
Abstract
Obstructive sleep apnea (OSA), present in 5–15% of adults, is strongly associated with the incidence and poor outcome of hypertension, coronary artery disease, arrhythmia, heart failure, and stroke. Treatment of OSA completely reverses its cardiovascular consequences. In this review, we discuss the clinical evidence for the strong association between OSA and cardiovascular disease and present an argument for approaching OSA as a cardiovascular disease. We particularly focus on the causative relationship between OSA and hypertension, and on the increasingly recognized relationship between OSA and heart failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obstructive sleep apnea (OSA) is present in up to 15% of middle-aged adults [1], and has a strong association with obesity. Therefore, OSA is poised to gain increasing public health importance with the rising prevalence of obesity [2]. Several well-conducted human and animal studies provided compelling evidence that OSA is a cause of hypertension [3–5]. Furthermore, in patients with existing hypertension, OSA worsens blood pressure control [6, 7]. OSA is also linked to increased prevalence and worse outcome in coronary artery disease (CAD) [8], arrhythmia [9, 10], and diabetes [11]. Additionally, there is now evidence that OSA can, independent of hypertension, induce left ventricular dysfunction [12, 13]. In patients with established heart failure, OSA is likely to be a detrimental co-morbidity that may severely impact outcomes [14]. Treatment of underlying OSA in patients with cardiac dysfunction improves systolic function [15]. Taken together, these data start to describe a critical role for OSA in inducing cardiovascular disease, or accelerating the progression of existing cardiovascular disease toward heart failure.
In this review, we will discuss the clinical evidence for a causative relationship between OSA and cardiovascular disease with focus on hypertension as the most important and best established cardiovascular consequence of OSA. We will also attempt to present a conceptual framework that explains the potential causative relation between OSA and heart failure.
OSA is a cause of hypertension
Hypertension, the most prevalent cardiovascular disease, is also the best established cardiovascular consequence of obstructive sleep apnea [4, 5, 16–18]. Hypertension affects approximately 30% of adults in the United States [19, 20] with an impact on public health that cannot be overstated. The World Health Organization estimates that even suboptimal blood pressure levels are responsible for 62% of cerebrovascular disease and 49% of ischemic heart disease [21]. On the other hand, OSA affects between 9% and 25% of middle-aged adults [1, 22]. OSA is estimated to be present in 30–40% of patients with hypertension, while 40% of patients with OSA are thought to have hypertension [23–25]. Since both disorders are very prevalent in middle-aged individuals, and coexist in a large portion of the population it had been difficult to elucidate the cause–effect relationship between the two disorders. However, in recent years, mounting evidence from large epidemiological studies, along with human and animal experiments, was able to confirm the presence of this causal relationship between OSA and hypertension. The mechanism of hypertension in patients with OSA remains only partially understood so far. In this article, we will focus on the clinical and epidemiological evidence for the causative relationship between OSA and hypertension. The basic mechanisms of the association between OSA and hypertension were reviewed in the first part of this series.
Epidemiological studies evaluating the relationship between OSA and hypertension
An association between snoring and systemic hypertension was first described in the early 80s [26]. As the understanding of OSA was evolving, a relation between OSA and vascular morbidity became more apparent [27]. The absence of the normal decrease in blood pressure during sleep, termed as “nondipping” may be the earliest sign of OSA-related hypertension [28, 29]. Some studies suggested that an isolated increase in diastolic blood pressure may be the earliest hypertensive change associated with OSA [30, 31]. Others observed that isolated systolic hypertension was uncommon in patients with OSA [32, 33]. However, a high prevalence of systolic hypertension was described in patients with OSA and chronic heart failure [34].
The earliest compelling description of a dose–response relation between OSA and hypertension was provided by the Wisconsin Sleep Cohort [5, 35]. In this landmark population-based study, 1,060 asymptomatic individuals underwent nocturnal polysomnography (in-laboratory sleep studies) to evaluate the presence and severity of OSA. The investigators found a dose–response relation between the severity of OSA and the odds ratio of having hypertension in this cohort. The dose–response relation was still present after correction for known risk factors of hypertension. Later, the Sleep Heart Health Study was the largest cross-sectional study to identify OSA as an independent risk factor for hypertension [36]. In this large multi-center study of cardiovascular risk factors, 6,424 patients were evaluated with home sleep studies. The investigators also observed a linear relationship between the severity of sleep apnea and the risk of having hypertension. The most convincing epidemiological evidence for a causal relation between OSA and hypertension was again provided by the Wisconsin Sleep Cohort Study [4]. In a four-year follow-up of 709 individuals enrolled in this study, the odds ratio of developing hypertension over the follow-up period increased linearly with increasing apnea–hypopnea index (AHI). This linear association persisted after correction to known causes of hypertension. Another striking finding in this study was that the relation between OSA and risk of hypertension was still present at levels of AHI that are considered in the normal range (0.1–4.9 events/h). These findings supported a very strong cause–effect relation between any degree of OSA, including ranges considered normal, and the risk of developing hypertension.
Human and animal models of OSA
Experimental animal models of OSA were developed to evaluate the cardiovascular consequences of OSA. Brooks et al. [37] developed an elaborate dog model of OSA with an instrumented control group. Dogs with experimental OSA consistently developed hypertension compared to their instrumented controls. Furthermore, these investigators identified the intermittent hypoxia pattern of OSA as the critical stimulus for the blood pressure increase in the dog model. Fletcher et al. developed a rat model of OSA in which rats were exposed to a protocol of intermittent hypoxia that mimiced the pattern of hypoxia seen in patients with OSA. Animals exposed to the intermittent hypoxia protocol consistently developed increased blood pressure compared to control rats [38–41]. This increase in blood pressure resolved after withdrawal of the intermittent hypoxia stimulus.
Several studies evaluated the effect of intermittent hypoxia in humans who were free of cardiovascular disease or OSA. Brief exposures to intermittent hypoxia consistently produced surges in sympathetic activity and blood pressure that persisted after recovery of hypoxia [42–44]. Arabi et al. [45] described an increase in morning diastolic blood pressures in healthy individuals exposed to nocturnal hypoxia. These human experiments further confirmed that the intermittent hypoxia pattern of OSA produces sympathetic-mediated hypertensive response, and may be the earliest abnormality leading to persistent hypertension in patients with OSA.
Effect of treatment of OSA on blood pressure
Several studies evaluated the effect of treatment of OSA on blood pressure. While many of these studies enrolled small numbers of patients, they all showed a consistent effect on improving blood pressure control. Logan et al. observed that occult OSA was present in up to 83% of a small group of patients with refractory hypertension [46]. Becker et al. [7] randomized patients with moderate to severe OSA to either therapeutic or sub-therapeutic Continuous Positive Airway Pressure (CPAP). Mean arterial blood pressure decreased by 10 mm Hg in the group that received effective CPAP treatment. In the sub-therapeutic CPAP arm, and despite a 50% reduction in the severity of OSA, no significant improvement in blood pressure was noted. This drop in mean arterial blood pressure by 10 mm Hg would be predicted to reduce coronary heart disease event risk by 37% and stroke risk by 56% [47]. Pepperell et al. [48] in a randomized trial, evaluated the blood pressure response in 118 men with obstructive sleep apnea and excessive daytime sleepiness to treatment with therapeutic versus sub-therapeutic CPAP. Therapeutic CPAP significantly reduced mean ambulatory arterial blood pressure, whereas sub-therapeutic CPAP increased the mean blood pressure. The benefit of therapeutic CPAP was seen in both systolic and diastolic blood pressure, and during both sleep and wakefulness. The CPAP benefit was most pronounced in patients with severe sleep apnea and in patients on anti-hypertensive medications. It emerged from these two studies that the treatment effect was most pronounced when blood pressure was more severe at baseline and when OSA was almost completely eliminated. In a subsequent trial by Campos-Rodrigues et al. [49], the effect of CPAP on blood pressure was more modest than in the two preceding trials; but also more pronounced in patients with severe hypertension and those with better adherence to with CPAP.
Sleepiness is an important clinical consequence of OSA and an indicator of severity of illness that describes an aspect of the disease not necessarily expressed by AHI alone. The presence of daytime sleepiness may also affect the adherence and benefit of CPAP. Therefore, it was reasonable to investigate whether the presence of underlying sleepiness would explain the difference in the degree of blood pressure responsiveness to CPAP. Robinson et al., in a randomized sham-placebo-controlled crossover trial [50], studied 35 non-sleepy hypertensive patients with OSA. Participants were treated with CPAP for 1 month then crossed over to the sham-placebo arm. The investigators found no significant difference in mean 24-h BP in the two groups. Generally, studies evaluating the effect of CPAP on blood pressure seem to suggest overall a consistent effect of reducing blood pressure, albeit to different degrees in varied subpopulations of patients with OSA. Large studies were able to show an effect of treatment of OSA in reducing fatal and nonfatal cardiovascular [3]. This effect of CPAP may be mediated by its favorable effect on blood pressure control.
Combined together, studies of human and animal models of OSA, along with the clinical studies evaluating the effect of treatment on blood pressure, confirm that OSA induces hypertension, and that hypertension is ameliorated by treatment of OSA.
Mechanism of hypertension in OSA
As the understanding of the pathophysiology of OSA was evolving, it emerged that several consequences of OSA and intermittent hypoxia are also etiological factors of hypertension. A detailed discussion of these mechanistic pathways is presented in an earlier article of this special issue. An important link in the cause–effect relationship between OSA and hypertension is the sympathetic activation in patients with OSA [51]. Intermittent hypoxia is a unique stimulus that is distinct from other forms of hypoxia [52, 53], and appears to be the only required stimulus for this sympathetic activation in patients with OSA [42, 44, 54]. In animal models of OSA, intact sympathetic system was required for the animals to manifest increased blood pressure [37, 44, 45, 54, 55]. Another recently emerging link between OSA and hypertension is oxidative stress, also present in both disorders [56, 57]. Endothelial dysfunction is an important precursor to cardiovascular disease and hypertension [58–62] and patients with OSA manifest oxidative stress-mediated endothelial dysfunction [63]. Another link between the mechanism of hypertension and OSA is the renin–angiotensin system. Patients with OSA demonstrate increased renin–angiotensin activity that improves with treatment [64, 65]. In an animal model of intermittent hypoxia, the renin–angiotensin pathway was critical for the hypertensive response to intermittent hypoxia [66, 67].
In summary, OSA is a cause of hypertension, and treatment of OSA improves blood pressure control and cardiovascular outcomes. The mechanism of OSA induced hypertension involves sympathetic activation and oxidative stress.
OSA and coronary artery disease
Coronary artery disease (CAD) is an important cause of morbidity and mortality in the United States [68], in addition to being a major risk factor for heart failure [69]. The pathophysiological basis for the relation between OSA and CAD may be similar to the basis for the relation between OSA and hypertension. OSA causes endothelial dysfunction [70], inflammation [71], and lipid dysfunction [72]. Patients with OSA have silent preclinical atherosclerosis that improves with treatment of OSA [73]. The causative relation between OSA and CAD was underscored in a recent report describing a relationship between OSA and preclinical coronary disease [74].
It is on this pathological background, that the sympathetic activation associated with OSA may promote ischemic events. Increased incidence of ST segment changes during sleep was reported in OSA patients with CAD and correlated with the severity of the respiratory events and sleep fragmentation. This OSA-related ST depression improved with nasal CPAP treatment [75–79]. A detrimental effect of chronic intermittent hypoxia on worsening myocardial infarcts was recently described [80, 81].
In a large observational study, OSA was an independent risk factor for developing CAD [82]. Peker et al. found an increased incidence of CAD in patients with OSA and no apparent cardiovascular disease over 7 years of follow-up [8, 23]. Patients with existing CAD and even mild to moderate OSA (Apnea Hypopnea Index > 10 events/h) were much more likely to experience cardiovascular death over a 5-year period than those with no OSA (37.5% vs. 9.3%, respectively) [83]. Similarly, Mooe et al. found an independent association between OSA and worse prognosis in patients with CAD [84]. This relation between untreated OSA and cardiovascular events, fatal and nonfatal, was recently demonstrated in several large studies [3, 16–18].
In summary, patients with OSA have increased risk of developing CAD [8] and having worse outcomes of CAD [83, 85]. Treatment with CPAP reverses this negative relationship between OSA and CAD [86].
OSA and arrhythmia
A hallmark of OSA is increased baseline sympathetic activity [51] which may be the mechanism of myocardial irritability and subsequent arrhythmia [87]. In a large cross-sectional study [88], individuals with severe OSA had 2- to 4-fold odds ratio of complex arrhythmias compared with those without OSA. This increased risk persisted even after adjustment for potential confounders. A significant relation was also observed between OSA events and ventricular irritability [89]. Treatment with CPAP reversed this ventricular irritability [87, 89]. Furthermore, treatment of OSA improves cardiac autonomic control measured by heart rate variability in patients with both heart failure and OSA [90].
The relation between OSA and atrial fibrillation is a particularly important and only recently recognized link between OSA and cardiovascular morbidity. Obesity and the magnitude of nocturnal oxygen desaturation were previously shown to be independent risk factors for incident atrial fibrillation [91]. In a prospective study of patients referred to a general cardiology practice, a strong association between OSA and atrial fibrillation was found (49% in patients with OSA compared to 32% in patients of general cardiology) [9]. OSA was also associated with recurrent atrial fibrillation after cardioversion [10].
Gami et al. found that patients with OSA were more likely to experience sudden death during the night compared with patients without OSA, further confirming the presence of increased arrhythmia in this patient population [92]. The relation between OSA and atrial fibrillation may also contribute to the increased risk of stroke observed in patients with OSA [93].
Probably due to increased sympathetic activation, OSA is associated with a spectrum of cardiac arrhythmias that complicate the cardiovascular consequences of OSA and further contribute to morbidity and mortality [94].
OSA and heart failure
Given the strong etiological relationship between OSA, on one hand, and CAD [83, 85], hypertension and arrhythmia [9, 10] on the other, it is not surprising that OSA is also an independent risk factor for heart failure [82]. The prevalence of OSA in heart failure patients is far higher than in the general middle-aged population [95–99].
Patients with OSA have high prevalence of hypertension creating difficulty in making strong conclusions about a direct relation between OSA and left ventricular abnormality independent of hypertension [100]. However, several studies have suggested a causative relation between OSA and left ventricular remodeling, probably via intermittent hypoxia-induced oxidative stress [12, 101, 102]. A large portion of patients with OSA have either systolic dysfunction or subclinical echocardiographic left ventricular abnormalities [12, 101, 102]. In an important study of children with OSA who are free of cardiovascular disease [13], Amin et al. found evidence of asymptomatic early left ventricular remodeling in these children.
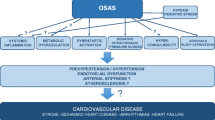
The available evidence, therefore, supports that OSA can produce cardiac dysfunction independent of hypertension [102]. It is, however, in patients with existing heart failure that the effect of OSA events may be most profound. The mechanism for these effects on the failing heart include increased cardiac muscle work index due to the increased negative intrathoracic pressure during obstructed breaths [103]. Changes in venous return due to the negative intrathoracic pressure generated from the inspiratory effort may affect preload and stroke volume [104]. Sympathetic activation secondary to hypoxia increases blood pressure and reduces myocardial perfusion [105, 106]. This increase in cardiac afterload and myocardiac ischemia during obstructive apneas was demonstrated in the normal heart in animal models [102], but its consequences are probably most striking in the failing heart. Studies showed that CPAP immediately reverses these effects [103]. Figure 1 summarizes several of the known pathways by which OSA may lead directly or indirectly to cardiac dysfunction and heart failure.

Chronic cardiovascular consequences of OSA; LV = left ventricle, RV = right ventricle, SVR = systemic vascular resistance, ↑ = increased
OSA compounds heart failure by potentiating sympathetic activation. Patients with heart failure and sleep apnea show higher daytime sympathetic nerve activity compared with patients with heart failure but no sleep apnea [107]. CPAP treatment in patients with heart failure and OSA decreased daytime sympathetic activity, systolic blood pressure, and heart rate [108], suggesting a significant contribution of OSA to increased sympathetic outflow. Randomized controlled studies in patients with heart failure and OSA demonstrated that CPAP treatment for a few weeks increased left ventricular ejection fraction and decreased blood pressure and sympathetic activation, strongly suggesting a pathogenic role of OSA in worsening cardiac function [15, 109, 110].
There is now compelling evidence that CPAP improves left ventricular ejection fraction in patients with heart failure and OSA. Whether the positive effects of CPAP on the cardiovascular system may also imply an improvement in long-term prognosis of patients with OSA and heart failure is still unknown [111]. Improved quality of life after CPAP treatment has only been documented in OSA patients with daytime sleepiness [110]. Data are lacking in heart failure patients with mild to moderate OSA, with or without daytime sleepiness. Additionally, no effect of treatment on survival has been shown to date.
OSA, pulmonary hypertension, and right ventricular dysfunction
The evidence for a relation between OSA and pulmonary hypertension is less compelling than that for the relation between OSA and systemic hypertension. Nevertheless, the reported prevalence of pulmonary arterial hypertension in OSA varies from 20% to 41% [112–114]. However, most studies in this area were limited by the presence of concomitant cardiovascular diseases which could have affected their pulmonary pressures [115, 116]. Acute hemodynamic changes involving the pulmonary artery and the right ventricle during apneic episodes are well recognized. The mechanism of the effect of OSA on pulmonary circulation may be intermittent hypoxia. Hypoxemia-induced endothelial cell dysfunction is a critical factor in pulmonary artery remodeling [117]. Hypoxia induces up-regulation of vascular endothelial growth factor, which is a mediator of angiogenesis, resulting in vascular remodeling [118, 119].
Several studies have evaluated right ventricular function and structure in OSA patients and demonstrated a decrease in ejection fraction and right ventricular hypertrophy [120, 121]. Right ventricular dysfunction in patients with OSA may be due to pulmonary hypertension. However, it is not possible so far to rule that the strong effect of OSA on left ventricular function is not the cause of the reported right ventricular dysfunction. Treatment with CPAP reverses right ventricular dysfunction in individuals with OSA [122]. Similarly, CPAP has been shown to decrease pulmonary pressures in OSA patients with either high or normal pulmonary pressures [123, 124]. The current consensus, with the available data, is that OSA may modestly increase pulmonary arterial pressures, and that evaluation for OSA should be part of the initial work-up in patients with pulmonary hypertension [123].
OSA and stroke
A recent study found that severe OSA was associated with diabetes, night-time stroke onset, and macro-angiopathy as a cause of stroke [125]. Intermittent hypoxia is probably the critical factor in the cerebrovascular abnormalities predisposing OSA patients to stroke. Patients with OSA have impaired cerebrovascular response to hypoxia [126] consistent with underlying abnormal endothelial function. Furthermore, animal models of intermittent hypoxia developed impaired cerebrovascular response to hypoxia [127]. Additionally, there was a direct relationship between the severity of nocturnal oxygen desaturations and carotid artery intimal thickness and atherosclerotic plaques in the carotid arteries of OSA patients, which was independent of the presence of hypertension [128, 129]. These early signs of atherosclerosis were reversible with treatment of OSA, supporting a causal relation between OSA and atherosclerosis [130], and subsequently stroke.
The prevalence of OSA in patients with acute stroke was estimated between 44% and 72% [131, 132]. It is intriguing that the type of sleep apnea that is most common after acute stroke is actually obstructive and not central apnea. Central but not obstructive sleep apneas decrease during recovery after a stroke or a transient ischemic attack, suggesting that obstructive events are most likely to have preceded the cerebrovascular event [133]. The relationship between OSA on one hand, and hypertension and stroke on the other, may become critical in the acute poststroke phase when control of blood pressure is very important. In a recent report, the mean 24-h blood pressure level in patients with acute stroke positively correlated with severity of sleep apnea [134].
OSA increases severity of stroke leading to increased incidence of death as well as poststroke mortality and morbidity. Increased severity of OSA correlated with increasing incidence of stroke and death in a cohort of OSA patients after a median follow-up of 3.4 years [93].
Increased mortality was noted in patients with severe OSA (AHI > 30) after stroke [125, 135]. CPAP treatment after stroke improved 18 month survival [136]. OSA also increases the risk of recurrence of ischemic stroke [137]. Patients with stroke and OSA show more severe functional impairment and longer hospitalization during rehabilitation compared to patients without OSA [138].
In summary, OSA is an independent risk factor for developing stroke [93, 139], and for poor outcome once stroke has occurred [125, 140]. Subsequently, OSA is associated with profound impact on public health. To date, surveillance and treatment of OSA in patients at risk for stroke, or even patients who had a stroke, is not part of the standard of care.
Conclusion
OSA is strongly associated with the incidence and poor outcome of hypertension, CAD, arrhythmia, heart failure, and stroke. In addition, obesity and aging, both on the rise in the general population, are risk factors for both OSA and heart failure. Treatment of OSA completely reverses its cardiovascular consequences. Therefore, OSA should be approached as an important modifiable cardiovascular risk factor.
References
Young T et al (1993) The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328(17):1230–1235. doi:10.1056/NEJM199304293281704
Center for Disease Control and Prevention. N.C.f.h.S., Health, U.D.o.H.a.H. Services, Editor (2006) National Health Interview Survey: US Census
Marin JM et al (2005) Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365(9464):1046–1053
Peppard PE et al (2000) Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 342(19):1378–1384. doi:10.1056/NEJM200005113421901
Young T et al (1997) Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med 157(15):1746–1752. doi:10.1001/archinte.157.15.1746
Wilcox I et al (1993) Effect of nasal continuous positive airway pressure during sleep on 24-hour blood pressure in obstructive sleep apnea. Sleep 16(6):539–544
Becker HF et al (2003) Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation 107(1):68–73. doi:10.1161/01.CIR.0000042706.47107.7A
Peker Y, Carlson J, Hedner J (2006) Increased incidence of coronary artery disease in sleep apnoea: a long-term follow-up. Eur Respir J 28(3):596–602. doi:10.1183/09031936.06.00107805
Gami AS et al (2004) Association of atrial fibrillation and obstructive sleep apnea. Circulation 110(4):364–367. doi:10.1161/01.CIR.0000136587.68725.8E
Kanagala R et al (2003) Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 107(20):2589–2594
Reichmuth KJ et al (2005) Association of sleep apnea and type II diabetes: a population-based study. Am J Respir Crit Care Med 172(12):1590–1595. doi:10.1164/rccm.200504-637OC
Alchanatis M et al (2002) Evidence for left ventricular dysfunction in patients with obstructive sleep apnoea syndrome. Eur Respir J 20(5):1239–1245. doi:10.1183/09031936.02.00278002
Amin RS et al (2002) Left ventricular hypertrophy and abnormal ventricular geometry in children and adolescents with obstructive sleep apnea. Am J Respir Crit Care Med 165(10):1395–1399. doi:10.1164/rccm.2105118
Wang H et al (2007) Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol 49(15):1625–1631. doi:10.1016/j.jacc.2006.12.046
Kaneko Y et al (2003) Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med 348(13):1233–1241
McNicholas WT, Bonsigore MR (2007) Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. Eur Respir J 29(1):156–178. doi:10.1183/09031936.00027406
Campos-Rodriguez F et al (2005) Mortality in obstructive sleep apnea-hypopnea patients treated with positive airway pressure. Chest 128(2):624–633. doi:10.1378/chest.128.2.624
Doherty LS et al (2005) Long-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndrome. Chest 127(6):2076–2084. doi:10.1378/chest.127.6.2076
Hajjar I, Kotchen TA (2003) Trends in prevalence, awareness, treatment, and control of hypertension in the United States, 1988–2000. JAMA 290(2):199–206. doi:10.1001/jama.290.2.199
Fields LE et al (2004) The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension 44(4):398–404. doi:10.1161/01.HYP.0000142248.54761.56
World Health Report 2002: Reducing risks, promoting healthy life. Geneva, Switzerland: World Health Organization, 2002. http://who.int/whr/2002
Young T, Skatrud J, Peppard PE (2004) Risk factors for obstructive sleep apnea in adults. JAMA 291(16):2013–2016. doi:10.1001/jama.291.16.2013
Peker Y et al (2002) Increased incidence of cardiovascular disease in middle-aged men with obstructive sleep apnea: a 7-year follow-up. Am J Respir Crit Care Med 166(2):159–165. doi:10.1164/rccm.2105124
Silverberg DS, Oksenberg A (2001) Are sleep-related breathing disorders important contributing factors to the production of essential hypertension? Curr Hypertens Rep 3(3):209–215. doi:10.1007/s11906-001-0040-8
Hedner J et al (2006) Hypertension prevalence in obstructive sleep apnoea and sex: a population-based case-control study. Eur Respir J 27(3):564–570. doi:10.1183/09031936.06.00042105
Lugaresi E et al (1980) Some epidemiological data on snoring and cardiocirculatory disturbances. Sleep 3(3–4):221–224
Partinen M, Guilleminault C (1990) Daytime sleepiness and vascular morbidity at seven-year follow-up in obstructive sleep apnea patients. Chest 97(1):27–32. doi:10.1378/chest.97.1.27
Davies CW et al (2000) Case-control study of 24 hour ambulatory blood pressure in patients with obstructive sleep apnoea and normal matched control subjects. Thorax 55(9):736–740. doi:10.1136/thorax.55.9.736
Suzuki M et al (1996) Blood pressure “dipping” and “non-dipping” in obstructive sleep apnea syndrome patients. Sleep 19(5):382–387
Baguet JP et al (2005) Night-time and diastolic hypertension are common and underestimated conditions in newly diagnosed apnoeic patients. J Hypertens 23(3):521–527. doi:10.1097/01.hjh.0000160207.58781.4e
Sharabi Y et al (2003) Diastolic blood pressure is the first to rise in association with early subclinical obstructive sleep apnea: lessons from periodic examination screening. Am J Hypertens 16(3):236–239. doi:10.1016/S0895-7061(02)03250-8
Grote L, Hedner J, Peter JH (2001) Mean blood pressure, pulse pressure and grade of hypertension in untreated hypertensive patients with sleep-related breathing disorder. J Hypertens 19(4):683–690. doi:10.1097/00004872-200104000-00004
Haas DC et al (2005) Age-dependent associations between sleep-disordered breathing and hypertension: importance of discriminating between systolic/diastolic hypertension and isolated systolic hypertension in the Sleep Heart Health Study. Circulation 111(5):614–621. doi:10.1161/01.CIR.0000154540.62381.CF
Sin DD et al (2003) Relationship of systolic BP to obstructive sleep apnea in patients with heart failure. Chest 123(5):1536–1543. doi:10.1378/chest.123.5.1536
Hla KM et al (1994) Sleep apnea and hypertension. A population-based study. Ann Intern Med 120(5):382–388
Nieto FJ et al (2000) Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 283(14):1829–1836. doi:10.1001/jama.283.14.1829
Brooks D et al (1997) Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest 99(1):106–109. doi:10.1172/JCI119120
Fletcher EC (2001) Invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol 90(4):1600–1605
Fletcher EC (2000) Cardiovascular consequences of obstructive sleep apnea: experimental hypoxia and sympathetic activity. Sleep 23(Suppl 4):S127–S131
Bao G et al (1997) Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol 83(1):95–101
Fletcher EC, Bao G (1996) The rat as a model of chronic recurrent episodic hypoxia and effect upon systemic blood pressure. Sleep 19(10, Suppl):S210–S212
Xie A et al (2000) Neurocirculatory consequences of intermittent asphyxia in humans. J Appl Physiol 89(4):1333–1339
Khayat RN et al (2003) Cardiorespiratory effects of added dead space in patients with heart failure and central sleep apnea. Chest 123(5):1551–1560. doi:10.1378/chest.123.5.1551
Katragadda S et al (1997) Neural mechanism of the pressor response to obstructive and nonobstructive apnea. J Appl Physiol 83(6):2048–2054
Arabi Y et al (1999) Daytime blood pressure elevation after nocturnal hypoxia. J Appl Physiol 87(2):689–698
Logan AG et al (2001) High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 19(12):2271–2277. doi:10.1097/00004872-200112000-00022
MacMahon S et al (1990) Blood pressure, stroke, and coronary heart disease. Part 1. Prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet 335(8692):765–774. doi:10.1016/0140-6736(90)90878-9
Pepperell JC et al (2002) Ambulatory blood pressure after therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised parallel trial. Lancet 359(9302):204–210. doi:10.1016/S0140-6736(02)07445-7
Campos-Rodriguez F et al (2007) Long-term effect of continuous positive airway pressure on BP in patients with hypertension and sleep apnea. Chest 132(6):1847–1852. doi:10.1378/chest.07-1478
Robinson GV et al (2006) Continuous positive airway pressure does not reduce blood pressure in nonsleepy hypertensive OSA patients. Eur Respir J 27(6):1229–1235. doi:10.1183/09031936.06.00062805
Somers VK et al (1995) Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96(4):1897–1904. doi:10.1172/JCI118235
Adhikary G et al (2001) Gene regulation during intermittent hypoxia: evidence for the involvement of reactive oxygen species. Adv Exp Med Biol 499:297–302
Prabhakar NR et al (2001) Intermittent hypoxia: cell to system. Am J Physiol Lung Cell Mol Physiol 281(3):L524–L528
Xie A et al (2001) Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol 91(4):1555–1562
Fletcher EC (1997) Sympathetic activity and blood pressure in the sleep apnea syndrome. Respiration 64(Suppl 1):22–28
Lavie L, Vishnevsky A, Lavie P (2004) Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 27(1):123–128
Carpagnano GE et al (2003) 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest 124(4):1386–1392. doi:10.1378/chest.124.4.1386
Deanfield JE, Halcox JP, Rabelink TJ (2007) Endothelial function and dysfunction: testing and clinical relevance. Circulation 115(10):1285–1295
Prior JO et al (2005) Coronary circulatory dysfunction in insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus. Circulation 111(18):2291–2298. doi:10.1161/01.CIR.0000164232.62768.51
Bugiardini R et al (2004) Endothelial function predicts future development of coronary artery disease: a study of women with chest pain and normal coronary angiograms. Circulation 109(21):2518–2523. doi:10.1161/01.CIR.0000128208.22378.E3
De Vriese AS et al (2004) Endothelium-derived hyperpolarizing factor-mediated renal vasodilatory response is impaired during acute and chronic hyperhomocysteinemia. Circulation 109(19):2331–2336. doi:10.1161/01.CIR.0000129138.08493.4D
MacCarthy PA, Shah AM (2000) Impaired endothelium-dependent regulation of ventricular relaxation in pressure-overload cardiac hypertrophy. Circulation 101(15):1854–1860
Grebe M et al (2006) Antioxidant vitamin C improves endothelial function in obstructive sleep apnea. Am J Respir Crit Care Med 173(8):897–901. doi:10.1164/rccm.200508-1223OC
Bostrom KB et al (2007) Interaction between the angiotensin-converting enzyme gene insertion/deletion polymorphism and obstructive sleep apnoea as a mechanism for hypertension. J Hypertens 25(4):779–783
Moller DS et al (2003) Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am J Hypertens 16(4):274–280. doi:10.1016/S0895-7061(02)03267-3
Fletcher EC, Orolinova N, Bader M (2002) Blood pressure response to chronic episodic hypoxia: the renin-angiotensin system. J Appl Physiol 92(2):627–633
Fletcher EC, Bao G, Li R (1999) Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension 34(2):309–314
Rosamond W et al (2007) Heart disease and stroke statistics-2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 115(5):e69–e171. doi:10.1161/CIRCULATIONAHA.106.179918
Nieminen MS et al (2006) EuroHeart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur Heart J 27(22):2725–2736. doi:10.1093/eurheartj/ehl193
Ip MS et al (2004) Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med 169(3):348–353. doi:10.1164/rccm.200306-767OC
Schulz R et al (2000) Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 162(2 Pt 1):566–570
Barcelo A et al (2000) Abnormal lipid peroxidation in patients with sleep apnoea. Eur Respir J 16(4):644–647. doi:10.1034/j.1399-3003.2000.16d13.x
Minoguchi K et al (2005) Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med 172(5):625–630. doi:10.1164/rccm.200412-1652OC
Sorajja D et al (2008) Independent association between obstructive sleep apnea and subclinical coronary artery disease. Chest 133(4):927–933
Hanly P et al (1993) ST-segment depression during sleep in obstructive sleep apnea. Am J Cardiol 71(15):1341–1345. doi:10.1016/0002-9149(93)90552-N
Alonso-Fernandez A et al (2005) Cardiac rhythm disturbances and ST-segment depression episodes in patients with obstructive sleep apnea-hypopnea syndrome and its mechanisms. Chest 127(1):15–22. doi:10.1378/chest.127.1.15
Peled N et al (1999) Nocturnal ischemic events in patients with obstructive sleep apnea syndrome and ischemic heart disease: effects of continuous positive air pressure treatment. J Am Coll Cardiol 34(6):1744–1749. doi:10.1016/S0735-1097(99)00407-6
Peters RW (2005) Obstructive sleep apnea and cardiovascular disease. Chest 127(1):1–3. doi:10.1378/chest.127.1.1
Schafer H et al (1997) Sleep-related myocardial ischemia and sleep structure in patients with obstructive sleep apnea and coronary heart disease. Chest 111(2):387–393. doi:10.1378/chest.111.2.387
Beguin PC et al (2005) Acute intermittent hypoxia improves rat myocardium tolerance to ischemia. J Appl Physiol 99(3):1064–1069. doi:10.1152/japplphysiol.00056.2005
Joyeux-Faure M et al (2005) Chronic intermittent hypoxia increases infarction in the isolated rat heart. J Appl Physiol 98(5):1691–1696. doi:10.1152/japplphysiol.01146.2004
Shahar E et al (2001) Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med 163(1):19–25
Peker Y et al (2000) Respiratory disturbance index: an independent predictor of mortality in coronary artery disease. Am J Respir Crit Care Med 162(1):81–86
Mooe T et al (2001) Sleep-disordered breathing and coronary artery disease: long-term prognosis. Am J Respir Crit Care Med 164(10 Pt 1):1910–1913
Yumino D et al (2007) Impact of obstructive sleep apnea on clinical and angiographic outcomes following percutaneous coronary intervention in patients with acute coronary syndrome. Am J Cardiol 99(1):26–30. doi:10.1016/j.amjcard.2006.07.055
Milleron O et al (2004) Benefits of obstructive sleep apnoea treatment in coronary artery disease: a long-term follow-up study. Eur Heart J 25(9):728–734. doi:10.1016/j.ehj.2004.02.008
Javaheri S (2000) Effects of continuous positive airway pressure on sleep apnea and ventricular irritability in patients with heart failure. Circulation 101(4):392–397
Mehra R et al (2006) Association of nocturnal arrhythmias with sleep-disordered breathing: the Sleep Heart Health Study. Am J Respir Crit Care Med 173(8):910–916. doi:10.1164/rccm.200509-1442OC
Ryan CM et al (2005) Effect of continuous positive airway pressure on ventricular ectopy in heart failure patients with obstructive sleep apnoea. Thorax 60(9):781–785. doi:10.1136/thx.2005.040972
Gilman MP et al (2008) Continuous positive airway pressure increases heart rate variability in heart failure patients with obstructive sleep apnoea. Clin Sci (Lond) 114(3):243–249. doi:10.1042/CS20070172
Gami AS et al (2007) Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol 49(5):565–571. doi:10.1016/j.jacc.2006.08.060
Gami AS et al (2005) Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 352(12):1206–1214. doi:10.1056/NEJMoa041832
Yaggi HK et al (2005) Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 353(19):2034–2041. doi:10.1056/NEJMoa043104
Mehra R, Benjamin EJ, Shahar E et al (2006) Association of nocturnal arrhythmias with sleep-disordered breathing: the sleep heart health study. Am J Respir Crit Care Med 173(8):910–916
Ferrier K et al (2005) Sleep-disordered breathing occurs frequently in stable outpatients with congestive heart failure. Chest 128(4):2116–2122. doi:10.1378/chest.128.4.2116
Javaheri S (2006) Sleep disorders in systolic heart failure: a prospective study of 100 male patients. The final report. Int J Cardiol 106(1):21–28. doi:10.1016/j.ijcard.2004.12.068
Javaheri S et al (1998) Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations. Circulation 97(21):2154–2159
Mossey P, Singh GD, Smith ME (1999) More extensive analysis is needed when assessing facial structure in SIDS. BMJ (Clin Res Ed.) 318(7180):396–397
Sin DD et al (1999) Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 160(4):1101–1106
Hedner J, Ejnell H, Caidahl K (1990) Left ventricular hypertrophy independent of hypertension in patients with obstructive sleep apnoea. J Hypertens 8(10):941–946. doi:10.1097/00004872-199010000-00009
Laaban JP et al (2002) Left ventricular systolic dysfunction in patients with obstructive sleep apnea syndrome. Chest 122(4):1133–1138. doi:10.1378/chest.122.4.1133
Parker JD et al (1999) Acute and chronic effects of airway obstruction on canine left ventricular performance. Am J Respir Crit Care Med 160(6):1888–1896
Tkacova R et al (1998) Effects of continuous positive airway pressure on obstructive sleep apnea and left ventricular afterload in patients with heart failure. Circulation 98(21):2269–2275
Chen L, Shi Q, Scharf SM (2000) Hemodynamic effects of periodic obstructive apneas in sedated pigs with congestive heart failure. J Appl Physiol 88(3):1051–1060
Scharf SM, Graver LM, Balaban K (1992) Cardiovascular effects of periodic occlusions of the upper airways in dogs. Am Rev Respir Dis 146(2):321–329
Chen L, Scharf SM (1998) Systemic and myocardial hemodynamics during periodic obstructive apneas in sedated pigs. J Appl Physiol 84(4):1289–1298
Spaak J et al (2005) Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension 46(6):1327–1332. doi:10.1161/01.HYP.0000193497.45200.66
Usui K et al (2005) Inhibition of awake sympathetic nerve activity of heart failure patients with obstructive sleep apnea by nocturnal continuous positive airway pressure. J Am Coll Cardiol 45(12):2008–2011. doi:10.1016/j.jacc.2004.12.080
Malone S et al (1991) Obstructive sleep apnoea in patients with dilated cardiomyopathy: effects of continuous positive airway pressure. Lancet 338(8781):1480–1484. doi:10.1016/0140-6736(91)92299-H
Mansfield DR, Naughton MT (2004) Sleep apnea and congestive heart failure. Minerva Med 95(4):257–280
Roebuck T et al (2004) Increased long-term mortality in heart failure due to sleep apnoea is not yet proven. Eur Respir J 23(5):735–740. doi:10.1183/09031936.04.00060404
Bady E et al (2000) Pulmonary arterial hypertension in patients with sleep apnoea syndrome. Thorax 55(11):934–939. doi:10.1136/thorax.55.11.934
Sajkov D et al (1994) Pulmonary hypertension and hypoxemia in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 149(2 Pt 1):416–422
Sanner BM et al (1997) Pulmonary hypertension in patients with obstructive sleep apnea syndrome. Arch Intern Med 157(21):2483–2487. doi:10.1001/archinte.157.21.2483
Chaouat A et al (1996) Pulmonary hemodynamics in the obstructive sleep apnea syndrome. Results in 220 consecutive patients. Chest 109(2):380–386. doi:10.1378/chest.109.2.380
Weitzenblum E et al (1988) Daytime pulmonary hypertension in patients with obstructive sleep apnea syndrome. Am Rev Respir Dis 138(2):345–349
Atwood CW Jr et al (2004) Pulmonary artery hypertension and sleep-disordered breathing: ACCP evidence-based clinical practice guidelines. Chest 126(1 Suppl):72S–77S. doi:10.1378/chest.126.1_suppl.72S
Gozal D, Lipton AJ, Jones KL (2002) Circulating vascular endothelial growth factor levels in patients with obstructive sleep apnea. Sleep 25(1):59–65
Lavie L et al (2002) Plasma vascular endothelial growth factor in sleep apnea syndrome: effects of nasal continuous positive air pressure treatment. Am J Respir Crit Care Med 165(12):1624–1628. doi:10.1164/rccm.20110-040OC
Berman EJ et al (1991) Right ventricular hypertrophy detected by echocardiography in patients with newly diagnosed obstructive sleep apnea. Chest 100(2):347–350. doi:10.1378/chest.100.2.347
Sanner BM et al (1997) Right ventricular dysfunction in patients with obstructive sleep apnoea syndrome. Eur Respir J 10(9):2079–2083. doi:10.1183/09031936.97.10092079
Nahmias J, Lao R, Karetzky M (1996) Right ventricular dysfunction in obstructive sleep apnoea: reversal with nasal continuous positive airway pressure. Eur Respir J 9(5):945–951. doi:10.1183/09031936.96.09050945
Alchanatis M et al (2001) Daytime pulmonary hypertension in patients with obstructive sleep apnea: the effect of continuous positive airway pressure on pulmonary hemodynamics. Respiration 68(6):566–572. doi:10.1159/000050574
Sajkov D et al (2002) Continuous positive airway pressure treatment improves pulmonary hemodynamics in patients with obstructive sleep apnea. Am J Respir Crit Care Med 165(2):152–158
Bassetti CL, Milanova M, Gugger M (2006) Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
Foster GE et al (2007) Effects of continuous positive airway pressure on cerebral vascular response to hypoxia in patients with obstructive sleep apnea. Am J Respir Crit Care Med 175(7):720–725. doi:10.1164/rccm.200609-1271OC
Phillips SA et al (2004) Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol 286(1):H388–H393. doi:10.1152/ajpheart.00683.2003
Baguet JP et al (2005) The severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrence. Chest 128(5):3407–3412. doi:10.1378/chest.128.5.3407
Suzuki T et al (2004) Obstructive sleep apnea and carotid-artery intima-media thickness. Sleep 27(1):129–133
Drager LF et al (2007) Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 176(7):706–712. doi:10.1164/rccm.200703-500OC
Yaggi H, Mohsenin V (2004) Obstructive sleep apnoea and stroke. Lancet Neurol 3(6):333–342. doi:10.1016/S1474-4422(04)00766-5
Turkington PM, Elliott MW (2004) Sleep disordered breathing following stroke. Monaldi Arch Chest Dis 61(3):157–161
Parra O et al (2000) Time course of sleep-related breathing disorders in first-ever stroke or transient ischemic attack. Am J Respir Crit Care Med 161(2 Pt 1):375–380
Selic C et al (2005) Blood pressure evolution after acute ischemic stroke in patients with and without sleep apnea. Stroke 36(12):2614–2618. doi:10.1161/01.STR.0000189689.65734.a3
Parra O et al (2004) Sleep-related breathing disorders: impact on mortality of cerebrovascular disease. Eur Respir J 24(2):267–272. doi:10.1183/09031936.04.00061503
Martinez-Garcia MA et al (2005) Continuous positive airway pressure treatment in sleep apnea prevents new vascular events after ischemic stroke. Chest 128(4):2123–2129. doi:10.1378/chest.128.4.2123
Dziewas R et al (2005) Increased prevalence of sleep apnea in patients with recurring ischemic stroke compared with first stroke victims. J Neurol 252(11):1394–1398. doi:10.1007/s00415-005-0888-7
Kaneko Y et al (2003) Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 26(3):293–297
Arzt M et al (2005) Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med 172(11):1447–1451. doi:10.1164/rccm.200505-702OC
Good DC et al (1996) Sleep-disordered breathing and poor functional outcome after stroke. Stroke 27(2):252–259
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Devulapally, K., Pongonis, R. & Khayat, R. OSA: the new cardiovascular disease. Heart Fail Rev 14, 155–164 (2009). https://doi.org/10.1007/s10741-008-9101-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-008-9101-2