Fe oxides formed at significantly lower concentrations (see Fig. 3). What might be the reason for this discrepancy? Under natural conditions, the capillary rise of Fe2+-rich shallow groundwater ensured a constant supply of the reductant into the topsoil for a period of 30 days, considerably different from the artificial setup in the laboratory, in which Fe2+ concentrations in the solution were presumably auto-oxidized (by O2) within 24 h. This pH-dependent step has been reported to take place from <1 min to hours (Kosman 2013) and provides the most suitable explanation of the observed incongruity.

Dithionite-citrate-bicarbonate (DCB) extractable Fe content of Mn redox bars in duplicate protruded into FeCl2-solution containing 0.05 (black squares), 5 (black circles), 100 (white squares), and 500 (white circles) mg Fe2+ L−1 at defined time intervals (a), and pH of the corresponding solutions after bar removal (b). Error bars represent the standard deviation

Images of Mn redox bars protruded into FeCl2-solutions of varying Fe2+ concentration after defined time intervals

Image of a Mn redox bar before (a) and after (b) protrusion into acidified hydroxylamine hydrochloride (AAH) solution for 10 min
Another important observation made during the experiment was that tri- and tetravalent manganese as an oxidant for Fe2+ is an essential precursor to the formation of a durable Fe oxide coating along the PVC surface. This assumption is interesting, because we neither observed Fe precipitates along uncoated PVC bars protruded into solutions of various Fe2+ concentrations, nor along uncoated PVC bars installed during the monitoring campaign.
3.2 Extractable element contents along redox bars
Figure 3 shows an Mn redox bar before and after protrusion into AAH solution for 10 min. The original dark brown Mn oxide coating (Fig. 3a) dissolved instantaneously, showing the white PVC surface underneath (Fig. 3b). Boundaries of the remaining Fe oxides were easy to identify. Hence, it is possible to differentiate and analyze elements potentially bonded to either Mn or Fe oxide along the Mn redox bar surface using a selective chemical extraction procedure.
Figure 4 illustrates the mean contents of various elements bonded to synthesized Mn oxides (AAH-extractable; Fig. 4a), to Fe oxides naturally precipitated along Mn redox bars (DCB-extractable; Fig. 4b), and elements bonded to synthesized Fe oxides along Fe redox bars (DCB-extractable; Fig. 4c). For reasons of simplicity and further discussion, in situ formed Fe oxides are termed “field”-Fe oxides, and those used for the manufacture of Fe redox bars as “lab”-Fe oxides. Along Mn redox bars, Mn was the major AAH-extractable element, at 1.84 μM. In accordance with the percentage oxide removal determined via digital analysis (Dorau et al. 2015), the highest levels of Mn were found in July (10.38 μM) when the WT was low, favoring aerobic conditions and no reductive dissolution of the coating, and lowest in June (0.02 μM) when reducing conditions were intensified. Iron was the main adsorbate for Mn oxide, followed by elements in the following order: Zn > P > Cr > Cu > As > V > Pb > Ni > Mo (Fig. 4a). A positive and highly significant relationship between Mn and element content was found for Pb (r = 0.751***), V (r = 0.730***), and Mo (r = 0.682***), while a weaker relationship was found for Zn (r = 0.401*); no statistically significant relationship was present for the other elements (Table 1). The sink function of Mn oxides has been reported for Pb (McKenzie 1980), Mo (Barling and Anbar 2004), Zn (Komárek et al. 2013), and V (Takematsu et al. 1985; Yin et al. 2015). Matern and Mansfeldt (2015) demonstrated the strong effect of molybdate (MoO4 2−) adsorption onto the surface of a synthetic birnessite. The synthesis of birnessite in this latter study is equivalent to the procedure employed here for the manufacture of the Mn redox bars, with the correlation indicating the coherence between laboratory batch experiments (Matern and Mansfeldt 2015) and the present field experiments. Manganese oxides have been also identified as the most efficient stabilizing agent for Cu, Co, Cd, and Zn (Komárek et al. 2013). However, concurrency of sorption sites along the Mn oxide coating and the presence of DOM are only two possible factors that could result in weak correlation with Cu and Zn (Table 1). Han et al. (2006) confirmed the presence of competitive adsorption and found a decrease in Cu adsorption in the presence of Pb ions. When retrieving the redox bars from the soil, organoleptic assessment revealed the unique smell of H2S; metal precipitation with S2− is therefore an additional process that may cause inconsistent sorption of elements from the soil solution to the redox bar coatings. However, the stronger correlation between Mn oxide and Pb compared to that between Mn oxide and Cu (Table 1) indicates a stronger affinity for Pb sorption onto Mn oxide surfaces. These results are in agreement with data by Feng et al. (2007).

Acidified hydroxylamine hydrochloride (AAH) and dithionite-citrate-bicarbonate (DCB) extractable mean content of elements bonded to Mn oxide along Mn redox bars (a), to Fe oxides along Mn redox bars (b), and to Fe oxides along Fe redox bars (c). The averaged element contents include data covering the period from March to July for the top and bottom of Mn and Fe redox bars in duplicate (n = 20). Error bars represent the standard deviation
Visual assessment of Mn redox bars revealed zones of field-Fe oxides along the PVC surface, an observation in agreement with the content of the DCB extracts. Indeed, Fe was the main DCB-extractable element, at 4.97 μM (Fig. 4b). The content of adsorbate bonded to Fe oxide followed the order P > As > Mn > Zn > V > Cr > Ni > Pb > Mo > Cu (Fig. 4b). Among these elements, As (r = 0.941***) and P (r = 0.789***) exhibited a strong relationship with extractable Fe (Table 1), with both elements showing very strong sorption characteristics probably forming an inner-sphere bidentate binuclear complex (Manning and Goldberg 1997) with the FeIII oxide surface. The DCB-extractable element composition of lab-Fe oxides (Fig. 4c) revealed similar sorption behavior to that of field-Fe oxides (Fig. 4b). Nevertheless, the iron content of 5.90 μM was slightly higher than the 4.97 μM Fe found along Mn redox bars, with minor differences also occurring in the order of adsorbate content: P > As > Zn > V > Mn > Cr > Ni > Mo > Pb > Cu (Fig. 4c). Nickel (r = 0.920***), P (r = 0.819***), V (r = 0.749***), As (r = 0.678***), and Zn (r = 0.663***) were strongly correlated, Mo (r = 0.523*) was slightly less correlated, and no statistically significant correlation was recorded for Cr, Cu, and Pb with Fe content along Fe redox bars (Table 1). The adsorption of Ni, Zn (Brümmer et al. 1988), V (Peacock and Sherman 2004), P (Strauss et al. 1997), As (Giménez et al. 2007), and Mo (Goldberg et al. 2009) to the surface of goethite minerals has been reported previously. The consistency of the applied Fe oxide minerals in the suspension used for Fe redox bar manufacture might explain the stronger correlation between many elements and lab-Fe oxides compared to field-Fe oxides. We characterized the initial Fe oxide suspension using both an oxalate (Feo) and a DCB (Fed) extraction. Whereas the former selectively extracts short-range-ordered and nano-crystalline Fe phases (e.g., ferrihydrite, but also nano-goethite), the latter extracts all Fe oxides from soils. The Fe oxide suspension possessed a Feo/Fed ratio of 0.76, indicating that 76 % was ferrihydrite and 24 % goethite that remained stable when applied to the PVC bars (Rabenhorst et al. 2008). The defined goethite and ferrihydrite content of lab-Fe oxides is therefore a unique characteristic. Whereas the synthesis of Fe oxides is widespread and commonly adapted in the laboratory, the mechanisms involved in their formation under natural conditions are complex and diverse, leading to a broad range of in situ formed Fe oxides (Jolivet et al. 2004). At the study site, most of the in situ formed FeIII oxides were composed of ferrihydrite (Mansfeldt et al. 2012). The key factor altering oxide form and crystallinity is the rate of Fe2+ contribution to the initial nuclei and the rate of Fe2+ oxidation (Cornell and Schwertmann 2003; Wang et al. 2013a). Boundary conditions leading to characteristic pathways of Fe oxide formation include pH, concentration of Fe3+, and temperature. Due to the presence of variable WT depths and water content changes in the capillary fringe, the hydraulic gradient varies and the supply of Fe2+ into the topsoil is not constant. Hence, the formation of FeIII via redox reaction with the Mn oxide coating will also vary. Furthermore, soil temperature at the study site increased from 3.8 °C in March to 16.8 °C in July at 25-cm depth (Dorau and Mansfeldt 2015). It thus seems reasonable that variations in the mineral structure and further in the sorption capacity of field-Fe oxides resulted in the observed reduction in statistical correlation compared with the constant conditions experienced by lab-Fe oxides along Fe redox bars (Table 1).
3.3 Preference for element sorption to Mn or Fe oxides
We categorized quotients of mean AAH- to DCB-extractable element contents above 1.5 as indicating element preference for bonding to Mn oxide, values between 1.5 and 0.5 as indicating similar sorption behavior toward Mn and Fe oxides, and values below 0.5 as indicating a preference for bonding to Fe oxides (Table 2). Manganese oxide was found to sorb 19.7 times more Cu and was associated with slightly elevated Pb (3.6) and Zn (2.9) content compared to field-Fe oxides, a result in agreement with numerous studies in the literature (reviewed by Komárek et al. 2013). Whereas chromium (0.94) and Ni (0.72) sorbed equally to Mn and Fe oxide surfaces, preferential sorption to Fe oxides was evident in the following order: As (0.02), P (0.04), Mo (0.16), and V (0.23). The latter elements occur as oxyanions in the soil solution under the assumed EH-pH conditions of 400 mV (Mn and Fe oxides remain stable and play the role of adsorbents) and pH 4.5 (the soil pH at the study site). The high point of zero charge (PZC) of ferrihydrite (pH 7.9) and goethite (pH 9.7) favors functional group protonation under the acidic soil conditions present at the study site, in contrast to birnessite (pH 2.6). This enables oxyanions in the soil solution to be electrostatically bonded, e.g., to positively charged OH groups (Cornell and Schwertmann 2003; Bradl 2004). Although only a limited number of studies have dealt with the role of Mn oxides as stabilizing amendments, successful element immobilization has been reported for Cu, Pb, and Zn (Della Puppa et al. 2013; Komárek et al. 2013), a finding that we can verify here in the sorption behavior of these predominantly cationic elements. Our results suggest that differences in the characteristics of the Mn and Fe oxide coatings (e.g., PZC, cation exchange capacity, and specific surface area) lead to selective sorption behavior for dissolved elements present in the soil solution to the redox bar coatings under field conditions.
3.4 Sorption capacities: field-Fe oxides versus lab-Fe oxides
The crystallinity of Fe oxides in soil varies widely from short-range-ordered to long-range-ordered phases due to changes in geochemical conditions in an open system, a process that is especially relevant for soils with changing redox conditions. This phenomenon was previously shown via an artificial setup in which biweekly soil redox oscillations from 200 to 700 mV led to an increase in Fe oxide crystallinity (Thompson et al. 2006). As these oxides are important interfaces for the sorption of dissolved elements in the soil solution, sorption capacity is also a function of crystallinity (Wang et al. 2013b).
Assuming ferrihydrite (Fe5HO8·4H2O) with a corresponding specific surface area of 200 m2 g−1 (Cornell and Schwertmann 2003) to be the major Fe oxide along the Mn redox bar coating, the recorded DCB-extractable Fe contents of 1.97 to 17.05 μM Fe are equal to a total surface area of 0.19 to 1.64 m2. Exclusively for Fe redox bars, the goethite (FeOOH) content of 24 % in the initial Fe oxide suspension is considered to have a total surface area of 50 m2 g−1 (Cornell and Schwertmann 2003), with the remaining 74 % being ferrihydrite. This assumption yields total surface areas for lab-Fe oxides along Fe redox bars in the range of 0.12 to 0.90 m2. Figure 5 shows the development of element loading for As (Fig. 5a) and P (Fig. 5b) on a hypothetical Fe oxide surface from March to July along Mn and Fe redox bars. These two elements are known to strongly sorb onto Fe oxides, and here were associated with the strongest correlation between DCB-extractable field- and lab-Fe oxides (Table 1); hence, the two are suitable for the comparison of element loading between naturally formed and synthesized Fe oxides. Element loading under natural conditions can be considered “actual” sorption (AS). In contrast to this, potential sorption (PS) can be derived from laboratory batch experiments and is equivalent to the sorption capacity of a distinct mineral for an adsorbent. The AS values of As and P with respect to the Fe oxide surface varied throughout the study period, but were, in general, lower at the top than at the bottom of redox bars (Fig. 5a, b), a finding which we attribute to elevated As and P concentrations in the capillary fringe (Mansfeldt and Overesch 2013). Increased levels of As and P in the topsoil (Table 3) seem to play a minor role regarding sorption to the redox bar coating. Besides this, the top and bottom of field-Fe oxides sorbed on average 0.69 μM As m−2 and 2.29 μM P m−2, levels significantly higher than the 0.38 and 1.04 μM As and P m−2, respectively, sorbed by lab-Fe oxides (Fig. 5a, b). Since goethite has lower sorption capacities for H2AsO4 − and H2PO4 − m−2 oxide surface (1.33 and 2.5 μM) compared to ferrihydrite (15.5 and 3.1 μM) (Borggaard 1983; Strauss et al. 1997; Raven et al. 1998; Giménez et al. 2007), we assume that in situ-formed Fe oxide phases are solely short-range-ordered. Considering the AS values recorded at the top and bottom of redox bars, field-Fe oxides sorbed on average 1.8 times more As and 2.2 times more P compared to lab-Fe oxides. It should be noted that the development of monthly element loading onto field- and lab-Fe oxides varied in magnitude, likely due to variations in As and P concentrations in the soil solution (Fig. 5). Mansfeldt et al. (2012) previously found high δ57Fe values in the topsoil at the study site, caused by the rapid precipitation of ferrihydrite when Fe2+ was exposed to O2. These findings underline the previous assumption and also that the goethite content of lab-Fe oxides seems to be responsible for the lower AS. Finally, a number of discrepancies were observed because the AS of field-Fe oxides with P (Fig. 5b) generally exceeded that with As (Fig. 5a). This contradicts the PS data reported in the literature, in which the adsorption capacity of ferrihydrite for As (Raven et al. 1998) is reported to exceed that for P (Borggaard 1983). Hence, differences are apparent between the PS results derived in laboratory batch experiments using synthesized minerals and those obtained in the present study via the use of naturally formed minerals. Nevertheless, the AS values of field- and lab-Fe oxides are below the PS values reported in the literature, which supports the validity of studying the in situ sorption behavior of elements in wet soils using redox bars.

Element loading of field-Fe oxides along Mn redox bars (open symbols) and of lab-Fe oxides along Fe redox bars (solid symbols) with As (a) and P (b) for the top (white squares, black squares) and bottom (white circles, black circles) sections of previously installed redox bars, for the period from March to July. Dithionite-citrate-bicarbonate (DCB) extractable Fe contents were transformed to Fe oxide surfaces by assuming a ferrihydrite-(FH) formula of Fe5HO8·4H2O and a goethite-(GT) formula of FeOOH with a corresponding surface area of 200 (50) m2 g−1 FH (GT). We assume that field-Fe oxides are solely composed of FH while lab-Fe oxides are composed of 76 % FH and 24 % GT. The corresponding As and P contents are transformed to molarities
3.5 Energy-dispersive x-ray spectroscopy of redox bars
Figure 6 shows typical color patterns observed on redox bars of the original Mn oxide coating (A), Fe precipitates (B), and complete oxide removal (C) along Mn redox bars (a), as well as of the partial depletion of Fe oxides (D), and the original Fe oxide coating (E) on Fe redox bars (b). The former color pattern displays an EH gradient ranging from oxidizing soil conditions (MnIII,IV oxides remain stable, A) at the edge to strongly reducing soil conditions (MnIII,IV and FeIII oxides are reduced, C) in the center. The partial depletion of Fe oxides (D) was often observed in combination with pale yellow zones reflecting the preferential dissolution of ferrihydrite over goethite (Rabenhorst et al. 2008). Energy-dispersive x-ray spectroscopy of the uncoated PVC chip revealed 11 % O, 81 % Cl, and no incorporation of Mn and Fe within the plastic material (Table 4). As Cl is the major component of the polymer material and the penetration depth of the emitted electrons is approximately 15 μm, we analyzed the Cl content of samples A to E, with background values ranging from 35 to 71 %. Hence, the oxide coating clearly attenuated the Cl signal. Chemical extraction already revealed that Fe oxides are precipitated along the Mn oxide coating without triggering a change in color, a finding further confirmed at spot A (Fig. 6a) with 3 % Fe and 10 % Mn. The higher Fe content at spot B (15 % Fe) favored a change in color accompanied by complete dissolution of remaining Mn oxides (Table 4). To the center of the depletion spot, Fe content further decreased to 2 %. Soil-reducing conditions intensified reductive dissolution of the Fe oxide coating at spot D with only 8 % Fe, compared to 26 % Fe at spot E, where oxidizing conditions prevailed. Hence, EDX enabled the determination of element distribution along the Mn and Fe redox bars at very small scale (<1 mm2).

Detailed image of a Mn (a) and Fe (b) redox bar previously installed in June. The rectangles (A to E) denote areas used for EDX point measurements
4 Conclusions
The presented selective chemical extraction procedure enables the study of the in situ sorption behavior of elements to either Mn or Fe oxide-coated redox bars. Both the laboratory experiments and field observations highlight the importance of Mn oxide as a precursor favoring the likelihood of oxidizing ferrous Fe from the soil solution. Whereas the Mn oxide coating showed preferential sorption behavior for cationic elements (Cu, Zn, and Pb), the Fe oxide coating was enriched in As, P, Mo, and V. The latter elements occur as oxyanions in the soil solution and show a bonding preference for positively charged Fe oxide surfaces as favored at the study site (pH 4.5). Arsenic as a pollutant and P as a nutrient exhibited strong correlation with DCB-extractable Fe oxides along redox bars. Field-Fe oxides precipitated along Mn redox bars sorbed 1.8 (2.2) times more As (P) than did synthesized lab-Fe oxides along Fe redox bars, indicating that naturally precipitated oxides have a higher sorption capacity due to their lower crystallinity. In general, various elements showed distinct affinities for bonding to either Mn or Fe oxides, with EDX spectroscopy enabling the analysis of the oxide coating at a very small scale (<1 mm2). Besides the characterization of soil redox status, Mn and Fe redox bars can be an additional tool with which to study and monitor the element sorption behavior of nutrients and pollutants in wet soils.
References
Abd-Elfattah ALY, Wada K (1981) Adsorption of lead, copper, zinc, cobalt, and cadmium by soils that differ in cation-exchange materials. J Soil Sci 32:271–283
Adriano DC (2001) Trace elements in terrestrial environments: biogeochemistry, bioavailability, and risks of metals, 2nd edn. Springer, Heidelberg
Ajouyed O, Hurel C, Ammari M, Allal LB, Marmier N (2010) Sorption of Cr(VI) onto natural iron and aluminum (oxy)hydroxides: effects of pH, ionic strength and initial concentration. J Hazard Mater 174:616–622
Arai Y (2008) Spectroscopic evidence for Ni(II) surface speciation at the iron oxyhydroxides−water interface. Environ Sci Technol 42:1151–1156
Atta SK, Mohammed SA, Van Cleemput O, Zayed A (1996) Transformations of iron and manganese under controlled EH, EH-pH conditions and addition of organic matter. Soil Technol 9:223–237
Barling J, Anbar AD (2004) Molybdenum isotope fractionation during adsorption by manganese oxides. Earth Planet Sci Lett 217:315–329
Borggaard OK (1983) Effects of surface area and mineralogy of iron oxides on their surface charge and anion-adsorption properties. Clay Clay Miner 31:230–232
Bradl HB (2004) Adsorption of heavy metal ions on soils and soils constituents. J Colloid Interface Sci 277:1–18
Brümmer GW, Gerth J, Tiller KG (1988) Reaction kinetics of the adsorption and desorption of nickel, zinc and cadmium by goethite. I. adsorption and diffusion of metals. J Soil Sci 39:37–52
Castenson KL, Rabenhorst MC (2006) Indicator of reduction in soil (IRIS): evaluation of a new approach for assessing reduced conditions in soil. Soil Sci Soc Am J 70:1222–1226
Chao TT (1972) Selective dissolution of manganese oxides from soils and sediments with acidified hydroxylamine hydrochloride. Soil Sci Soc Am J 36:764–768
Cornell RM, Schwertmann U (2003) The iron oxides: structure, properties, reactions, occurences, and uses, 2nd edn. Wiley-VCH, Weinheim
Della Puppa L, Komárek M, Bordas F, Bollinger JC, Joussein E (2013) Adsorption of copper, cadmium, lead and zinc onto a synthetic manganese oxide. J Colloid Interface Sci 399:99–106
Dorau K, Mansfeldt T (2015) Manganese-oxide-coated redox bars as an indicator of reducing conditions in soils. J Environ Qual 44:696–703
Dorau K, Eickmeier M, Mansfeldt T (2015) Comparison of manganese and iron oxide-coated redox bars for characterization of the redox status in wetland soils. WETLANDS revised version under review
Feng XH, Zhai LM, Tan WF, Liu F, He JZ (2007) Adsorption and redox reactions of heavy metals on synthesized Mn oxide minerals. Environ Pollut 147:366–373
Giménez J, Martínez M, de Pablo J, Rovira M, Duro L (2007) Arsenic sorption onto natural hematite, magnetite, and goethite. J Hazard Mater 141:575–580
Goldberg T, Archer C, Vance D, Poulton SW (2009) Mo isotope fractionation during adsorption to Fe (oxyhydr)oxides. Geochim Cosmochim Acta 73:6502–6516
Golden DC, Dixon JB, Chen CC (1986) Ion exchange, thermal transformations, and oxidizing properties of birnessite. Clay Clay Miner 34:511–520
Gotoh S, Patrick WH (1972) Transformation of manganese in a waterlogged soil as affected by redox potential and pH. Soil Sci Soc Am Proc 36:738–742
Gotoh S, Patrick WH (1974) Transformation of iron in a waterlogged soil as influenced by redox potential and pH. Soil Sci Soc Am Proc 38:66–71
Han R, Zou W, Li H, Li Y, Shi J (2006) Copper(II) and lead(II) removal from aqueous solution in fixed-bed columns by manganese oxide coated zeolite. J Hazard Mater 137:934–942
Hindersmann I, Mansfeldt T (2014) Trace element solubility in a multimetal-contaminated soil as affected by redox conditions. Water Air Soil Pollut 225:1–20
Hooda PS (2010) Trace elements in soils, 1st edn. Wiley, West Sussex
Jenkinson BJ, Franzmeier DP (2006) Development and evaluation of iron-coated tubes that indicate reduction in soils. Soil Sci Soc Am J 70:183–191
Jolivet JP, Chanéac C, Tronc E (2004) Iron oxide chemistry. From molecular clusters to extended solid networks. Chem Commun 5:481–483
Komárek M, Vaněk A, Ettler V (2013) Chemical stabilization of metals and arsenic in contaminated soils using oxides—a review. Environ Pollut 172:9–22
Kosman DJ (2013) Iron metabolism in aerobes: managing ferric iron hydrolysis and ferrous iron autoxidation. Coord Chem Rev 257:210–217
Manning BA, Goldberg S (1997) Arsenic(III) and arsenic(V) adsorption on three california soils. Soil Sci 162:886–895
Mansfeldt T, Overesch M (2013) Arsenic mobility and speciation in a Gleysol with petrogleyic properties: a field and laboratory approach. J Environ Qual 42:1130–1141
Mansfeldt T, Schuth S, Häusler W, Wagner F, Kaufhold S, Overesch M (2012) Iron oxide mineralogy and stable iron isotope composition in a Gleysol with petrogleyic properties. J Soils Sediments 12:97–114
Matern K, Mansfeldt T (2015) Molybdate adsorption by birnessite. Appl Clay Sci 108:78–83
McKenzie R (1980) The adsorption of lead and other heavy metals on oxides of manganese and iron. Aust J Soil Res 18:61–73
Mehra OP, Jackson ML (1960) Iron oxide removal from soils and clays by a dithionite-citrate system buffered with sodium bicarbonate. Clay Clay Miner 7:317–327
Morse JW, Luther GW (1999) Chemical influences on trace metal-sulfide interactions in anoxic sediments. Geochim Cosmochim Acta 63:3373–3378
Nesbitt HW, Canning GW, Bancroft GM (1998) XPS study of reductive dissolution of 7Å-birnessite by H3AsO3, with constraints on reaction mechanism. Geochim Cosmochim Acta 62:2097–2110
Ottow JCG (2011) Microbiology of soils (In German), 1st edn. Springer, Heidelberg
Peacock CL, Sherman DM (2004) Vanadium(V) adsorption onto goethite (α-FeOOH) at pH 1.5 to 12: a surface complexation model based on ab initio molecular geometries and EXAFS spectroscopy. Geochim Cosmochim Acta 68:1723–1733
Peretyazhko T, Sposito G (2005) Iron(III) reduction and phosphorous solubilization in humid tropical forest soils. Geochim Cosmochim Acta 69:3643–3652
Rabenhorst MC, Ming DW, Morris RV, Golden DC (2008) Synthesized iron oxides used as a tool for documenting reducing conditions in soils. Soil Sci 173:417–423
Rai D, Eary LE, Zachara JM (1989) Environmental chemistry of chromium. Sci Total Environ 86:15–23
Raven KP, Jain A, Loeppert RH (1998) Arsenite and arsenate adsorption on ferrihydrite: kinetics, equilibrium, and adsorption envelopes. Environ Sci Technol 32:344–349
Rennert T, Mueller CW, Mansfeldt T, Lugmeier J (2013) Collecting in situ precipitated iron oxides in their natural soil environment. J Plant Nutr Soil Sci 176:497–499
Roberts DR, Scheinost AC, Sparks DL (2002) Zinc speciation in a smelter-contaminated soil profile using bulk and microspectroscopic techniques. Environ Sci Technol 36:1742–1750
Scheinost AC, Kretzschmar R, Pfister S, Roberts DR (2002) Combining selective sequential extractions, x-ray absorption spectroscopy, and principal component analysis for quantitative zinc speciation in soil. Environ Sci Technol 36:5021–5028
Smedley PL, Kinniburgh DG (2002) A review of the source, behaviour and distribution of arsenic in natural waters. Appl Geochem 17:517–568
Strauss R, Brümmer GW, Barrow NJ (1997) Effects of crystallinity of goethite: II. Rates of sorption and desorption of phosphate. Euro J Soil Sci 48:101–114
Takematsu N, Sato Y, Okabe S, Nakayama E (1985) The partition of vanadium and molybdenum between manganese oxides and sea water. Geochim Cosmochim Acta 49:2395–2399
Thompson A, Chadwick OA, Rancourt DG, Chorover J (2006) Iron-oxide crystallinity increases during soil redox oscillations. Geochim Cosmochim Acta 701710–1727
Wang X, Liu F, Tan W, Feng X, Koopal LK (2013a) Transformation of hydroxycarbonate green rust into crystalline iron (hydr)oxides: influences of reaction conditions and underlying mechanisms. Chem Geol 351:57–65
Wang X, Liu F, Tan W, Li W, Feng X, Sparks DL (2013b) Characteristics of phosphate adsorption-desorption onto ferrihydrite: comparison with well-crystalline Fe (hydr)oxides. Soil Sci 178:1–11
Yin H, Feng X, Tan W, Koopal LK, Hu T, Zhu M, Liu F (2015) Structure and properties of vanadium(V)-doped hexagonal turbostratic birnessite and its enhanced scavenging of Pb2+ from solutions. J Hazard Mater 288:80–88
Zachara JM, Girvin DC, Schmidt RL, Resch CT (1987) Chromate adsorption on amorphous iron oxyhydroxide in the presence of major groundwater ions. Environ Sci Technol 21:589–594
Acknowledgments
This study was financially supported by Verein der Freunde und Förderer der Universität zu Köln. We would like to thank Karin Greef (University of Cologne) and Gerd Welp and Addi Kiener (University of Bonn) for analyzing the extracts, and Ruth Bruker and Stefan Roitsch (University of Cologne) for EDX analysis. Additionally, we are grateful to the Duke of Croy and Thomas Seine, who enabled the field measurements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Dong-Mei Zhou
Rights and permissions
About this article

Cite this article
Dorau, K., Mansfeldt, T. Manganese and iron oxide-coated redox bars as a tool to in situ study the element sorption in wet soils. J Soils Sediments 16, 976–986 (2016). https://doi.org/10.1007/s11368-015-1300-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-015-1300-6