
Abstract
Background
Sleep apnea (SA) may be linked to coronary artery disease (CAD). Both conditions have similar risk factors, confounding the analyses. Investigation of the lipid profile is routine in the adult population, even without symptoms or suspected cardiac ailment. SA, however, remains underdiagnosed even in the presence of unambiguous clinical manifestations.
Purpose
The aim of this study was to verify the association between SA and CAD, adjusting for usual CAD risk factors.
Methods
Patients who underwent diagnostic or therapeutic coronariography and portable type III polysomnography were studied. The severity of SA was determined by the apnea–hypopnea index (AHI). We measured classic CAD risk factors: fasting glucose; total, HDL, and LDL cholesterols; triglycerides; uric acid, and high-sensitivity C-reactive protein. We excluded patients older than 65 years, with body mass index higher than 40 kg/m2, with diabetes, and with history of smoking in the last year.
Results
Of 55 included patients, 28 had AHI > 14, showing an odds ratio of 8.7 for CAD. Patients without (n = 29) and with CAD (n = 26), showed AHI of, respectively, 11 ± 11 and 23 ± 14 per hour (P = 0.001). In a binary logistic regression to predict CAD, controlling for all the above risk factors, the only variables entered in the stepwise model were AHI (either as continuous or categorical variable) and uric acid.
Conclusion
In a sample without smokers, morbidly obese, or diabetic patients, AHI is the main predictor of CAD. SA should integrate the set of risk factors routinely assessed in clinical investigation for coronary disease risk stratification.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sleep apnea (SA), a relevant public health problem, is characterized by recurrent complete or partial upper airway obstruction during sleep. Each apneic episode leads to oxygen desaturation and arousal. Common consequences are daytime sleepiness and cardiovascular diseases [1, 2].
SA is associated with hypertension, coronary artery disease (CAD), heart failure, cardiac arrhythmias, reduced heart rate variability, atrial fibrillation, stroke, and sudden death [3–10]. The pathophysiological basis for the relationship between SA and CAD may be similar to that described for the SA–hypertension relationship [11].
While the possibility of SA as a causal mechanism in chronic cardiovascular disease is attractive, this relationship remains unclear. Gottlieb et al. failed to demonstrate a clear association between SA and incident CAD in 4,422 subjects of the Sleep Heart Health Study cohort [12]. Peker et al., in 62 patients, after controlling for age, gender, and body mass index (BMI), and adjusting for traditional risk factors, concluded that SA, current smoking, and diabetes mellitus were independently associated with CAD, while hypertension and hypercholesterolemia were not [13]. Considering that smoking and diabetes are powerful coronary aggressors, we hypothesized that excluding these factors would make the effect of hypertension and dyslipidemia more evident. The present study aims to verify the relationship between SA and CAD, controlling for the usual biochemical risk factors, but excluding cases with major well-established risk factors.
Methods
Patients
This is a secondary analysis of a sample screened to study oxidative stress in SA and CAD [14]. The study was performed at the catheterization laboratory, Division of Cardiology of our Institution, conducted between March 2007 and February 2008. Flow diagram of subject recruitment is displayed in Fig. 1. Consecutive patients complaining angina, between 35 and 65 years of age, were referred by their physicians for diagnostic coronary angiography. The exclusion criteria were smoking in the last year, clinical diagnosis of diabetes mellitus, BMI > 40 kg/m2; any physical, psychological, or social issue encumbering the attainment of the home polysomnographic test, and previous coronary intervention (myocardial revascularization or angioplasty). A full medical history was taken from all study participants. Dyslipidemia was diagnosed when cholesterol levels were greater than 200 mg/dL or the patient was under statin therapy. Hypertension was defined based on patients' reports. BMI was calculated as weight divided by height squared (kilograms per square meter). Subjects signed written informed consent forms, and the protocol was approved by the Institutional Ethics Committee.

Flow diagram of subject recruitment
Laboratory measurements
Blood collection
In the morning, 18-mL arterial blood samples were collected from each patient, fasted for at least 8 h, at the site of femoral artery puncture for catheterization. Blood was collected in three vials, containing coagulation activator, EDTA, or citrate. Immediately after the collection, the samples were refrigerated, centrifuged at 0°C for 10 min, aliquoted, and stored at −80°C. Hemolysates were prepared by lysing red blood cell with ethanol 2% (ratio 1:10) followed by centrifugation to obtain crude extracts. Total cholesterol, high-density lipoprotein, low-density lipoprotein, triglycerides, dyslipidemia, uric acid, high-sensitivity C-reactive protein (hs-CRP), and fasting glucose were measured in the routine clinical analysis laboratory.
Sleep study
The volunteers underwent portable polysomnography using a level III monitor (SomnoCheck, Weinmann, Germany), a procedure previously validated by our group [15]. In brief, air flow and snoring were detected through a nasal cannula connected to a pressure transducer; additionally, ventilatory effort, pulse oximetry, heart rate, and sleep position were evaluated. The recordings were made at the patient's home, usually between 11 pm and 7 am. The respiratory analyses of the polysomnography records were made by one board-certified sleep specialist in a different location, blind to the catheterization results.
Apneas were defined as airflow reduction to 10% or less of the baseline value for 10 s or more; hypopneas as airflow reduction of 50% or more, associated with at least 3% oxygen desaturation and/or autonomic arousal evidenced by an increase in heart rate greater than five beats per minute [16]. The severity of SA was assessed by the AHI, calculated by dividing the number of apneas and hypopneas by the number of hours of artifact-free recording.
Coronary angiography study
All patients were assessed by quantitative angiography, using the same equipment (SIEMENS D40) and projections, with the table and image intensifier kept at constant height. Image quantification was carried out in all cases by the same investigator, who was blinded to laboratory and polysomnography results. A magnification of 7 in. was used for all images. The cases were defined as with CAD when vessel lumen narrowing >50% of at least one coronary segment (CAD group). Patients with no lesion or with lesions ≤50% of luminal narrowing were considered as controls (non-obstructive CAD group).
Statistical analysis
Data were analyzed using SPSS v.16 (SPSS Inc., Chicago, IL, USA) to obtain descriptive statistics of frequency, mean value and standard deviation, or median and interquartile range. Comparisons of baseline characteristics between the groups were made by Student's t test or Mann–Whitney U test for nonparametric variables. Logistic regression models were employed to predict CAD, including stepwise forward model by likelihood ratio. The following regressors were used: gender, age, BMI, fasting glucose, hs-CRP, lowest O2, uric acid, high blood pressure, dyslipidemia, and AHI. AHI, indicating OSA severity, was analyzed as follows: (1) continuous variable; (2) two groups divided at the median value of 14 events per hour (AHI > 14); (3) four groups divided according the recommended OSA severity criteria (<5, 5–15, 15–30, and >30 events per hour; AHI-4). A P value < 0.05 was considered statistically significant. Odds ratios are presented with their 95% confidence intervals.
Results
Table 1 shows the characteristics of the total sample and of the groups formed by the angiographic result of catheterization. Male gender and age differ between cases and controls, but not BMI or ethnic background. Cases with 50% or greater coronary occlusion were using more medication, but, of 13 pharmaceutical classes analyzed, the difference was significant only for statins and alpha-adrenergic inhibitor. Controls with <50% coronary lesions exhibited significantly lower AHI.
Table 2 shows the sample characteristics of patient groups divided at median AHI, AHI ≤14 and >14. No significant difference was seen in BMI and biochemical markers for CAD risk. Basically, the same results were obtained utilizing the continuous variable AHI or categorizing AHI in four severity groups.
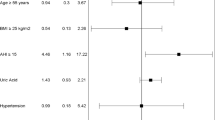
Figure 2 shows prediction of CAD by four binary logistic regression models. In model 1, classical cardiovascular risk factors were individually analyzed in the binary logistic regression, including one variable at a time. Gender, age, uric acid, and dyslipidemia were significant predictors, besides AHI. In model 2, two variables were included at a time, being one of them AHI. Using AHI > 14, uric acid persisted significant (P = 0.44), but neither using continuous AHI nor AHI-4. Therefore, including AHI > 14 caused variables that were significant in the univariate model, such as male gender, age, dyslipidemia, and BMI to become non-significant as risk factors for CAD.

Model 1 univariate analysis, model 2 bivariate analysis using AHI >14 with each regressor, model 3 full model multivariate binary logistic analysis, model 4 stepwise binary logistic analysis, CI confidence interval, OR odds ratio, AHI apnea–hypopnea index; *P = <0.05; **P = <0.01
In Fig. 2, model 3, including all regressors simultaneously in the binary logistic regression to predict CAD risk, only AHI > 14 and dyslipidemia showed statistical significance. All the three forms of including AHI in the model, AHI > 14 (P = 0.012), AHI-4 (P = 0.009), and continuous AHI (P = 0.034) were significant. The increment of one event per hour in the continuous AHI increases 11% the probability of CAD.
In Fig. 2, model 4, AHI > 14 and uric acid were the only factors entered in the stepwise forward model. Using AHI-4 instead of AHI > 14 in the model increases 3.2 times the probability of CAD for each increase in severity category (e.g., from mild to moderate); raising 1 mg/dL of uric acid concentration, increases 70% (with AHI > 14) and 66% (with AHI-4) the risk for CAD. Utilizing the continuous variable AHI, dyslipidemia enters the model with an odds ratio (OR) of 4.8 (1.06–22; P = 0.042).
Discussion
Our study suggests that AHI, measured by validated portable home monitoring, is the most important risk factor for coronary lesion in a sample of angina patients when individuals with diabetes, morbid obesity, smoking history, and age >65 years are excluded. The results are particularly interesting because even with a reduced sample size, the statistical significance of AHI to predict CAD resisted multivariate analysis, controlling for most risk factors used in checkups for CAD prevention. One implication of this finding is that, if SA really explains the risk imposed by hypertension, cholesterol, C-reactive protein, future studies on risk factors for cardiovascular disease will be incomplete if lacking information on SA. At the present time, few studies include SA in the listing of cardiovascular risk factors.
The fact that we screened 519 patients arriving at the catheterization laboratory to obtain a sample of only 55 cases shows the importance of smoking and age >65 years in causing CAD (Fig. 1). In spite of the predominance of middle-aged men in our sample, after controlling the CAD risk for age and gender in the regression model, AHI persisted significant, suggesting that SA is directly involved in the genesis of CAD, in spite of gender and age.
There is evidence that SA may contribute to obesity and, in particular, to deposition of visceral fat [17]. There is also evidence of correlation between BMI and hs-CRP levels in SA patients [18, 19]. The relationship between hs-CRP and sleep-disordered breathing is still controversial [20, 21], due mainly to the fact that hs-CRP levels are directly correlated with obesity [22]. The hs-CRP levels were similar between groups either divided by percentage of coronary occlusion or by median AHI.
It is difficult to assess the importance of inflammation and dyslipidemia as risk factor in this sample due to the higher proportion of individuals under statins among cases (68%) than among controls (31%). Besides lowering lipids, statins may have lowered the hs-CRP. There is increasing evidence that SA is independently associated with dyslipidemia. Other cross-sectional studies suggested that SA is independently associated with increased levels of total cholesterol, low-density lipoprotein, and triglycerides [23–25].
In our analysis, the frequency of cases with hypertension was significantly higher in the group with AHI > 14 per hour. The measured blood pressure was not different between groups, because virtually, all hypertensive cases were receiving antihypertensive medication. Our results are in agreement with the literature showing association of SA with hypertension, especially in the resistant form [26, 27].
In our study, uric acid was the second predictor for CAD in the regression model. A recent systematic review showed that hyperuricemia may marginally increase the risk of cardiovascular outcomes, independently of other traditional cardiovascular risk factors [28]. This OR is significant only when the remaining variables do not enter the model. The increased formation of free radicals in SA happens via the conversion of xanthine dehydrogenase into its oxidase form during hypoxia, followed by the activation of the oxidase form during reoxygenation by the hypoxanthine formed during hypoxia. This xanthine oxidase activity generates superoxide radical, superoxide radical, and uric acid [29]. Uric acid may be involved in platelet aggregation and adhesiveness, in inflammatory processes, as well as in the genesis of hypertension [30, 31]. The actual effect of this antioxidant and its relationship with atherosclerosis and SA remains uncertain.
The high covariance among the risk factors in such a small and selected sample may be interfering in the significance of the various tested models. Uric acid remaining in our model does not favor uric acid being a better predictor of CAD than, for instance, hs-CRP. AHI, however, enters and resists combinations with all risk factors. Sleep apnea is, therefore, a risk factor to be taken seriously.
The small sample size is a clear limitation. We attempted to circumvent this lack of power by excluding powerful risk factors, such as old age and smoking, and by doing multivariate analysis with a limited number of variables or in a stepwise fashion. The design of this study prevents us from identifying causal relationship. We report an association between CAD and SA, but not proof that SA leads to CAD. Not every known CAD risk factor was included in the analysis, limiting the adjustments to the aspects analyzed.
The main finding in the present study is that AHI, a marker of SA severity, proved to be a strong risk factor for CAD, independent of age, gender, BMI, and other traditional risk factors. Being our results limited by a small sample does not disqualify the importance of identifying sleep-disordered breathing in patients at risk for cardiovascular disease. Effective treatment of the SA may decelerate the atherogenesis process, as evidenced by intima-media thickness decrease after 4 months of CPAP treatment [32].
Another well-known confounder in the SA–CAD relationship is obesity. Cases and controls had non-significant difference in BMI. The mean BMI around 27 kg/m2 indicates that the sample was mostly overweight, with just one third of the sample, 17 patients, exhibiting BMI between 30 and 36 kg/m2. The OR of BMI did not reach significance in any attempt to control for this important variable. Morbid obesity is a known inflammatory state [33], and its exclusion may have helped to show the effect of SA on CAD risk in the present study.
The relationship between OSA and CAD is complex [34]. The association has been researched in large samples [12] and even using the Berlin Questionnaire [35]. Inflammation happens in the core of the atherogenic process, and patients with OSA have higher levels of inflammatory markers that are reversed by CPAP therapy [32]. Endothelial damage in OSA can be caused by mechanisms as shear stress, due to blood pressure oscillations, and oxidative stress, due to hypoxia/reoxygenation [36, 37]. Platelets also play a central role in CAD. Patients with OSA have increased platelet activation and it is reduced after CPAP therapy [38]. Cardiac dysfunction, however, has to be considered as cause of obstructive and central sleep apnea [39]. CAD may impair hemodynamics causing fluid retention. Rostral fluid displacement from the lower limbs increases peripharyngeal and pulmonary liquid content. This increases upper-airway collapsibility and ventilation, facilitating the occurrence of obstructive and of central apneas [39].
This small study provides evidence that SA is a better predictor of coronary lesions than recognized risk factors when aging, smoking, morbid obesity, and diabetes are excluded. Future studies should be performed to confirm the data in unselected populations. The present findings, although preliminary, are potentially useful if used by clinicians for risk stratification and risk factor modification in clinical practice.
References
Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, Daniels S, Floras JS, Hunt CE, Olson LJ, Pickering TG, Russell R, Woo M, Young T (2008) Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. J Am Coll Cardiol 8:686–717
Crummy F, Piper AJ, Naughton MT (2008) Obesity and the lung: 2. Obesity and sleep-disordered breathing. Thorax 8:738–746
Devulapally K, Pongonis R Jr, Khayat R (2009) OSA: the new cardiovascular disease: part II: overview of cardiovascular diseases associated with obstructive sleep apnea. Heart Fail Rev 3:155–164
Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ, Heart N, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, National High Blood Pressure Education Program Coordinating Committee (2003) The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC VII). J Am Med Assoc 289:2560–2572
Lüthje L, Andreas S (2008) Obstructive sleep apnea and coronary artery disease. Sleep Med Rev 1:19–31
Nelson CA, Wolk R, Somers VK (2005) Sleep-disordered breathing: implications for the pathophysiology and management of cardiovascular disease. Compr Ther 1:21–27
Leung RS (2009) Sleep-disordered breathing: autonomic mechanisms and arrhythmias. Prog Cardiovasc Dis 4:324–338
Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V (2005) Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 19:2034–2041
Gami AS, Howard DE, Olson EJ, Somers VK (2005) Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 12:1206–1214
McNicholas WT, Bonsigore MR (2007) Management Committee of EU COST ACTION B26. Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. Eur Respir J 1:156–178
Butt M, Dwivedi G, Khair O, Lip GY (2010) Obstructive sleep apnea and cardiovascular disease. Int J Cardiol 1:7–16
Gottlieb DJ, Yenokyan G, Newman AB, O'Connor GT, Punjabi NM, Quan SF, Redline S, Resnick HE, Tong EK, Diener-West M, Shahar E (2010) Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure. The Sleep Heart Health Study. Circulation 4:352–360
Peker Y, Kraiczi H, Hedner J, Löth S, Johansson A, Bende M (1999) An independent association between obstructive sleep apnoea and coronary artery disease. Eur Respir J 1:179–184
Klein C, Martinez D, Hackenhaar FS, Medeiros TM, Marcolin ML, Silveira FS, Wainstein MV, Gonçalvez SC, Benfato MS (2010) Carbonyl groups: bridging the gap between sleep disordered breathing and coronary artery disease. Free Radic Res 8:907–912
Tonelli de Oliveira AC, Martinez D, Vasconcelos LF, Gonçalves SC, Lenz MC, Fuchs SC, Gus M, Abreu-Silva EO, Moreira LB, Fuchs FD (2009) Diagnosis of obstructive sleep apnea syndrome and its outcomes with home portable monitoring. Chest 2:330–336
American Academy of Sleep Medicine (2005) International classification of sleep disorders. Diagnostic and coding manual (ICSD-2). 2nd ed. Westchester, IL
Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL (2002) Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med 5:677–682
Lear SA, Chen MM, Birmingham CL, Frohlich JJ (2003) The relationship between simple anthropometric indices and C-reactive protein: ethnic and gender differences. Metabolism 12:1542–1546
Ryan S, Taylor CT, McNicholas WT (2009) Systemic inflammation: a key factor in the pathogenesis of cardiovascular complications in obstructive sleep apnoea syndrome? Thorax 7:631–636
Steiropoulos P, Kotsianidis I, Nena E, Tsara V, Gounari E, Hatzizisi O, Kyriazis G, Christaki P, Froudarakis M, Bouros D (2009) Long-term effect of continuous positive airway pressure therapy on inflammation markers of patients with obstructive sleep apnea syndrome. Sleep 4:537–543
Taheri S, Austin D, Lin L, Nieto FJ, Young T, Mignot E (2007) Correlates of serum C-reactive protein (CRP) -no association with sleep duration or sleep disordered breathing. Sleep 8:991–996
Kao TW, Lu IS, Liao KC, Lai HY, Loh CH, Kuo HK (2009) Associations between body mass index and serum levels of C-reactive protein. S Afr Med J 5:326–330
Kono M, Tatsumi K, Saibara T, Nakamura A, Tanabe N, Takiguchi Y, Kuriyama T (2007) Obstructive sleep apnea syndrome is associated with some components of metabolic syndrome. Chest 5:1387–1392
Drager LF, Bortolotto LA, Maki-Nunes C, Trombetta IC, Alves MJ, Fraga RF, Negrão CE, Krieger EM, Lorenzi-Filho G (2010) The incremental role of obstructive sleep apnoea on markers of atherosclerosis in patients with metabolic syndrome. Atherosclerosis 2:490–495
Roche F, Sforza E, Pichot V, Maudoux D, Garcin A, Celle S, Picard-Kossovsky M, Gaspoz JM, Barthélémy JC, PROOF Study Group (2009) Obstructive sleep apnoea/hypopnea influences high-density lipoprotein cholesterol in the elderly. Sleep Med 8:882–886
Gonçalves SC, Martinez D, Gus M, de Abreu-Silva EO, Bertoluci C, Dutra I, Branchi T, Moreira LB, Fuchs SC, de Oliveira AC, Fuchs FD (2007) Obstructive sleep apnea and resistant hypertension: a case–control study. Chest 6:1858–1862
Lopez-Jimenez F, Sert Kuniyoshi FH, Gami A, Somers VK (2008) Obstructive sleep apnea: implications for cardiac and vascular disease. Chest 3:793–804
Kim SY, Guevara JP, Kim KM, Choi HK, Heitjan DF, Albert DA (2010) Hyperuricemia and coronary heart disease: a systematic review and meta-analysis. Arthritis Care Res 2:170–180
Lavie L, Hefetz A, Luboshitzky R, Lavie P (2003) Plasma levels of nitric oxide and L-arginine in sleep apnea patients: effects of nCPAP treatment. J Mol Neurosci 1:57–63
Gagliardi AC, Miname MH, Santos RD (2009) Uric acid: a marker of increased cardiovascular risk. Atherosclerosis 1:11–17
Tsai WC, Huang YY, Lin CC, Li WT, Lee CH, Chen JY, Chen JH (2009) Uric acid is an independent predictor of arterial stiffness in hypertensive patients. Heart Vess 5:371–375. doi:10.1007/s00380-008-1127-9
Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi GF (2007) Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 7:706–712
De Luis DA, González Sagrado M, Conde R, Aller R, Izaola O (2010) Resistin levels and inflammatory markers in patients with morbid obesity. Nutr Hosp 4:630–634
Chami HA, Resnick HE, Quan SF, Gottlieb DJ (2011) Association of incident cardiovascular disease with progression of sleep-disordered breathing. Circulation 12:1280–1286
Martinez D, da Silva RP, Klein C, Fiori CZ, Massierer D, Cassol CM, Bos AJ, Gus M (2011) High risk for sleep apnea in the Berlin questionnaire and coronary artery disease. Sleep and Breathing 6. doi:10.1007/s11325-010-0460-2
Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP (2010) Pathophysiology of sleep apnea. Physiol Rev 1:47–112
Kohli P, Balachandran JS, Malhotra A (2011) Obstructive sleep apnea and the risk for cardiovascular disease. Curr Atheroscler Rep 2:138–146
Hui DS, Ko FW, Fok JP, Chan MC, Li TS, Tomlinson B, Cheng G (2004) The effects of nasal continuous positive airway pressure on platelet activation in obstructive sleep apnea syndrome. Chest 5:1768–1775
Yumino D, Redolfi S, Ruttanaumpawan P, Su MC, Smith S, Newton GE, Mak S, Bradley TD (2010) Nocturnal rostral fluid shift: a unifying concept for the pathogenesis of obstructive and central sleep apnea in men with heart failure. Circulation 14:1598–1605
Acknowledgments
Drs. Jorge Pinto Ribeiro and Marco Vugman Wainstein are thanked for their valuable contribution in providing data from the cineangiocoronariography.
Financial support
Students received grants from the Brazilian government through CAPES and CNPq. The main support was offered by the Research Incentive Fund (FIPE) of the Hospital de Clínicas de Porto Alegre (Porto Alegre, Rio Grande do Sul, Brazil).
Conflict of interest
The authors declare that they have no conflict of interest regarding the present study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Martinez, D., Klein, C., Rahmeier, L. et al. Sleep apnea is a stronger predictor for coronary heart disease than traditional risk factors. Sleep Breath 16, 695–701 (2012). https://doi.org/10.1007/s11325-011-0559-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11325-011-0559-0