
Abstract
Introduction
PKCδ is ubiquitously expressed in mammalian cells and its dysregulation plays a key role in the onset of several incurable diseases and metabolic disorders. However, much remains unknown about the metabolic pathways and disturbances induced by PKC deficiency, as well as the metabolic mechanisms involved.
Objectives
This study aims to use metabolomics to further characterize the function of PKC from a metabolomics standpoint, by comparing the full serum metabolic profiles of PKC deficient mice to those of wild-type mice.
Methods
The serum metabolomes of PKCδ knock-out mice were compared to that of a wild-type strain using a GCxGC-TOFMS metabolomics research approach and various univariate and multivariate statistical analyses.
Results
Thirty-seven serum metabolite markers best describing the difference between PKCδ knock-out and wild-type mice were identified based on a PCA power value > 0.9, a t-test p-value < 0.05, or an effect size > 1. XERp prediction was also done to accurately select the metabolite markers within the 2 sample groups. Of the metabolite markers identified, 78.4% (29/37) were elevated and 48.65% of these markers were fatty acids (18/37). It is clear that a total loss of PKCδ functionality results in an inhibition of glycolysis, the TCA cycle, and steroid synthesis, accompanied by upregulation of the pentose phosphate pathway, fatty acids oxidation, cholesterol transport/storage, single carbon and sulphur-containing amino acid synthesis, branched-chain amino acids (BCAA), ketogenesis, and an increased cell signalling via N-acetylglucosamine.
Conclusion
The charaterization of the dysregulated serum metabolites in this study, may represent an additional tool for the early detection and screening of PKCδ-deficiencies or abnormalities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Incurable diseases, metabolic disorders (such as diabetes, hypertension, dyslipidemia, newborn inborn errors of metabolism, and ethylmalonic encephalopathy), infectious diseases, and a plethora of other autoimmune diseases, are becoming increasingly prevalent globally. According to available data, chronic noncommunicable diseases continue to be a global health concern. The world health organization (WHO) estimates that about 41 million deaths are due to non-communicable diseases (NCDs), accounting for 71% of all global deaths per annum (WHO, 2021b). Additionally, the Covid-19 pandemic has recently exacerbated the health-care system’s multimorbidity burden. (Azarpazhooh et al., 2020; WHO, 2021a; Wood et al., 2021).
Many enzyme deficiencies/abnormalities have been associated with the aforementioned metabolic disorders or metabolism-related diseases, plaguing humanity, none more so, however, than protein kinase C delta (PKCδ), a member of the PKC serine or threonine kinases family (Brodie & Blumberg, 2003). To date, three categories of ten PKC isozymes/families have been identified (conventional, novel, and atypical), and their activation differs significantly. (Fields & Gustafson, 2003; Zhao et al., 2012). These PKC isozymes frequently perform overlapping, but sometimes opposing, functions (Black & Black, 2013; Dempsey et al., 2000), as they differ in structure, function, and biochemical properties (e.g. requirements for co-factors and calcium) (Brodie & Blumberg, 2003; Newton, 2001). Diacylglycerol, DAG (a lipid second messenger), and calcium, activate the conventional PKCs (PKCα, PKCβI and PKCβ, and PKCγ); the novel PKCs (PKCδ, PKCε, PKCθ, and PKCη) are activated solely by DAG, whereas the atypical PKCs (PKCζ and PKCι/λ) are neither activated by lipid second messengers or calcium. (Black & Black, 2013; Poole et al., 2004; Zhao et al., 2012). PKCs have been implicated in numerous cellular processes, including transcriptional regulation, membrane structure modulation, immune mediation, and cell growth regulation (Carey et al., 2020; Lien et al., 2021; Poole et al., 2004; Salzer et al., 2016). Although the full functionality of PKCδ is yet to be determined, it is the first identified member of the novel PKC subfamily to be largely expressed in mammalian cells, including macrophages (Wang et al., 2020; Zhao et al., 2012). While PKCδ generally functions as a pro-apoptotic protein during DNA damage-induced apoptosis, it also acts as an anti-apoptotic protein during receptor-initiated cell death (Salzer et al., 2016). PKCδ’s functionality depends on various factors, including its localization, tyrosine phosphorylation, and the presence of other pro- and anti-apoptotic signaling molecules (Basu & Pal, 2010).
PKCδ dysregulation contributes to the onset and aggravation of several non-communicable disease conditions including (1) LPS-induced osteolysis and human lupus [systemic lupus erythematosus (SLE)], (Gorelik et al., 2015; Khor et al., 2013), (2) diabetes and other obesity-related complications (Lien et al., 2021; Sparks et al., 2019), (3) Alzheimer’s (AD) and Parkinson’s disease (PD), (Du et al., 2018; Gordon et al., 2016). Remarkably, people with autoimmune diseases, such as a PKCδ-deficiency or defect, also typically have other invasive, recurring, and/or severe infections. In particular, PKCδ-deficient patients are more susceptible to viral, bacterial, and fungal infections, mostly affecting the lung, gastrointestinal tract, and lymph nodes (Kuehn et al., 2013; Nanthapisal et al., 2017; Salzer et al., 2016; Sharifinejad et al., 2020). PKCδ’s conflicting roles in cell survival and cell death signaling (acting either pro-survival and/or pro-apoptotic during cellular activities), makes it a potential target in the treatment and diagnosis of many disease conditions, including the aforementioned. PKCδ inactivation has been shown in vivo to (1) protect organs, (2) improve pathogen clearance, and (3) improve survival during sepsis. (Liverani et al., 2020). Hence PKCδ inhibition may represent a valuable target for innovative therapeutic strategies, for example, in the study by Choi et al., (2013), PKCδ inhibition reversed all pathophysiologic symptoms of asthma. Dysregulated PKCδ expression has also been shown to play a regulatory role in tumorigenesis (tumor suppression and cancer proliferation) (Allen-Petersen et al., 2014; Berardi et al., 2016; Chen et al., 2011). Herein, PKCδ isozyme inactivation may also represent a new molecular target for cancer stem cell populations (CSC-like populations).
Most PKCδ research to date (cellular process regulation, interference, inhibition, etc.) has been done using various histological, molecular, genomics, proteomics, and transcriptomics techniques (Berardi et al., 2016; Contreras et al., 2012; Gorelik et al., 2015; Nanthapisal et al., 2017). The paucity of metabolomics investigations into the functionality of PKC, either independently and/or in conjunction with other omics techniques, however, necessitates this study. This also follows the understanding that the potential use of PKC isoforms, including PKCδ, as a target for therapeutic intervention, still faces diverse challenges. Furthermore, to better understand the effects or role of PKCδ on the mammalian metabolome, the myriad of metabolites and metabolic changes associated with its dysregulation should firstly be evaluated, as was done in the current investigation. Since various host-directed therapeutic approaches continue to show promising advantages in the management of many non-communicable (e.g. PKCδ deficiency and PKCδ-comorbidities) and communicable diseases (e.g. tuberculosis, covid-19, HIV), the identification of new biomarkers is now readily recognized as important not only for diagnostics but also to better disease characterization, and in so doing, directing the development of new therapies against many of these incurable diseases plaguing humanity (Adeniji et al., 2020; Baindara et al., 2020; Bausch et al., 2021; Mahon & Hafner, 2017).
Metabolomics is a rather new, yet well-established research technology, employed towards clinical biomarker discovery, for better elucidating pathological processes (Beyoğlu et al., 2013; Bonte et al., 2019) and the dynamic metabolic responses of substances in the mammalian metabolome (Stander et al., 2020). These important clinical-metabolomic investigations range from elucidating differences between healthy and diseased states (Dahabiyeh et al., 2021; Hao et al., 2017), to the quantitative measurement of endogenous disruptions induced by exogenous substances introduced to a host system (Knoll et al., 2021). Considering this, PKCδ is a host factor important for disease control, while also serving as a marker of disease progression in humans (Parihar et al., 2018; Wang et al., 2020). This study seeks to use metabolomics in order further characterize the functionality of PKCδ from a metabolomics perspective, by comparing the full serum metabolic profiles of naïve PKCδ deficient mice with that of its wild-type mouse counterpart. The metabolomic analysis presented here not only corroborates the findings of other omics strategies but also reveals novel PKCδ functionality.
2 Methods
The experiment and metabolomics analysis were conducted following the procedures previously described by Williams et al. (2006), Du Preez and Loots (2013), and Parihar et al. (2018). PKCδ deficient and wild-type mice on the 129 Sv genetic background (8–12 weeks), were maintained under specific-pathogen-free conditions in individually ventilated cages. Five animals per group were used to collect serum samples from naïve mice. All experiments were performed in accordance with the South African National Guidelines and University of Cape Town practice for laboratory animal procedures. The protocol (AEC: 012/036) was approved by the Animal Ethics Committee, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa.
2.1 Reagents and chemicals
Deionised water from a Millipore MilliQ purification system was used in all cases. Acetonitrile, Optima grade, was purchased from Fisher Scientific (Pittsburgh, USA). All other reagents and organic solvents used for this investigation were purchased from Sigma–Aldrich, St. Louis, MO, USA, unless specified otherwise.
2.2 GC x GC-TOFMS metabolome analysis of PKCδ deficient and wild-type mice serum
2.2.1 Sample preparation and extraction
Extraction of the collected serum samples from PKCδ deficient and wild-type mice were performed as previously described by Williams et al. (2006), using 50 µL of 0.45 µg/mL 3-phenylbutyric acid (Sigma-Aldrich), as an internal standard, before GC x GC-TOFMS analyses. Briefly, the serum samples (100 mL) were protein precipitated using 3 volumes of acetonitrile and the sample was then centrifuged at 3000 rpm for 10 min at room temperature, then 100 mL of each supernatant was evaporated to dryness under vacuum, prior to derivatization. In addition, before analysis, 50 mL of each sample were pooled to generate a quality control (QC) sample stock solution and, following protein precipitation, 100 mL of this pooled sample was also evaporated to dryness. All the dried samples were derivatized with 20 mL methoxylamine hydrochloride (40 mg/mL in pyridine) at 28 °C for 90 min followed by 180 mL MSTFA at 37 °C for 30 min. All experimental samples were randomly assigned to, and analyzed in batches of 10 samples, together with 1 aliquot of pooled QC sample per batch, injected in the beginning, middle and end of each batch.
2.2.2 GC x GC-TOFMS analysis
Chromatographic analyses of the derivatized samples were performed using a Pegasus GC x GC-TOFMS (Leco Corporation), utilizing an Agilent 7890 A GC (Agilent) coupled to a time of flight mass spectrometer (TOFMS) (Leco Corporation) and a Gerstel Multi-Purpose Sampler (MPS) (Gerstel GmbH & co. KG) as described previously, with minor modifications. This included that a split ratio of 1:2 was used, a Restek Rxi-17 (1 m, 0.25 mm i.d., 0.25 mm d.f.) column for the second dimension separation, and the secondary oven programmed with an offset of + 15 °C, increasing at 4.5 °C per min to a final temperature of 300 °C. Cryogenic modulations and a hot pulse of nitrogen gas of 0.5 s, every 3 s was used to control the effluent emerging from the primary column onto the secondary column. Detection was achieved by using MS detection in full scan mode (m/z 50–800). Leco Corporation ChromaTOF software (v4.5) was used for peak finding and mass spectral deconvolution at an S/N ratio of 50, with a minimum of 2 apexing peaks. Peak identification and alignment was done as previously described by Du Preez and Loots (2013). To select metabolites, a comparison of mass fragment patterns of metabolites identified with those in the commercially available databases (National Institute of Standards and Technology (NIST) 2020 and Whiley v12), containing previously injected standards was done. All compounds were normalized relative to the internal standard by calculating the relative concentrations of each.
2.3 Statistical analysis
The dataset presented for statistical analysis comprised the relative concentrations of 581 compounds for QC, WT, and KO samples. To more closely match the assumptions made by the various statistical techniques used, the data were pre-treated using a standardized metabolomics data clean-up procedure (Gromski et al., 2014; Sullivan & Feinn, 2012; Wei et al., 2018). As stated above, analytes and compounds with more than 50% zero values in each group were excluded from further analysis. The remaining zero values were replaced by random numbers generated from the left tail of a beta distribution fitted to the data. The data were log-transformed to improve normality for parametric univariate analysis and subsequently converted to z-scores for multivariate analysis, ensuring the abundance of a compound does not dictate its importance, as abundance does not necessarily imply biological importance (van den Berg et al., 2006). Multivariate models and univariate statistics were used in a complementary manner, as univariate analyses are not affected by uninformative, yet competing variables, while multivariate approaches leverage the joint ability of variables to explain variation (Saccenti et al., 2014).
Univariately, both the statistical (independent t-test p-values adjusted for multiple testing to control the false discovery rate) and practical significance (Cohen’s d-value) of group differences were evaluated. However, given the large number of compounds and the necessity to correct for multiple testing, great emphasis was placed on practical significance. Furthermore, the impact of different transformations can be dramatic yet difficult to evaluate, therefore, XERp, a univariate method requiring no pre-processing, was also included (Van Reenen et al., 2017). The primary model for multivariate analysis of group differences was partial least squares–discriminant analysis (PLS-DA). As this supervised method is a known struggle given unseen data, different validation strategies were implemented alongside. Besides a leave-one-out cross-validation of the model’s predictive stability, principal components analysis (PCA) (Brereton, 2003) was also performed to identify the compounds associated with the largest amount of variability observed and how that relates to the groups. PCA was also used to evaluate the repeatability of GC x GC-TOFMS analysis by comparing the variability between QC aliquots and research samples. All data preparation and analyses were performed in Matlab using the Statistics toolbox (MATLAB, 2012b) and the PLS packages from Eigenvector (Eigenvector, 2016).
3 Results
3.1 Data overview
The GC x GC-TOFMS system detected on average over 1000 compounds per sample. This data set was cleaned/reduced by removing “unknowns” (compounds with no corresponding mass spectral matches in the libraries), column and/or reagent-related compounds (non-biological), and merging duplicate peaks of putative compounds (e.g. sugars and amino acids), 581 usable compounds remained. This data set containing the 581 compounds (excluding internal standard) were then subjected to the aforementioned processing steps [i.e. quality control coefficient of variation (QC-CV) filter etc.] and further statistical analyses.
3.2 Quality control
A PCA model was constructed for all data including QC repeated observations of a sample. A scores plot labelled as research samples and QC observations was drawn (Fig. 1). A PCA model was constructed for all data including QC repeated observations of a sample. A scores plot labelled as research samples and QC observations was drawn (Fig. 1). The differentiating grouping of the QC observations with respect to the experimental samples is most likely due to the high number of metabolites detected in low abundance, which would have been further reduced due to pooling to construct the pooled QC sample. Almost half of the QC observations were zeroes (49%), which is why zero filtering was approached with care (refer to the section on Statistical Analysis) in this instance. In evaluating the quality of the raw data, we are therefore content to see a good clustering of QC samples, which indicates negligeable shifts between batches and machine drift within batches. Furthermore, all samples were randomised to batches as well as within batches, therefore any analytical variation would have been present in both experimental groups, and as such is accounted for.

Principal component analysis scores plot for all experimental and quality control samples
3.3 Multivariate statistical results
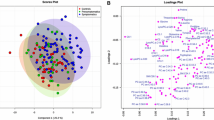
Partial least-squares discriminant analysis (PLS-DA) showed excellent separation (Fig. 2a) and predictive accuracy, however, due to the small group sizes, the model was unstable. Principal component analysis (PCA) showed comparable separation (Fig. 2b) and considering that this is an unsupervised method, the variable rankings underpinning the PCA model was preferred for variable selection. PCA ranks variables in terms of their role in explaining the variability captured by selected latent variables, in these cases, latent variables 1 through 3. The ranking is quantified into a power metric which was used for variable selection. The rule-of-thumb is that variables with power values exceeding 0.5 contribute sufficiently to the model to be of interest. Considering the small group sizes and the fact that a variable’s contribution to a PCA model does not necessarily correlate to its predictive ability (PCA models are unsupervised and so do not attempt to explain group membership, but rather general variability in the data), the rule was made stricter (power > 0.9) and the focus shifted to univariate statistics.

Multivariate models for wild-type (NWT) versus PKCδ deficient mice (NKO) after gene deletion. a Partial least-squares discriminant analysis (PLS-DA) scores plot of the first three latent variables (LV). Percentages represent the proportion of variation explained in the observed data (X) and the group assignment (Y) by the specific LV. b Principal component analysis (PCA) scores plot for the first three principal components (PC). Percentages in brackets represent the proportion of variation explained in the observed data by the specific PC
3.4 Univariate statistical results
Two univariate approaches were used for variable selection: (1) Cohen’s d-value in its strictest form, i.e. using the maximum standard deviation as the denominators, and (2) variables identified using XERp showing predictive potential. The most common approach in this setting would be the independent samples t-test, which was also applied here. However, the design lacked power and so limited significance was retained after correcting for multiple testing over 500 variables (Fig. 3). Effect sizes can still be informative when statistical power is insufficient (Sullivan & Feinn, 2012) and as such was the focus for variable selection. A cut-off of > 0.8 was set for Cohen’s d-values which is indicative of a practically significant difference between groups.

A scatter plot of log scaled p-values from independent samples t-tests for each compound against Cohen’s d-value where the sign is indicative of up- or down-regulating effect. P-values have not been adjusted for multiple testing
Cohen’s d-value assumes normality and even though these calculations were done using log-transformed data after zero replacement, it is not possible to ensure this assumption is met. Therefore, Cohen’s d was supplemented by XERp (van Reenen et al., 2017), a method making no distributional assumptions with the ability to also assess the importance of differences in the proportion of zeros. XERp follows a leave-one-out approach, selecting variables for each iteration based on their predictive accuracy within each group. Compounds selected in all iterations, with a classification accuracy of at least 80% for both groups, were considered informative.
3.5 Marker selection
The metabolite markers (n = 37) best describing the variance between PKCδ deficient and wild-type mice were selected based on compliance with the following criteria: a PCA power value > 0.9, a t-test p-value < 0.05, or an effect size > 1. The selected metabolite markers are listed according to their metabolic pathway activity Table 1, along with their respective average concentrations and univariate test outcomes. The metabolome disruptions (Table 1) affected several metabolites related to (i). amino acid metabolism and ketogenesis (α-ketoisocaproic acid, α-ketoisovaleric acid, leucinic acid, serine, and β-hydroxybutyric acid), (ii). carbohydrate metabolism (glucose, 2-phosphogylceric acid, pyruvic acid, mannose, N-acetyl-d-glucosamine, galactose, β-d-glucopyranuronic acid, arabitol, 5-deoxyribitol, erythritol, ribitol, and xylitol), (iii). the tricarboxylic acid [TCA] cycle (malic acid), (iv). fatty acid metabolism (citraconic acid, dodecanedioic acid, ethylmalonic acid, glycerol 1-palmitic acid, hexadecanedioic acid, 3-hydroxydodecanedioic acid, 3-hydroxysebacic acid, linolenic acid, malonic acid, palmitic acid, sebacic acid, and tetradecanedioic acid), (v). cholesterol and steroid hormone synthesis (cholesteryl acetate, mevalonic acid, 4α-methylzymosterol, and 17 α-hydroxypregnenolone), and (vi). fatty acid amides (dodecanamide and oleamide) (Fig. 4). Of the total, 78.4% (29/37) of the markers were elevated; and 48.65% of these were associated with fatty acid metabolism (18/37). Biological functions and metabolic pathways of these metabolites were deduced using databases such as SMPDB (The Small Molecule Pathway Database), Human Metabolome Database (HMDB), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, MetaCyc Metabolic Pathway Database, PubChem, and Chemspider.

Figure created with https://biorender.com/
4 Discussion
The biochemical analysis of tissue, serum, plasma, whole blood, urine, sweat, and salivary samples, holds great promise for the discovery of biomarkers of disease progression and response to therapy (Tounta et al., 2021; Zhang et al., 2015). Whilst some studies have revealed that a PKCδ-deficiency/inactivation results in an increased susceptibility to other invasive, recurrent, and/or severe infections, other in vivo investigations have shown that PKCδ inactivation was associated with: (1) enhanced organ protection, (2) rapid pathogen eradication, and (3) improved sepsis survival (Liverani et al., 2020). In this investigation, several metabolites were significantly altered in the PKCδ knock-out mice serum, which when interpreted in the context of the previously published literature, gives clues to better elucidate the function of PKCδ, and its possible role in many communicable and non-communicable diseases. As seen in Fig. 2, our results show a clear group separation and sample clustering between the PKCδ deficient and wild-type mice. The changes to the metabolites detected in the PKCδ-deficient mice serum (Table 1), included dodecanedioic acid, tetradecanedioic acid, sebacic acid, ethylmalonic acid, leucinic acid, mannose, galactose, 3-hydroxysebacic acid, serine, ribitol, pyruvic acid, cholesteryl acetate, glucose, alpha-ketoisovaleric acid and malonic acid, alterations of which have been previously implicated in many metabolic deficiencies and diseases. However, before we describe in detail the metabolic changes induced in the PKCδ knockout (Fig. 4), a general observation needs to be mentioned. PKC activation has been previously associated with increased breakdown of glucose, and both elevated ATP and insulin production (Salway, 2017). PKCδ has also been implicated in the functionality of NADPH oxidase 5 (Nitti et al., 2007, 2010; Thamilselvan et al., 2009)—therewithal, NADPH oxidase 5 catalyses the conversion of NADPH to NADP (Fisher & Zhang, 2006). Consequently, PKCδ deactivation, as was the case in this investigation, would be expected to result in a reduced NADH:NAD ratio (supported by a study done by Mayr et al. (2004), and an elevated NADPH:NADP ratio. Since these cofactors are associated with several enzyme reactions, a change in their rations will result in a change in the metabolic flux of certain enzyme reactions or even more so, entire metabolic pathways. Considering this in the context of a general metabolic overview, one would expect to see inhibition of glycolysis and fatty acid synthesis, and subsequently activation of alternative metabolic pathways, to supply the necessary intermediates (acetyl-CoA to the TCA cycle) for energy production, such as via elevated β-oxidation, ketogenesis, and branched chain amino acid metabolism.
Apart from this general change in metabolic flux expected due to the change in the aforementioned ratios, PKCδ has been directly associated with the expression of many specific enzymes. Although the elevated levels of glucose (and additionally mannose and galactose), 2-phosphoglycerate, and pyruvate observed in the PKCδ deficient mice serum in this study supports the aforementioned inhibition of glycolysis, Mayr et al. (2004), additionally indicated that a PKCδ deficiency results in a marked decrease in the enzyme activities of glycerol-3-phosphate dehydrogenase, pyruvate kinase 3, and lactate dehydrogenase, which directly would result in the elevated glucose (and additionally mannose and galactose), 2-phosphoglycerate, and pyruvate seen in our investigation. The increased activity of a malic enzyme, as previously reported by Mayr et al. (2004), further contributes to the elevated pyruvic acid and the increased NADPH:NADP ratio, previously mentioned. Furthermore, the accumulation of pyruvate is also an indication of inhibition of the TCA cycle in the PKCδ deficient mice as previously mentioned. This is most likely due to the inhibition of α-ketoglutarate dehydrogenase as previously reported by Mayr et al. (2004), which would result in inhibition of the TCA cycle post α-ketoglutarate, and possibly also result in an accumulation of the latter metabolite (although not detected as a marker in this investigation using the strict stats criteria for marker selection). Elevated levels of another TCA cycle intermediate; malic acid, was however observed and can be ascribed to be derived from the elevated pyruvate, via oxaloacetate, catalysed via the enzymes pyruvate carboxylase and malate dehydrogenase (Garrett & Grisham, 2016), and also via other complementary pathways feeding into the TCA cycle at succinic acid (e.g. oxidative glutamine metabolism) (Yang et al., 2014). During glucose metabolism impairment (such as during a mitochondrial pyruvate import blockage), cell survival is dependent on glutamate dehydrogenase (Yang et al., 2009), whilst glutamine-derived malate can then be converted into pyruvate via malic enzyme 2 (Salway, 2017; Yang et al., 2014), which was previously also seen to be altered in a PKCδ deficient model (Mayr et al., 2004). Furthermore, single carbon metabolism and sulphur-containing metabolic pathways (serine-cysteine metabolism) can also account for the elevated pyruvic acid in this study, driven by the elevated concentrations of the aforementioned 2 phosphoglyceric acid and serine detected in this investigation (Salway, 2017), and also a source for the elevated concentrations of the ethylmalonic acid detected in the PKCδ deficient mice (Barth et al., 2010).
Out of the 5 sugar alcohols or polyols (erythritol, arabitol, ribitol, xylitol, and 5-deoxyribitol) detected in the serum of the PKCδ deficient mice, only ribitol showed a marked decrease. Although detection of polyhydric alcohols (e.g. polyols) (in the human metabolome), has been mostly attributed to dietary sources (e.g. fruits, vegetables, and nuts), polyols are derived from aldoses and ketoses through NADPH reduction (Gao et al., 2012). PKC deactivation causes elevated NADPH (Bey et al., 2004), with activation of the polyol pathway, which is NADPH dependent (Wolf et al., 2008). Limited information exists on 5-deoxyribitol, however, in rats, it has been produced from the metabolic degradation of 5-deoxyribose 1-phosphate (dR1P) (Ichihara et al., 1985; Sekowska et al., 2019). In this study, the increased polyols were possibly due to the aforementioned general glycolytic system dysfunction, which correlates with the study of Gao et al. (2012) and Sato et al. (1999). The elevated concentrations of galactose corroborate the increased concentrations of the polyols. Elevated polyols with glucose, mannose, and oleamide, along with decreased urinary sugars (fructose and galactose) have been associted with amplified aldose reductase (AR) activity (Semmo et al., 2015). Naturally, AR has a low affinity for glucose (limiting the conversion of glucose to polyols) (Tanenberg & Donofrio, 2008), however, in high glucose levels/hyperglycaemia conditions, the polyol pathway converts glucose into sugar polyols using aldose reductase and is accompanied by NADPH oxidation to NADP+ (Laaksonen & Sen, 2000). Polyol pathway stimulation vis-à-vis elevated polyol levels, could promote oxidative stress/hyperosmotic stress in cells and possibly PKC reactivation as well (Jeng-Miller & Baumal, 2018; Lorenzi, 2007), however reactivation of this wouldn’t be possible in the PKCδ knock out mice used in this investigation.
Significantly evaluated levels of N-acetyl glucosamine were also detected and have previously been associated with induction of the galactose metabolic pathway (Kamthan et al., 2013), and both metabolites are rate-limiting intermediates in the biosynthesis of glycoproteins and proteoglycans (Dani & Broadie, 2012). N-Acetyl-d-glucosamine is also considered important for the formation of articular cartilage glycosaminoglycans, glycoproteins, and proteoglycans (Dunsmore, 2006; Wagermaier & Fratzl, 2012), hence, its use as a dietary supplement in osteoarthritis therapy (Jain et al., 2016; Kubomura et al., 2017). Furthermore, N-acetyl-d-glucosamine is a key component of the extracellular matrix of animal cells, influencing cell signalling through the post-translational modification by glycosylation, of a variety of intracellular proteins, including NFκB, c-myc, and p53 (Konopka, 2012).
The fatty acids as a group were the most significantly dysregulated metabolite class in the study and this indicates the strong role of PKCδ in fatty acid metabolism or the upregulation of β-oxidation as previously described (Greene et al., 2014; Schmitz-Peiffer, 2013; Yang et al., 2013). Various enzymes associated with lipid metabolism (e.g. phosphorylated isoforms of acyl-CoA dehydrogenases) were also significantly altered in a proteomics study done by Mayr et al. (2004) in PKCδ-deficient heart tissue, confirming this finding (Fig. 4). Apart from the aforementioned mechanisms, in the bid to downregulate the elevated concentrations of pyruvate in the PKCδ deficient mice, citramalate can be synthesized from both pyruvate and acetyl CoA, in a reaction catalysed by citramalate synthase (Sugimoto et al., 2021). Thereafter, isopropylmalate isomerase (2-methylmalate hydrolase) converts citramalate to citraconic acid, which was also elevated in our study. Lastly, α-linolenic acid [a n-3 polyunsaturated fatty acid (PUFAs)] functions as an energy substrate when carbohydrate reserves are depleted (Lyudinina et al., 2020). Its involvement in the efficient supply of energy in comparison to other n-3 PUFAs, has also been documented (Baker et al., 2016). α-Linolenic acid was reduced in the PKCδ-deficient mice in our study, suggesting the metabolite serves as an energy substrate, agreeing with the report by Tsukamoto and Sugawara (2018).
Additionally, it is also well described, that under conditions of reduced energy production via glycolysis, elevated branched-chain amino acid (BCAA) catabolism exists, and subsequently ketone body formation (Garrett & Grisham, 2016). As previously mentioned, a PKCδ deficiency has been associated with an inhibition of α-ketoglutarate dehydrogenase (Mayr et al., 2004), which would be expected to result in elevated concentrations of α-ketoglutarate, which would further drive BCAA catabolism (Fig. 4). Confirmation of this in our investigation was the elevated concentrations of α-ketoisocaproic acid (from elevated leucine metabolism in particular). Leucine metabolism is also strongly linked to ketogenesis and cholesterol metabolism, and the elevated concentrations of both β-hydroxybutyric acid and mevalonic acid (Fig. 4), indicate that a PKCδ deficiency not only results in increased β-oxidation, and leucine catabolism, but also ketogenesis and cholesterol synthesis. Of the 3 cholesterol/steroid intermediates (cholesteryl acetate, 4α-methylzymosterol, and 17 α-hydroxypregnenolone), cholesteryl acetate (a cholesterol ester synthesized by formal acylation of the hydroxyl group of cholesterol by acetic acid) was the only one of the 3 to be elevated. Cholesteryl esters are the key transport and storage form of cholesterol in lipoproteins and several cell types (Kuivenhoven et al., 1998), and hence the elevated cholesteryl acetate levels indicate that knocking out the PKCδ gene, results in elevated cholesterol transport and storage as opposed to it serving as a substrate for hormone production (Fig. 4), and hence the reduced levels of 4α-methylzymosterol, and 17 α-hydroxypregnenolone in the PKCδ knock-out mice. This is additionally confirmed by Mayr et al. (2004), showing an elevated activity of Apolipoprotein A1 activity in their PKCδ knock-out model, which is responsible for reverse cholesterol transport.
Two fatty acid amides (FAMS) (oleamide and dodecanamide) with hypocholesterolemic, anti-inflammatory, and anticancer activity (Li et al., 2014; Lo et al., 2001), were down-regulated in the PCKδ deficient mice in this investigation. Whilst, limited information exists on dodecanamide, FAMS generally (e.g. oleamide, and anandamide) exhibit vasorelaxant potentials (Hoi & Hiley, 2006; Hopps et al., 2012), and are reportedly a competitive substrate for the brain enzyme; fatty acid amide hydrolase (FAAH) (Cravatt et al., 1996; Lichtman et al., 2002). FAAH catalyses the hydrolytic degradation of these FAM chemical messengers. Therefore, functional impairment of the hydrolase (suspected hyperactivity) caused the downregulation of the signalling lipids. Waluk et al. (2014), previously showed that downregulation of oleamide in mice ensued with active FAAH. The mechanism of FAMS synthesis in the body is unclear (Farrell et al., 2012; Mueller & Driscoll, 2009). Some investigations show that FAMS can disrupt the intestinal absorption of cholesterol via the inhibition of the enzyme acyl-CoA:cholesterol acyltransferase (Roth et al., 1992; Xu et al., 2004). Besides this, it’s also been reported that FAMS are synthesised following a two-step catalytic metabolism of N-fatty acylglycine, by peptidylamidoglycolate lyase (Farrell et al., 2012; Merkler et al., 1999; Wilcox et al., 1999). Considering this, a deficiency (loss of activity) of this lyase would also result in the downregulation of the FAMS. The upregulation of the monoglycerol (MG); glycerol 1-palmitic acid (1-monopalmitin) here, is consistent with the study by Su et al. (2020). This upregulation signifies dysregulation of monoacylglycerol pathway and monoglyceride lipase deficiency (MGL). MGL influences energy metabolism by hydrolysis of MG into fatty acids and glycerol, and degradation of 2-arachidonoyl glycerol (Taschler et al., 2011; Vujic et al., 2016). Hence, glycerol 1-palmitic acid upregulation would be towards compensating for energy losses, serving as an alternative substrate for β-oxidation. 1-Monopalmatin is a serum biomarker in obesity and diabetes (Hameed et al., 2020; Park et al., 2015; Taschler et al., 2011), and as a potential salivary biomarker of hepatocellular carcinoma (Hershberger et al., 2021).
Lastly, the largely increased levels of serum palmitic acid, accompanied by the elevations in glycerol 1-palmitic acid, are indicative of a metabolic flux towards phospholipid synthesis, in particular, the glycerophospholipids (Carta et al., 2017), in the PKCδ knock-out model. Considering the functionality of these phospholipids is for regulating membrane permeability (Rajasekaran et al., 2011; Yu & Rasenick, 2012), fat absorption from the intestine (Murota, 2020), elevated mitochondrial function (Hishikawa et al., 2014), cholesterol transport and accumulation in the cells (Blanco & Blanco, 2017; Carta et al., 2017; Castro-Gómez et al., 2015), and preventing fat accumulation in the liver (Carta et al., 2017; Wang et al., 2013), supports the aforementioned observations.
5 Conclusion
Metabolic biomarkers associated with various metabolic disorders, infectious diseases, and many other autoimmune diseases, need to be specific enough to predict the presence of their corresponding conditions and also explain the mechanisms associated with the pathophysiology of the disease. In this study, the metabolite markers that were significantly altered in the PKCδ knock-out mice, accurately coincided with the changes to the specific enzymes detected in the proteomics study done by Mayr et al. (2004), proving the accuracy of not only metabolomics as a standalone technology, but also that of proteomics. That said, the use of multiple omics techniques, does have value, for surety, when it comes to better characterizing a perturbation. From the results of this metabolomics investigation, it is clear that total loss of PKCδ results in an inhibition of glycolysis and the TCA cycle, accompanied by upregulation of the pentose phosphate pathway, fatty acids oxidation, cholesterol transport/storage, single carbon and sulfur-containing amino acid synthesis, branched-chain amino acids (BCAA), ketogenesis, phospholipid synthesis, and cell signalling. The measurement of the dysregulated serum metabolites in this study may represent an additional tool for the early detection and screening of PKCδ-deficiencies.
References
Adeniji, A. A., Knoll, K. E., & Loots, D. T. (2020). Potential anti-TB investigational compounds and drugs with repurposing potential in TB therapy: A conspectus. Applied Microbiology and Biotechnology, 104, 5633–5662.
Allen-Petersen, B. L., Carter, C. J., Ohm, A. M., & Reyland, M. E. (2014). Protein kinase Cδ is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene, 33, 1306–1315.
Azarpazhooh, M. R., Morovatdar, N., Avan, A., Phan, T. G., Divani, A. A., Yassi, N., Stranges, S., Silver, B., Biller, J., Tokazebani Belasi, M., Kazemi Neya, S., Khorram, B., Frydman, A., Nilanont, Y., Onorati, E., & Di Napoli, M. (2020). COVID-19 pandemic and burden of non-communicable diseases: An ecological study on data of 185 countries. Journal of Stroke and Cerebrovascular Diseases: The Official Journal of National Stroke Association, 29, 105089.
Baindara, P., Agrawal, S., & Mandal, S. M. (2020). Host-directed therapies: A potential solution to combat COVID-19. Expert Opinion on Biological Therapy, 20, 1117–1120.
Baker, E. J., Miles, E. A., Burdge, G. C., Yaqoob, P., & Calder, P. C. (2016). Metabolism and functional effects of plant-derived omega-3 fatty acids in humans. Progress in Lipid Research, 64, 30–56.
Barth, M., Ottolenghi, C., Hubert, L., Chrétien, D., Serre, V., Gobin, S., Romano, S., Vassault, A., Sefiani, A., Ricquier, D., Boddaert, N., Brivet, M., de Keyzer, Y., Munnich, A., Duran, M., Rabier, D., Valayannopoulos, V., & de Lonlay, P. (2010). Multiple sources of metabolic disturbance in ETHE1-related ethylmalonic encephalopathy. Journal of Inherited Metabolic Disease, 33 Suppl(3), S443–S453.
Basu, A., & Pal, D. (2010). Two faces of protein kinase Cδ: The contrasting roles of PKCδ in cell survival and cell death. The Scientific World Journal, 10, 2272–2284.
Bausch, F. J., Beran, D., Hering, H., Boulle, P., Chappuis, F., Dromer, C., Saaristo, P., & Perone, S. A. (2021). Operational considerations for the management of non-communicable diseases in humanitarian emergencies. Conflict and Health, 15, 9.
Berardi, D. E., Flumian, C., Rodriguez, C. E., Bessone, M. I., Cirigliano, S. M., Joffé, E. D., Fiszman, G. L., Urtreger, A. J., & Todaro, L. B. (2016). PKCδ inhibition impairs mammary cancer proliferative capacity but selects cancer stem cells, involving autophagy. Journal of Cellular Biochemistry, 117, 730–740.
Bey, E. A., Xu, B., Bhattacharjee, A., Oldfield, C. M., Zhao, X., Li, Q., Subbulakshmi, V., Feldman, G. M., Wientjes, F. B., & Cathcart, M. K. (2004). Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. The Journal of Immunology, 173, 5730–5738.
Beyoğlu, D., Imbeaud, S., Maurhofer, O., Bioulac-Sage, P., Zucman‐Rossi, J., Dufour, J. F., & Idle, J. R. (2013). Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology, 58, 229–238.
Black, A., & Black, J. (2013). Protein kinase C signaling and cell cycle regulation. Frontiers in Immunology. https://doi.org/10.3389/fimmu.2012.00423
Blanco, A., & Blanco, G. (2017). Chap. 5—Lipids. In A. Blanco & G. Blanco (Eds.), Medical Biochemistry (pp. 99–119). Academic Press.
Bonte, R., Bongaerts, M., Demirdas, S., Langendonk, J. G., Huidekoper, H. H., Williams, M., Onkenhout, W., Jacobs, E. H., Blom, H. J., & Ruijter, G. J. G. (2019). Untargeted metabolomics-based screening method for inborn errors of metabolism using semi-automatic sample preparation with an UHPLC-orbitrap-MS platform. Metabolites, 9, 289.
Brereton, R. G. (2003). Chemometrics: Data analysis for the laboratory and chemical plant. Wiley.
Brodie, C., & Blumberg, P. M. (2003). Regulation of cell apoptosis by protein kinase c δ. Apoptosis, 8, 19–27.
Carey, B. D., Akhrymuk, I., Dahal, B., Pinkham, C. L., Bracci, N., Finstuen-Magro, S., Lin, S. C., Lehman, C. W., Sokoloski, K. J., & Kehn-Hall, K. (2020). Protein Kinase C subtype δ interacts with Venezuelan equine encephalitis virus capsid protein and regulates viral RNA binding through modulation of capsid phosphorylation. PLoS Pathogens, 16, e1008282.
Carta, G., Murru, E., Banni, S., & Manca, C. (2017). Palmitic acid: Physiological role, metabolism and nutritional implications. Frontiers in Physiology. https://doi.org/10.3389/fphys.2017.00902
Castro-Gómez, P., Garcia-Serrano, A., Visioli, F., & Fontecha, J. (2015). Relevance of dietary glycerophospholipids and sphingolipids to human health. Prostaglandins Leukotrienes and Essential Fatty Acids, 101, 41–51.
Chen, Z., Forman, L. W., Miller, K. A., English, B., Takashima, A., Bohacek, R. A., Williams, R. M., & Faller, D. V. (2011). Protein kinase Cδ inactivation inhibits cellular proliferation and decreases survival in human neuroendocrine tumors. Endocrine-Related Cancer, 18, 759–771.
Choi, Y. H., Jin, G. Y., Li, L., & Yan, G. H. (2013). Inhibition of protein kinase C delta attenuates allergic airway inflammation through suppression of PI3K/Akt/mTOR/HIF-1 Alpha/VEGF pathway. PLoS ONE, 8, e81773.
Contreras, X., Mzoughi, O., Gaston, F., Peterlin, M. B., & Bahraoui, E. (2012). Protein kinase C-delta regulates HIV-1 replication at an early post-entry step in macrophages. Retrovirology, 9, 37.
Cravatt, B. F., Giang, D. K., Mayfield, S. P., Boger, D. L., Lerner, R. A., & Gilula, N. B. (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature, 384, 83–87.
Dahabiyeh, L. A., Mujammami, M., Arafat, T., Benabdelkamel, H., Alfadda, A. A., & Rahman, A. (2021). A metabolic pattern in healthy subjects given a single dose of metformin: A metabolomics approach. Frontiers in Pharmacology, 12, 705932–705932.
Dani, N., & Broadie, K. (2012). Glycosylated synaptomatrix regulation of trans-synaptic signaling. Developmental Neurobiology, 72, 2–21.
Dempsey, E. C., Newton, A. C., Mochly-Rosen, D., Fields, A. P., Reyland, M. E., Insel, P. A., & Messing, R. O. (2000). Protein kinase C isozymes and the regulation of diverse cell responses. American Journal of Physiology-Lung Cellular and Molecular Physiology, 279, L429–L438.
Du Preez, I., & Loots, D. (2013). New sputum metabolite markers implicating adaptations of the host to Mycobacterium tuberculosis, and vice versa. Tuberculosis, 93, 330–337.
Du, Y., Zhao, Y., Li, C., Zheng, Q., Tian, J., Li, Z., Huang, T. Y., Zhang, W., & Xu, H. (2018). Inhibition of PKCδ reduces amyloid-β levels and reverses Alzheimer disease phenotypes. Journal of Experimental Medicine, 215, 1665–1677.
Dunsmore, S. E. (2006). Extracellular matrix | Collagens. In G. J. Laurent & S. D. Shapiro (Eds.), Encyclopedia of Respiratory Medicine (pp. 168–175). Academic Press.
Eigenvector (2016). PLS_Toolbox 8.2.1 Eigenvector Research Inc., Manson, USA v. 8.2.1.
Farrell, E. K., Chen, Y., Barazanji, M., Jeffries, K. A., Cameroamortegui, F., & Merkler, D. J. (2012). Primary fatty acid amide metabolism: Conversion of fatty acids and an ethanolamine in N18TG2 and SCP cells1. Journal of Lipid Research, 53, 247–256.
Fields, A. P., & Gustafson, W. C. (2003). Protein kinase C in disease. In A. C. Newton (Ed.), Protein Kinase C Protocols (pp. 519–537). Humana Press.
Fisher, A. B., & Zhang, Q. (2006). NADPH and NADPH oxidase. In G. J. Laurent & S. D. Shapiro (Eds.), Encyclopedia of Respiratory Medicine (pp. 77–84). Academic Press.
Garrett, R. H., & Grisham, C. M. (2016). Biochemistry, Sixth Edition edn. Cengage Learning, 20 Channel Center Street Boston, MA, 02210, USA.
Gao, X., Chen, W., Li, R., Wang, M., Chen, C., Zeng, R., & Deng, Y. (2012). Systematic variations associated with renal disease uncovered by parallel metabolomics of urine and serum. BMC Systems Biology, 6, S14.
Gordon, R., Singh, N., Lawana, V., Ghosh, A., Harischandra, D. S., Jin, H., Hogan, C., Sarkar, S., Rokad, D., Panicker, N., Anantharam, V., Kanthasamy, A. G., & Kanthasamy, A. (2016). Protein kinase Cδ upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiology of Disease, 93, 96–114.
Gorelik, G., Sawalha, A. H., Patel, D., Johnson, K., & Richardson, B. (2015). T cell PKCδ kinase inactivation induces lupus-like autoimmunity in mice. Clinical Immunology, 158, 193–203.
Greene, M. W., Burrington, C. M., Lynch, D. T., Davenport, S. K., Johnson, A. K., Horsman, M. J., Chowdhry, S., Zhang, J., Sparks, J. D., & Tirrell, P. C. (2014). Lipid metabolism, oxidative stress and cell death are regulated by PKC delta in a dietary model of nonalcoholic steatohepatitis. PLoS ONE, 9, e85848.
Gromski, P., Xu, Y., Kotze, H., Correa, E., Ellis, D., Armitage, E., Turner, M., & Goodacre, R. (2014). Influence of missing values substitutes on multivariate analysis of metabolomics data. Metabolites, 4, 433–452.
Hameed, A., Mojsak, P., Buczynska, A., Suleria, H. A. R., Kretowski, A., & Ciborowski, M. (2020). Altered metabolome of lipids and amino acids species: A source of early signature biomarkers of T2DM. Journal of Clinical Medicine, 9, 2257.
Hao, J., Yang, T., Zhou, Y., Gao, G. Y., Xing, F., Peng, Y., Tao, Y. Y., & Liu, C. H. (2017). Serum metabolomics analysis reveals a distinct metabolic profile of patients with primary biliary cholangitis. Scientific Reports, 7, 784.
Hershberger, C. E., Rodarte, A. I., Siddiqi, S., Moro, A., Acevedo-Moreno, L. A., Brown, J. M., Allende, D. S., Aucejo, F., & Rotroff, D. M. (2021). Salivary metabolites are promising non-invasive biomarkers of hepatocellular carcinoma and chronic liver disease. Liver Cancer International, 2, 33–44.
Hishikawa, D., Hashidate, T., Shimizu, T., & Shindou, H. (2014). Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. Journal of Lipid Research, 55, 799–807.
Hoi, P. M., & Hiley, C. R. (2006). Vasorelaxant effects of oleamide in rat small mesenteric artery indicate action at a novel cannabinoid receptor. British Journal of Pharmacology, 147, 560–568.
Hopps, J. J., Dunn, W. R., & Randall, M. D. (2012). Enhanced vasorelaxant effects of the endocannabinoid-like mediator, oleamide, in hypertension. European Journal of Pharmacology, 684, 102–107.
Ichihara, S., Ichihara, Y., Tomisawa, H., Fukazawa, H., & Tateishi, M. (1985). Identification of 5-deoxy-D-ribitol as a major metabolite of 5’-deoxy-5-fluorouridine in rats. Drug Metabolism and Disposition, 13, 520–521.
Jain, T., Kumar, H., & Dutta, P. K. (2016). D-glucosamine and N-acetyl D-glucosamine: Their potential use as regenerative medicine. In P. K. Dutta (Ed.), Chitin and Chitosan for Regenerative Medicine (pp. 279–295). Springer.
Jeng-Miller, K. W., & Baumal, C. R. (2018). Chap. 5—Genetics of diabetic retinopathy. In C. R. Baumal & J. S. Duker (Eds.), Current Management of Diabetic Retinopathy (pp. 37–40). Elsevier.
Kamthan, M., Kamthan, A., Ruhela, D., Maiti, P., Bhavesh, N. S., & Datta, A. (2013). Upregulation of galactose metabolic pathway by N-acetylglucosamine induced endogenous synthesis of galactose in Candida albicans. Fungal Genetics and Biology, 54, 15–24.
Khor, E. C., Abel, T., Tickner, J., Chim, S. M., Wang, C., Cheng, T., Ng, B., Ng, P. Y., Teguh, D. A., Kenny, J., Yang, X., Chen, H., Nakayama, K. I., Nakayama, K., Pavlos, N., Zheng, M. H., & Xu, J. (2013). Loss of protein kinase C-δ protects against LPS-induced osteolysis owing to an intrinsic defect in osteoclastic bone resorption. PLoS ONE, 8, e70815.
Knoll, K. E., Lindeque, Z., Adeniji, A. A., Oosthuizen, C. B., Lall, N., & Loots, D. T. (2021). Elucidating the antimycobacterial mechanism of action of decoquinate derivative RMB041 using metabolomics. Antibiotics, 10, 693.
Konopka, J. B. (2012). N-acetylglucosamine functions in cell signaling. Scientifica. https://doi.org/10.6064/2012/489208
Kubomura, D., Ueno, T., Yamada, M., Tomonaga, A., & Nagaoka, I. (2017). Effect of N-acetylglucosamine administration on cartilage metabolism and safety in healthy subjects without symptoms of arthritis: A case report. Experimental and Therapeutic Medicine, 13, 1614–1621.
Kuehn, H. S., Niemela, J. E., Rangel-Santos, A., Zhang, M., Pittaluga, S., Stoddard, J. L., Hussey, A. A., Evbuomwan, M. O., Priel, D. A. L., Kuhns, D. B., Park, C. L., Fleisher, T. A., Uzel, G., & Oliveira, J. B. (2013). Loss-of-function of the protein kinase C δ (PKCδ) causes a B-cell lymphoproliferative syndrome in humans. Blood, 121, 3117–3125.
Kuivenhoven, J. A., Jukema, J. W., Zwinderman, A. H., de Knijff, P., McPherson, R., Bruschke, A. V. G., Lie, K. I., & Kastelein, J. J. P. (1998). The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. New England Journal of Medicine, 338, 86–93.
Laaksonen, D. E., & Sen, C. K. (2000). Part XI Chap. 38—Exercise and oxidative stress in diabetes mellitus. In C. K. Sen, L. Packer, & O. O. P. Hänninen (Eds.), Handbook of Oxidants and Antioxidants in Exercise (pp. 1105–1136). Elsevier Science B.V.
Li, Y., Song, X., Zhao, X., Zou, L., & Xu, G. (2014). Serum metabolic profiling study of lung cancer using ultra high performance liquid chromatography/quadrupole time-of-flight mass spectrometry. Journal of Chromatography B, 966, 147–153.
Lichtman, A. H., Hawkins, E. G., Griffin, G., & Cravatt, B. F. (2002). Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. Journal of Pharmacology and Experimental Therapeutics, 302, 73–79.
Lien, C. F., Chen, S. J., Tsai, M. C., & Lin, C. S. (2021). Potential role of protein kinase C in the pathophysiology of diabetes-associated atherosclerosis. Frontiers in Pharmacology. https://doi.org/10.3389/fphar.2021.716332
Liverani, E., Tursi, S. A., Cornwell, W. D., Mondrinos, M. J., Sun, S., Buttaro, B. A., Wolfson, M. R., Rogers, T. J., Tükel, Ç., & Kilpatrick, L. E. (2020). Protein kinase C-delta inhibition is organ-protective, enhances pathogen clearance, and improves survival in sepsis. The FASEB Journal, 34, 2497–2510.
Lo, Y. K., Tang, K. Y., Chang, W. N., Lu, C. H., Cheng, J. S., Lee, K. C., Chou, K. J., Liu, C. P., Chen, W. C., & Su, W. (2001). Effect of oleamide on Ca2 + signaling in human bladder cancer cells. Biochemical Pharmacology, 62, 1363–1369.
Lorenzi, M. (2007). The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Experimental Diabetes Research, 2007, 61038–61038.
Lyudinina, A. Y., Bushmanova, E. A., Varlamova, N. G., & Bojko, E. R. (2020). Dietary and plasma blood α-linolenic acid as modulators of fat oxidation and predictors of aerobic performance. Journal of the International Society of Sports Nutrition, 17, 57.
Mahon, R. N., & Hafner, R. (2017). Applying precision medicine and immunotherapy advances from oncology to host-directed therapies for infectious diseases. Frontiers in Immunology. https://doi.org/10.3389/fimmu.2017.00688
MATLAB. (2012). MATLAB and statistics toolbox release. The MathWorks Inc., Natick.
Mayr, M., Chung, Y. L., Mayr, U., McGregor, E., Troy, H., Baier, G., Leitges, M., Dunn, M. J., Griffiths, J. R., & Xu, Q. (2004). Loss of PKC-δ alters cardiac metabolism. American Journal of Physiology-Heart and Circulatory Physiology, 287, H937–H945.
Merkler, K. A., Baumgart, L. E., DeBlassio, J. L., Glufke, U., King 3, L., Ritenour-Rodgers, K., Vederas, J. C., Wilcox, B. J., & Merkler, D. J. (1999). A pathway for the biosynthesis of fatty acid amides. Advances in Experimental Medicine and Biology, 469, 519–525.
Mueller, G. P., & Driscoll, W. J. (2009). Chap. 3—Biosynthesis of oleamide, vitamins & hormones (pp. 55–78). Academic Press.
Murota, K. (2020). Digestion and absorption of dietary glycerophospholipids in the small intestine: Their significance as carrier molecules of choline and n-3 polyunsaturated fatty acids. Biocatalysis and Agricultural Biotechnology, 26, 101633.
Nanthapisal, S., Omoyinmi, E., Murphy, C., Standing, A., Eisenhut, M., Eleftheriou, D., & Brogan, P. A. (2017). Early-onset juvenile SLE associated with a novel mutation in protein kinase C δ. Pediatrics. https://doi.org/10.1542/peds.2016-0781
Newton, A. C. (2001). Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chemical Reviews, 101, 2353–2364.
Nitti, M., Furfaro, A. L., Cevasco, C., Traverso, N., Marinari, U. M., Pronzato, M. A., & Domenicotti, C. (2010). PKC delta and NADPH oxidase in retinoic acid-induced neuroblastoma cell differentiation. Cellular Signalling, 22, 828–835.
Nitti, M., Furfaro, A. L., Traverso, N., Odetti, P., Storace, D., Cottalasso, D., Pronzato, M. A., Marinari, U. M., & Domenicotti, C. (2007). PKC delta and NADPH oxidase in AGE-induced neuronal death. Neuroscience Letters, 416, 261–265.
Parihar, S., Ozturk, M., Marakalala, M., Loots, D., Hurdayal, R., Maasdorp, D. B., Van Reenen, M., Zak, D., Darboe, F., & Penn-Nicholson, A. (2018). Protein kinase C-delta (PKCδ), a marker of inflammation and tuberculosis disease progression in humans, is important for optimal macrophage killing effector functions and survival in mice. Mucosal Immunology, 11, 496–511.
Park, S., Sadanala, K. C., & Kim, E. K. (2015). A metabolomic approach to understanding the metabolic link between obesity and diabetes. Molecules and Cells, 38, 587.
Poole, A. W., Pula, G., Hers, I., Crosby, D., & Jones, M. L. (2004). PKC-interacting proteins: From function to pharmacology. Trends in Pharmacological Sciences, 25, 528–535.
Rajasekaran, S., Maxvold, N. J., & Bunchman, T. E. (2011). Chap. 71—Glomerulotubular dysfunction and acute kidney injury. In B. P. Fuhrman & J. J. Zimmerman (Eds.), Pediatric Critical Care (4th ed., pp. 1016–1035). Mosby.
Roth, B. D., Blankley, C. J., Hoefle, M. L., Holmes, A., Roark, W. H., Trivedi, B. K., Essenburg, A. D., Kieft, K. A., Krause, B. R., & Stanfield, R. L. (1992). Inhibitors of acyl-CoA: Cholesterol acyltransferase. 1. Identification and structure-activity relationships of a novel series of fatty acid anilide hypocholesterolemic agents. Journal of Medicinal Chemistry, 35, 1609–1617.
Saccenti, E., Hoefsloot, H. C., Smilde, A. K., Westerhuis, J. A., & Hendriks, M. M. (2014). Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics, 10, 361–374.
Salway, J. G. (2017). Diabetes II: Types I and II Diabetes, MODY and Pancreatic Beta-cell Metabolism | pg 120, Metabolism at a Glance Wiley-Blackwell, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK. pp. 144.
Salzer, E., Santos-Valente, E., Keller, B., Warnatz, K., & Boztug, K. (2016). Protein kinase C δ: A gatekeeper of immune homeostasis. Journal of Clinical Immunology, 36, 631–640.
Sato, S., Secchi, E. F., Lizak, M. J., Fukase, S., Ohta, N., Murata, M., Tsai, J. Y., & Kador, P. F. (1999). Polyol formation and NADPH-dependent reductases in dog retinal capillary pericytes and endothelial cells. Investigative Ophthalmology & Visual Science, 40, 697–704.
Schmitz-Peiffer, C. (2013). The tail wagging the dog—Regulation of lipid metabolism by protein kinase C. The FEBS Journal, 280, 5371–5383.
Sekowska, A., Ashida, H., & Danchin, A. (2019). Revisiting the methionine salvage pathway and its paralogues. Microbial Biotechnology, 12, 77–97.
Semmo, N., Weber, T., Idle, J. R., & Beyoğlu, D. (2015). Metabolomics reveals that aldose reductase activity due to AKR1B10 is upregulated in hepatitis C virus infection. Journal of Viral Hepatitis, 22, 617–624.
Sharifinejad, N., Azizi, G., Behniafard, N., Zaki-Dizaji, M., Jamee, M., Yazdani, R., Abolhassani, H., & Aghamohammadi, A. (2020). Protein kinase C-delta defect in autoimmune lymphoproliferative syndrome-like disease: First case from the national Iranian registry and review of the literature. Immunological Investigations. https://doi.org/10.1080/08820139.2020.1829638
Sparks, R., Lui, A., Bader, D., Patel, R., Murr, M., Guida, W., Fratti, R., & Patel, N. A. (2019). A specific small-molecule inhibitor of protein kinase CδI activity improves metabolic dysfunction in human adipocytes from obese individuals. The Journal of biological chemistry, 294, 14896–14910.
Stander, Z., Luies, L., Mienie, L. J., Van Reenen, M., Howatson, G., Keane, K. M., Clifford, T., Stevenson, E. J., & Loots, D. T. (2020). The unaided recovery of marathon-induced serum metabolome alterations. Scientific Reports, 10, 11060.
Su, K., Yi, B., Yao, B. Q., Xia, T., Yang, Y. F., Zhang, Z. H., & Chen, C. (2020). Liraglutide attenuates renal tubular ectopic lipid deposition in rats with diabetic nephropathy by inhibiting lipid synthesis and promoting lipolysis. Pharmacological Research, 156, 104778.
Sugimoto, N., Engelgau, P., Jones, A. D., Song, J., & Beaudry, R. (2021). Citramalate synthase yields a biosynthetic pathway for isoleucine and straight- and branched-chain ester formation in ripening apple fruit. Proceedings of the National Academy of Sciences, 118, e2009988118.
Sullivan, G. M., & Feinn, R. (2012). Using effect size—Or why the P value is not enough. Journal of Graduate Medical Education, 4, 279–282.
Tanenberg, R. J., & Donofrio, P. D. (2008). Chapter 3—Neuropathic problems of the lower limbs in diabetic patients. In J. H. Bowker & M. A. Pfeifer (Eds.), Levin and O’Neal’s The Diabetic Foot (7th ed., pp. 33–74). Mosby.
Taschler, U., Radner, F. P., Heier, C., Schreiber, R., Schweiger, M., Schoiswohl, G., Preiss-Landl, K., Jaeger, D., Reiter, B., Koefeler, H. C., Wojciechowski, J., Theussl, C., Penninger, J. M., Lass, A., Haemmerle, G., Zechner, R., & Zimmermann, R. (2011). Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. Journal of Biological Chemistry, 286, 17467–17477.
Thamilselvan, V., Menon, M., & Thamilselvan, S. (2009). Oxalate-induced activation of PKC-α and-δ regulates NADPH oxidase-mediated oxidative injury in renal tubular epithelial cells. American Journal of Physiology-Renal Physiology, 297, F1399–F1410.
Tounta, V., Liu, Y., Cheyne, A., & Larrouy-Maumus, G. (2021). Metabolomics in infectious diseases and drug discovery. Molecular Omics, 17, 376–393.
Tsukamoto, I., & Sugawara, S. (2018). Low levels of linoleic acid and α-linolenic acid and high levels of arachidonic acid in plasma phospholipids are associated with hypertension. Biomed Reports, 8, 69–76.
van den Berg, R. A., Hoefsloot, H. C., Westerhuis, J. A., Smilde, A. K., & van der Werf, M. J. (2006). Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics, 7, 1–15.
Van Reenen, M., Westerhuis, J. A., Reinecke, C. J., & Venter, J. H. (2017). Metabolomics variable selection and classification in the presence of observations below the detection limit using an extension of ERp. BMC Bioinformatics, 18, 1–13.
Vujic, N., Schlager, S., Eichmann, T. O., Madreiter-Sokolowski, C. T., Goeritzer, M., Rainer, S., Schauer, S., Rosenberger, A., Woelfler, A., Doddapattar, P., Zimmermann, R., Hoefler, G., Lass, A., Graier, W. F., Radovic, B., & Kratky, D. (2016). Monoglyceride lipase deficiency modulates endocannabinoid signaling and improves plaque stability in ApoE-knockout mice. Atherosclerosis, 244, 9–21.
Wagermaier, W., & Fratzl, P. (2012). 9.03—Collagen. In K. Matyjaszewski & M. Möller (Eds.), Polymer science: A comprehensive reference (pp. 35–55). Elsevier.
Waluk, D. P., Battistini, M. R., Dempsey, D. R., Farrell, E. K., Jeffries, K. A., Mitchell, P., Hernandez, L. W., McBride, J. C., Merkler, D. J., & Hunt, M. C. (2014). Chap. 9—Mammalian fatty acid amides of the brain and CNS. In R. R. Watson & F. De Meester (Eds.), Omega-3 fatty acids in brain and neurological health (pp. 87–107). Academic Press.
Wang, J., Sun, L., Nie, Y., Duan, S., Zhang, T., Wang, W., Ye, R. D., Hou, S., & Qian, F. (2020). Protein kinase C δ (PKCδ) attenuates bleomycin induced pulmonary fibrosis via inhibiting NF-κB signaling pathway. Frontiers in Physiology. https://doi.org/10.3389/fphys.2020.00367
Wang, T. Y., Liu, M., Portincasa, P., & Wang, D. Q. H. (2013). New insights into the molecular mechanism of intestinal fatty acid absorption. European Journal of Clinical Investigation, 43, 1203–1223.
Wei, R., Wang, J., Su, M., Jia, E., Chen, S., Chen, T., & Ni, Y. (2018). Missing value imputation approach for mass spectrometry-based metabolomics data. Scientific Reports, 8, 663.
WHO (2021a). World Health Organisation | Fact Sheets/Detail—Antimicrobial resistance. https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance
WHO (2021b). World Health Organisation | Key Facts/Fact sheets—Detail on Noncommunicable diseases. https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases
Wilcox, B. J., Ritenour-Rodgers, K. J., Asser, A. S., Baumgart, L. E., Baumgart, M. A., Boger, D. L., DeBlassio, J. L., deLong, M. A., Glufke, U., Henz, M. E., King 3, L., Merkler, K. A., Patterson, J. E., Robleski, J. J., Vederas, J. C., & Merkler, D. J. (1999). N-acylglycine amidation: Implications for the biosynthesis of fatty acid primary amides. Biochemistry, 38, 3235–3245.
Williams, R., Lenz, E., Wilson, A., Granger, J., Wilson, I., Major, H., Stumpf, C., & Plumb, R. (2006). A multi-analytical platform approach to the metabonomic analysis of plasma from normal and Zucker (fa/fa) obese rats. Molecular BioSystems, 2, 174–183.
Wolf, G., Sharma, K., & Ziyadeh, F. N. (2008). Chapter 78—Pathophysiology and pathogenesis of diabetic nephropathy in Alpern. In S. C. Hebert (Ed.), Seldin and Giebisch’s The Kidney (4th ed., pp. 2215–2233). Academic Press.
Wood, S., Harrison, S. E., Judd, N., Bellis, M. A., Hughes, K., & Jones, A. (2021). The impact of behavioural risk factors on communicable diseases: A systematic review of reviews. Bmc Public Health, 21, 2110.
Xu, M. Z., Lee, W. S., Kim, M. J., Park, D. S., Yu, H., Tian, G. R., Jeong, T. S., & Park, H. Y. (2004). Acyl-CoA: Cholesterol acyltransferase inhibitory activities of fatty acid amides isolated from Mylabris phalerate Pallas. Bioorganic & Medicinal Chemistry Letters, 14, 4277–4280.
Yang, C., Ko, B., Hensley, C. T., Jiang, L., Wasti, A. T., Kim, J., Sudderth, J., Calvaruso, M. A., Lumata, L., & Mitsche, M. (2014). Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Molecular Cell, 56, 414–424.
Yang, C., Sudderth, J., Dang, T., Bachoo, R. G., McDonald, J. G., & DeBerardinis, R. J. (2009). Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Research, 69, 7986–7993.
Yang, F., Cai, W., Yang, K., & Chen, M. (2013). PKCδ knockdown inhibits free fatty acid induction of endothelial cell apoptosis. Cell Biochemistry and Function, 31, 380–384.
Yu, J. Z., & Rasenick, M. M. (2012). Chap 2—Receptor signaling and the cell biology of synaptic transmission. In M. J. Aminoff, F. Boller, & D. F. Swaab (Eds.), Handbook of Clinical Neurology (pp. 9–35). Elsevier.
Zhang, A., Sun, H., Yan, G., Wang, P., & Wang, X. (2015). Metabolomics for biomarker discovery: Moving to the clinic. BioMed Research International, 2015, 354671.
Zhao, M., Xia, L., & Chen, G. Q. (2012). Protein kinase cδ in apoptosis: A brief overview. Archivum Immunolgiae et Therapiae Experimentalis, 60, 361–372.
Acknowledgements
We thank the University of Cape Town (UCT) Animal Unit staff, Rodney Lucas for technical assistance, Faried Abbas for maintenance of the BSL3 facility, Nazila Ghodsi, Munadia Ansari, and Fadwah Booley for genotyping of mice and maintenance of the laboratory, respectively. AAA appreciates North-West University for post-doctoral fellowship.
Funding
No funds, grants, or other support was received.
Author information
Authors and Affiliations
Contributions
DTL, SPP, MO, and FB: helped conceive the project, performed experiments, developed methods, provided equipment for experiments, and analyzed results. AAA: performed extensive literature review, curated data, analyzed results, and created metabolic map. MVR: helped conceive the project, curated data, and statistically analyzed results. All authors contributed to the manuscript, provided intellectual input, and approved it for submission.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
All experiments were carried out following South African National Guidelines and University of Cape Town laboratory animal procedures. The Animal Ethics Committee, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa, approved the protocol (AEC: 012/036).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Loots, D.T., Adeniji, A.A., Van Reenen, M. et al. The metabolomics of a protein kinase C delta (PKCδ) knock-out mouse model. Metabolomics 18, 92 (2022). https://doi.org/10.1007/s11306-022-01949-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-022-01949-w