
Abstract
Microplasminogen (μPlg), a truncated form of human plasminogen, has considerable potential as a direct-acting thrombolytic agent. To further develop μPlg into a thrombolytic agent with anti-thrombus properties, we constructed two μPlg variants containing tripeptide Arg-Gly-Asp (RGD) and tetrapeptide Gly-Pro-Arg-Pro (GPRP) by site-directed mutagenesis. The recombinant cDNAs were expressed in yeast (Pichia pastoris) and purified to high homogeneity by Ni–NTA affinity chromatography. The specific activities of RGD-μPlg and GPRP-μPlg were 7.7 and 13.3 U/mg, respectively, as determined using the fibrin-plate method. RGD-μPlg significantly inhibited ADP-induced platelet aggregation, which was 33.6- and 14.1-fold higher than the native μPlg and GPRP-μPlg, respectively. On the other hand, GPRP-μPlg prolonged thrombin-initialized fibrinogen polymerization in a concentration-dependent manner, which was 9.2- and 5.7-fold stronger than μPlg and RGD-μPlg, respectively. Under activation by urokinase, μPlg, RGD-μPlg, and GPRP-μPlg all showed over 80 % conversions to their active enzyme in 24 h. The structure models that docked RGD-μPlg and μPlg activation loops into the enzymatic active site of urokinase showed that Pro559 to Asp559 mutation of RGD-μPlg led to an alteration in the interaction, which possibly explains the slowed activation of RGD-μPlg by urokinase over an 80-min period. In conclusion, this study has presented two recombinant μPlg variants with anti-platelet aggregation and anti-fibrinogen clotting activity, thus suggesting the anti-thrombosis properties of these two μPlg derivatives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thromboembolism diseases, such as myocardial infarction, ischemic stroke, deep vein thrombosis and retinal blood vessel embolism, are prevalent causes of death and disability around the world. Thrombolytic therapy is a major strategy for thromboembolism disease treatment. However, the success of thrombolysis is limited by recurrent occlusion, which occurs in 10–20 % patients [1]. Thus, novel thrombolytic agents fused with anti-thrombotic elements are necessary.
The tetrapeptide Gly-Pro-Arg-Pro (GPRP) is an analogue of the amino-terminal GPRVV sequence of the human fibrin α-chain and can prevent fibrin monomers from polymerizing to insoluble clots [2]. Moreover, a PEGylated fibrin knob ‘A’ peptide containing GPRP exhibited a tenfold enhancement of anticoagulant activity than that of the non-PEGylated knob peptides [3]. On the other hand, Arg-Gly-Asp (RGD)-containing peptides can inhibit the binding of fibrinogen to integrin αIIb/β3 on activated platelets, thus inhibiting platelet aggregation [4]. Several indirectly-acting thrombolytic agents, such as staphylokinase and urokinase (UK), have been chimerized with anti-thrombosis peptides through gene recombination or chemical linkage, as reported in previous studies, and each presented anti-thrombosis characteristics [5–7]. However, the combination of anti-thrombosis peptides and direct-acting thrombolytic proteins has not yet been reported.
Plasmin (Plm) is the major fibrinolytic enzyme in human plasma and can be a potent direct-acting thrombolytic agent for local delivery [8]. It is noticeable that intravenous administration of a large amount of Plm, unlike tissue-type plasminogen activator (t-PA), appeared to be well tolerated without bleeding [9, 10]. In addition, two des-kringle variants of Plm, miniplasmin (mPlm) (Val442-Asn791) and microplasmin (μPlm) (Lys531-Asn791) are also well-tolerated and potent with pharmacological application [11–14]. Furthermore, μPlm was the only drug approved by FDA as a substitute for vitrectomy [15].
Plasminogen (Plg) is the precursor of Plm and contains 792 amino acids with a molecular weight of 92 kDa. Under physiological conditions, UK and t-PA convert inactive Plg to active Plm by hydrolyzing the Arg561-Val562 peptide bond. Under alkaline conditions (pH 10.0), Plm can autodegrade and degrade Plg, producing μPlm and microplasminogen (μPlg) respectively [16, 17]. It is difficult to express Plg due to its complex domains; however, a high production yield of μPlg has been obtained by its expression in yeast Pichia pastoris [18].
In this study, we constructed a novel μPlg variant, which comprised of Ala543 to Asn791 amino acids of human Plg. At the same time, the GPRP tetrapeptide and RGD tripeptide were each constructed into the μPlg by site-directed mutagenesis. Recombinant cDNAs of μPlg, RGD-μPlg, and GPRP-μPlg were expressed in yeast P. pastoris and purified to high homogeneity, and their biological activities were compared in vitro.
Materials and methods
Materials
Plasmid pDNR-LIB-hPLG containing full-length cDNA sequence of human Plg was obtained from Enogene biotech Co. Ltd (Nanjing, Jiangsu, China).The pPICZαA plasmid and yeast P. pastoris GS115 were purchased from Invitrogen (Carlsbad, CA, USA). The pGME®-T plasmid was obtained from Promega biotech (Shanghai, China). Xba-Ι, Xho-Ι, Sac-Ι, T4 DNA ligase, Premix Taq DNA polymerase and Escherichia coli TOP10 cells were obtained from Takara Bioengineering (Dalian, Liaoning, China). Primers were synthesized by Sangon biotech Co. Ltd (Shanghai, China). PCR Purification Kit was purchased from Omega Sciences (Germantown, MD, USA). Thrombin, fibrinogen, fibrin monomers, bovine serum Plg were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Ni2t-nitrilotriacetate (Ni–NTA) column was supplied by Bio-Rad (Hercules, CA, USA). DAPase was obtained from QiagenGEN (Valencia, CA, USA).
Construction of pGEM-T-μPLG plasmid
The cDNA fragment encoding μPlg (Ala543-Asn791) was subcloned by PCR from the plasmid pDNR-LIB-HPLG using forward primer F1 5′-CATCACCATCACCAT CACGCCCCTTCATTTGATTGTG G-3′ that contained a 6xHis tag and reverse primer D 5′-GTTTCTAGAAAGTTAATTATTTCTCATCACTCC-3′ containing an Xba-I site. In the second PCR, an Xho-I site followed by a KEX-2 cleavage site was added to the 5′ end with forward primer F2 5′-TCTCTCGAGAAAAGACATCACC ATCACCATCAC-3′. Both reactions were thermocycled as follows: one cycle at 94 °C, 5 min, 30 cycles at 94 °C, 30 s; 55 °C, 30 s and 72 °C, 7 min. The final PCR product was ligated into pGEM®-T Easy vector, sequenced with forward and reverse primers pUC/M13.
Site-directed mutagenesis
The cDNA sequences of RGD-μPLG and GPRP-μPLG were obtained by mutagenesis PCR. Briefly, the PCR for RGP to RGD mutation was performed on plasmid pGEM-T-μPLG with forward primer F2 and reverse primer M1 5′-CCTACAAC CCTTCCATCACATTTCTTCGGCT-3′. To amplify the RGD-μPLG sequence, the PCR product was used as forward primers together with reverse primer D on linearized plasmid pGEM-T-μPLG in a second PCR. The same double-PCR procedure was applied to obtain the cDNA sequence of GPRP-μPLG except using reverse primer M2: 5′-CACCATCACGGCCCTCGTCCTGATTGTGGG-3′ in the first PCR. The final PCR products were separated, purified and ligated into pGEM-T Easy vector. Sequencings were performed with primers pUC/M13 in both forward and reverse directions.
Expression and purification
The transformation, screening and expression of pGEM-T-μPLG and its mutants in yeast P. pastoris was referred to the operation manual of Invitrogen Kit. Briefly, these plasmids were digested with Xba-I and Xho-I. Purified cDNA fragments containing μPLG, RGD-μPLG and GPRP-μPLG were linked to pPICZαA plasmids, which was then amplified in E. coli TOP10 cells and transformed into competent cells P. pastoris GS115. Transformants were screened in sequence on YPDS plates containing 100, 200 and 800 µg/mL zeocin. High-expression strains were selected from YPDS plates containing 800 µg/mL zeocin.
Colonies were selected and inoculated in 100 mL BMGY medium and cultured for 16 h at 30 °C. The cells were pelleted, washed by sterilized water, re-suspended in 50 mL BMMY medium and cultured for 48 h. 1 % methanol was supplied every 24 h and 300 µL culture supernatant was retained in 0, 12, 24, 36 and 48 h. The expression of μPlg, RGD-μPlg or GPRP-μPlg in culture supernatant was identified by 12 % SDS-PAGE.
For batch purification, the culture supernatant was diluted 1:2 with column buffer (50 mM phosphate buffer, 500 mM NaCl, pH 7.4) and loaded on a Ni–NTA column at 1 mL/min. After loading, the column was washed with 10 columns of column buffer and 5 columns of column buffer supplemented with 10 mM imidazole at 2 mL/min to remove the unbounded proteins. μPlg, RGD-μPlg or GPRP-μPlg was eluted with column buffer supplemented with 500 mM imidazole, dialyzed against 150 mM NaCl, 50 mM phosphate buffer at pH 7.4 and concentrated by PEG 20 000. The 6xHis tag was cleaved by DAPase enzyme, and removed together with uncleaved proteins by Ni–NTA chromatography. The purified proteins with native N-terminus were assayed for protein concentration by BCA method and identified by western blot.
Fibrinolytic activity
The fibrinolytic activity was determined using fibrin plate method as described previously [19]. Briefly, 1 % agarose gel plates contained 0.15 M NaCl, 15 mg/mL human fibrinogen, 2.5 U/mL thrombin, 0.02 % NaN3 and 50 mM phosphate buffer, pH 7.4. On the solidified fibrin plates, wells of 2 mm diameter were punched and 10 μL samples were added and kept in moist box at 37 °C for 18 h. The diameter of the halo around the well was measured with vernier caliper to calculate the fibrinolytic activity of μPlm, RGD-μPlm and GPRP-μPlm by comparison with standard preparation for bovine plasmin.
Inhibiting ADP-induced platelet aggregation
The anti-platelet aggregation activity was measured by classic turbidity method [20]. Fresh blood obtained from rabbits was anti-coagulated by 110 mM sodium citrate at a ratio of 1:9 (v/v). Platelet rich plasma (PRP) was obtained by centrifugation at 800 rpm for 10 min, and a second centrifugation at 3500 rpm for 15 min was used to prepare platelet-poor plasma (PPP). The PRP was diluted by PPP to a platelet count of 400,000/µL. Two hundred microliter of PRP was added into colorimetric cup with continuous agitation and the reader was modified to “100 %”. 5 µL ADP (20 μM final concentration) and 5 µL 50 mM PBS was added to induce platelet aggregation. The relative turbidity at 350 nm wavelength in 200 s was recorded as PAG × blank. For sample assays, 20 μM μPlg, RGD-μPlg or RGD-μPlg was added instead of PBS and the relative turbidity was recorded as PAG sample. The percentage of aggregation inhibition (Ri %) was calculated by the following formula: Ri % = (PAG × sample – PAG × blank)/(100 % − PAG × blank) × 100 %. To determine the relative potencies of the tree proteins, the Ri % was measured as a function of protein concentration.
Inhibiting thrombin-fibrinogen polymerization
The fibrinogen clotting were induced by adding 2 U bovine thrombin to human fibrinogen solutions (2.0 mg/mL fibrinogen, 25 mM CaCl2, and 50 mM phosphate buffer, pH 7.4) and incubating at 37 °C for 2 min. For anti-fibrinogen polymerization assays, the fibrinogen solutions was pretreated with 50 mM PBS, 5 μM μPlg, RGD-μPlg or GPRP-μPlg respectively before thrombin. The scattering light at 450 nm wavelength was determined every 10 s, and scattering degree (SD) was recorded in 80 s. The percent inhibition rate was calculated as (SDPBS − SDsample)/(SDPBS) × 100 %. The functions of percent inhibition rates versus protein concentrations were also calculated to evaluate the relative inhibition potencies of μPlg, RGD-μPlg or GPRP-μPlg.
Urokinase activations kinetics
The activation of μPlg, RGD-μPlg and GPRP-μPlg by urokinase was performed at a molar ratio of 1 % of urokinase at 37 °C for 24 h in buffer (50 mM phosphate buffer, pH 7.4). Reaction mixture of 30 µL was taken in 20, 40, 60, 80 min, 6, 12, 18, 24 h and assayed by 12 % reduced SDS-PAGE. The gels were scanned and the optical densities of various bands were measured to calculate activation percentage.
RGD-μPlg modeling and docked into urokinase
The RGD-μPlg structure was modeled by software DS 2.5 (Accelrys Inc., CA, USA) based on μPlg crystal structure published previously [PDB No: 1DDJ] [21]. After artificial mutation of Pro559 to Asp559 in activation loop, energy minimization was performed for the loop domain using force field CHARMm. The crystal structure of urokinase used for the dock was also previously published [PDB No: 4MNY] [22]. Dock was conducted between RGD-μPlg activation loop and enzymatic active site of urokinase using ZDOCK module of DS 2.5. The Arg561 of RGD-μPlg and the Asp194 of urokinase were set as binding site residues for filtering the docked poses, which was arrayed according to the ZDOCK scores. The top 20 poses were selected for further refinement and rescored using RDOCK module under the force field CHARMm Polar H. Only the pose with the highest RDOCK scores was selected for protein–protein interaction analysis. A similar docking was made between native μPlg and urokinse. The best poses of μPlg and RGD-μPlg were superimposed and the RMSD was calculated for evaluating the structural alterations.
Statistical analysis
Data presented in figures and tables are expressed as mean ± S.D. of duplicate determinations from 3 to 4 independent experiments. Student’s t test was used where applicable. The differences with P values < 0.05 were considered statistically significant.
Results
Construction, expression, and purification
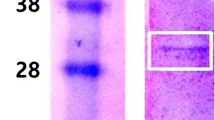
The successful constructions of three plasmids, pPICZαA-μPLG, pPICZαA-RGD-μPLG, and pPICZαA-GPRP-μPLG, were confirmed by double-enzyme cleavage and DNA sequencing. After expression in P. pastoris, SDS-PAGE showed a prominent band of 31 kDa in 12 h, which was consistent with the calculated molecular weight by amino acid sequences (Fig. 1a). Ni–NTA affinity chromatography showed these recombinant proteins reached to more than 93 % homogeneity as assayed by densitometric scanning. The averaged products of μPLG, RGD-μPLG, and GPRP-μPLG were 281 ± 25.6, 163 ± 19.0, and 198 ± 27.1 mg/L culture medium, respectively. Western blot assays confirmed the immunogenic reaction with anti-human Plg antibody (Fig. 1b).

Expression, purification and identification of RGD-µPlg and GPRP-µPlg. a Expression of RGD-µPlg and GPRP-µPlg in Pichia pastoris. Lane 1–5, RGD-µPlg expression 0, 12, 24, 36, and 48 h after menthol induction; Lane 6–10: GPRP-µPlg expression 0, 12, 24, 36, and 48 h after menthol induction. b Purification by Ni–NTA affinity chromatography and identification by western blot. Lane 1, culture supernatant; Lane 2, elution by column buffer; Lane 3–4, RGD-µPlg and GPRP-µPlg eluted by 500 mM imidazole, respectively; Lane 5–6, western blot assay of RGD-µPlg and GPRP-µPlg, respectively
Fibrinolytic activity
The fibrinolytic activities of μPlg, RGD-μPlg, and GPRP-μPlg were measured on fibrin plates in comparison with bovine plasma Plg standard (Fig. 2a). Regression analysis showed a significant linear correlation between the diameter squares of lysis zone and fibrinolytic activity (correlation coefficient = 0.9956; Fig. 2b). The specific fibrinolytic activities of μPlg, RGD-μPlg, and GPRP-μPlg were calculated according to the regression equation, which were 8.0, 7.7, and 13.3 U/mg, respectively.

Fibrinolysis assay of µPlg, RGD-µPlg, and GPRP-µPlg. a Fibrinolysis activity determined by fibrin plate method. Well 1, 50 mM phosphate buffer; Well 2–6, plasminogen standard gradients of 5.0, 2.5, 1.25, 0.63, and 0.31 U/mL respectively; Well 7–8, 50 and 100 μg/mL µPlg; Well 9–10, 50 and 100 μg/mL RGD-µPlg; Well 11–12, 50 and 100 μg/mL GPRP-µPlg. b Function of squares of halo diameters on plasminogen standard gradients. Fibrinolysis activities of samples were calculated according to the regression equation
Inhibiting ADP-induced platelet aggregation
The ADP-induced platelet aggregation was measured turbidimetrically. As shown in Fig. 3a, addition of ADP into PRP induced platelet aggregation and led to decreased turbidity. Incubating RGD-μPlg with PRP before ADP attenuated the turbidity decrease, indicating that RGD-μPlg inhibited platelet aggregation. The inhibition curve of RGD-μPlg confirmed a concentration-dependent inhibition of platelet aggregation. The slopes of linear regression of μPlg, RGD-μPlg and GPRP-μPlg were 0.008, 0.269, and 0.019, indicating that RGD-μPlg had a 33.6- and 14.2-fold stronger inhibition rate than μPlg and GPRP-μPlg, respectively (Fig. 3b).

Activity of inhibiting ADP-induced platelet aggregation. a Anti-platelet aggregation curves measured by turbidimetry. The turbidity of platelet rich plasma (PRP) without ADP was set as 100 %. After ADP addition, the relative turbidities of PRP for pretreatment with 50 mM phosphate buffer, 20 μM µPlg, RGD-µPlg, and GPRG-µPlg were recorded respectively. b percent inhibition functions of protein concentrations. Percent inhibitions were calculated as described in methods
Inhibiting thrombin-fibrinogen blotting
Thrombin-induced fibrinogen clotting transformed the fibrinogen solution into a fibrin gel, which increased astigmatism, meanwhile reducing transmittance. As shown in Fig. 4a, a progressive curve of astigmatic rate was observed during the thrombin-induced fibrin clotting process. The curve shifted to right significantly when the clotting system was pretreated with GPRP-μPlg and the clotting time was prolonged. The functions of protein concentration showed that the slopes of linear regression of μPlg, RGD-μPlg, and GPRP-μPlg were 0.178, 0.286 and 1.638, indicating that the inhibition rate of GPRP-μPlg was 9.2-fold, 5.7-fold stronger than μPlg and GPRP-μPlg, respectively (Fig. 4b).

Activity of inhibiting thrombin-induced fibrinogen polymerization. a Anti-fibrinogen polymerization curves measured by scattering turbidimetry. The fibrinogen polymerization increased the degree of light scattering, which was determined by scattering turbidimetry. The scattering ratios with time shift were recorded for pretreatment with 50 mM phosphate buffer, 5 μM µPlg, RGD-µPlg, and GPRG-µPlg. b Percent inhibition functions of protein concentrations. The relative inhibition activities were designed as slopes of the regression lines
Urokinase activation kinetics
The UK activation kinetics of μPlg, RGD-μPlg, and GPRP-μPlg were measured with reducing SDS-PAGE, in which the single-chain proenzymes, appearing as 31 kDa bands, were cleaved to form double-chain active enzymes that appeared as a 27 and a 4 kDa band under reducing conditions (Fig. 5a). When incubated with UK for 24 h, μPlg, RGD-μPlg, and GPRP-μPlg demonstrated over 80 % conversion as determined using photodensity assays. RGD-μPlg showed significantly slowed activation in 80 min but a similar activation percentage to μPlg and GPRP-μPlg in 24 h (Fig. 5b).

Activation kinetics of urokinase. a Reducing SDS-PAGE assays. µPlg, RGD-µPlg, and GPRP-µPlg of 1 μM were activated by 1 % urokinase at 37 °C for 24 h. b Urokinase activation curves. The relative activation ratios were measured by densitometry
RGD-μPlg modeling and docking with urokinase
To explain the effect of the RGD mutation on UK activation, we modeled the activation loop of RGD-μPlg and docked it into the active pocket of UK using ZDOCK. Figure 6 shows a surface representation of LMW UK docked with RGD-μPlg activation loop, clearly showing this active pocket, which provided an ideal complementary environment for Arg561 (Fig. 6a). The P559D modification led to formation of a hydrogen bond between the side-chain carbonyl oxygen of Asp559 and the main-chain amide nitrogen of Gly218 of UK, which substituted the hydrophobic interaction between native Pro559 and Gly218. This interaction promoted the moving of the whole activation loop towards the pocket wall near Gly218. At the same time, the cleavage site Arg561-Val562, which was on the opposite side of Asp559 in the activation loop, moved away from the pocket wall (Fig. 6c, d). Comparing the activation loop of RGD-μPlg with native μPlg showed that the root mean square deviation (RMSD) of main-chain atoms and side-chain atoms were 1.069 and 1.008 Å, respectively. This involved not only the translation of the framework but also the changes in the rotatable bonds of the proximate amino acid residents around Asp559 such as Gly560, Arg561, Val562, Val563, Cys566, and Val567 (Fig. 6b).

Molecular docking of µPlg and RGD-µPlg with urokinase. a Docking the activation loop of RGD-µPlg into the substrate pocket of urokinase. The RGD-µPlg activation loop was modeled using DS 2.5 based on the crystal structure of µPlg. After energy minimization, ZDOCK and RDOCK were performed to obtain the best position of the activation loop in the substrate pocket of urokinase. RGD-µPlg is displayed as sticks. The inset shows surface interaction between the activation loop and substance pocket. b Superposition of the activation loop of RGD-µPlg on µPlg. The best positions of the activation loops of RGD-µPlg and µPlg were superimposed to observe conformation changes. The green circle represented the differences of rotatable bonds between RGD-µPlg and µPlg activation loops. c, d, Interactive amino acids of urokinase with RGD-µPlg and µPlg. The activation loops of RGD-µPlg and µPlg are displayed as sticks, and the interacting amino acids of urokinase are displayed as lines. The amino acid numbers of RGD-µPlg and µPlg are referenced to full-length human plasminogen
Discussion
It has been proposed that several pathways are involved in rethrombosis after pharmacological thrombolysis. Firstly, after fibrinolytic treatment, there is a large amount of thrombin released from the degraded fibrin, which activates the coagulation system and promotes fibrinogen polymerization [23]. Secondly, the major fibrinolytic enzyme, Plm, could activate platelets and vascular endothelial cells in a receptor-dependent manner, leading to the release of arachidonate, which promotes platelet aggregation [24, 25]. Additionally, Plm could also immediately cleave and activate coagulation factor XII, and high molecular weight kininogen and complements, which activate an intrinsic coagulation cascade [26].
Given the mechanisms of rethrombosis, the present study constructed and expressed two human μPlg variants, RGD-μPlg and GPRP-μPlg. These recombinant proteins were bifunctional molecules containing the serine proteinase (SP) domain of human Plg for fibrinolysis, the RGD sequence for inhibiting platelet aggregation, and the GPRP sequence for blocking fibrinogen polymerization. The in vitro results revealed that RGD-μPlg and GPRP-μPlg demonstrated antithrombotic activity in addition to the fibrinolytic activity, whereas native μPlg only displayed fibrinolytic activity.
In RGD-μPlg, the RGD tripeptide was engineered into the activation loop of μPlg by P559D mutation (Fig. 7). The fibrinolytic activity of μPlg was not changed and the fused RGD tripeptide was able to bind to αIIb/β3 and inhibit platelet aggregation. The fibrin plate assay showed that RGD-μPlg was valid in hydrolyzing fibrin and the specific fibrinolytic activity was similar to native μPlg, although the UK activation kinetics showed a delayed activation. Molecular docking of RGD-μPlg with UK indicated that P559D did not change the binding model. However, the horizontal movement of the RGD-μPlg activation loop relative to the UK active pocket affected activation efficiency, leading to a delayed activation.

Schematic diagram of µPlg and the mutations constructed in the present study. The amino acid sequence of µPlg (Ala543-Asn791 of human plasminogen) are presented as solid lines; the six disulfide bonds are shown as dash lines; asterisks denote the catalytic triad of human plasminogen comprising His603-Asp646-Ser741; amino acids in text frames represent mutations constructed in the present study; the activation loop was locked by the Cys558-Cys566 disulfide bond; “PA” and arrow denote the activation site of plasminogen activator, including urokinase and tissue plasminogen activator. The amino acid numbers of µPlg are referenced to full-length human plasminogen
In GPRP-μPlg, the GPRP tetrapeptide was constructed using A543G, S545R, and F546P mutations at the N-terminus of μPlg (Fig. 7). The fibrinolytic activity of μPlg was enhanced, the constructed GPRP was able to inhibit fibrinogen polymerization, and the UK activation kinetics were almost unaffected compared to that with the native μPlg.
According to crystal structures of μPlg published previously [21], μPlg is folded into two β-barrels with a surface covered by various loops. The N-terminus and activation loop are exposed on the solvent-accessible surface and are far from the fibrinolytic pocket. Upon activation, the proteolysis-released N-terminus activation loop moves 12 Å to enter the fibrinolytic pocket, but the upstream cleavage site remains unchanged. This is consistent with findings that the mutations in this study had little effect on the fibrinolytic activity of μPlg, and that RGD-μPlg and GPRP-μPlg could respectively inhibit platelet aggregation and fibrinogen polymerization, indicating that GPRP and RGD in these variants play corresponding roles.
Additionally, low risk of bleeding is one of the superiorities of Plm to (recombinant tissue plasminogen activator, rt-PA), which is due to its quick neutralization by α2-antiplasmin once it separates from fibrin, leading to a short plasma half-life of 0.02 s [27]. Although μPlm has a plasma half-life 100-fold greater than Plm [28], the bleeding risk is still lower than that of rt-PA [29]. Moreover, since RGD and GPRP peptides can bind activated platelets and fibrin monomers, respectively, the addition of RGD and GPRP to μPlg may enhance platelet- and fibrin-targeted thrombolysis, which may explain the increased fibrinolytic activity of GPRP-μPlg than that of the native μPlg.
In conclusion, the present study presented two truncated plasminogen derivatives (543Ala-791Asn) which were integrated with the RGD tripeptide and the GPRP tetrapeptide, respectively. The recombinant chimeric proteins were expressed at high-level in P. pastoris and maintained similar fibrinolytic activity to native μPlg. In addition, two variants showed anti-platelet aggregation and anti-fibrin clotting properties, which provides a rationale for further investigation into their therapeutic potentials in animal models of thrombosis as well as into associated complications such as bleeding risk.
Abbreviations
- UK:
-
Urokinase
- Plm:
-
Plasmin
- t-PA:
-
Tissue-type plasminogen activator
- Plg:
-
Plasminogen
- μPlg:
-
Microplasminogen
References
Verheugt FW, Meijer A, Lagrand WK, Van Eenige MJ (1996) Reocclusion: the flip side of coronary thrombolysis. J Am Coll Cardiol 27:766–773
Laudano AP, Doolittle RF (1978) Synthetic peptide derivatives that bind to fibrinogen and prevent the polymerization of fibrin monomers. Proc Natl Acad Sci USA 75:3085–3089
Stabenfeldt SE, Aboujamous NM, Soon AS, Barker TH (2011) A new direction for anticoagulants: inhibiting fibrin assembly with PEGylated fibrin knob mimics. Biotechnol Bioeng 108:2424–2433
Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie GA, Ginsberg MH (1985) The effect of Arg-Gly-Asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc Natl Acad Sci USA 82:8057–8061
Bi Q, Cen X, Huang Y, Zhu S (2002) Construction and characterization of trifunctional single-chain urokinase-type plasminogen activators. Eur J Biochem 269:1708–1713
Bingxing S, Aiping Y, Yuying L, Li J, Jin J, Dong C, Wu C (2007) Locally activity-released bifunctional fusion protein enhances antithrombosis and alleviates bleeding risk. J Thromb Thrombolysis 24:283–292
Anmol K, Krishna Kanth P, Candasamy M, Kotra S, Rao KR (2013) Evaluation of a multifunctional staphylokinase variant with thrombin inhibition and antiplatelet aggregation activities produced from salt-inducible E. coli GJ1158. Can J Physiol Pharmacol 91:839–847
Novokhatny VV, Jesmok GJ, Landskroner KA, Marder VJ, Zimmerman TP (2004) Locally delivered plasmin: why should it be superior to plasminogen activators for direct thrombolysis. Trends Pharmacol Sci 25:72–75
Daphne S, Mansze K, Valery N, Jesmok G, Marder VJ (2003) Distinct dose-dependent effects of plasmin and TPA on coagulation and hemorrhage. Blood 101:3002–3007
Marder V (2008) Pre-clinical studies of plasmin: superior benefit-to-risk ratio of plasmin compared to tissue plasminogen activator. Thromb Res 1223:S9–S15
Thijs VN, Peeters A, Vosko M, Aichner F, Schellinger PD, Schneider D, Neumann-Haefelin T, Röther J, Davalos A, Wahlgren N, Verhamme P (2009) Randomized, placebo-controlled, dose-ranging clinical trial of intravenous microplasmin in patients with acute ischemic stroke. Stroke 40:3789–3795
Marder VJ, Manyak S, Gruber T, Goyal A, Moreno G, Hunt J, Bromirski J, Scuderi P, Petteway SR Jr, Novokhatny V (2010) Haemostatic safety of a unique recombinant plasmin molecule lacking kringles 2-5. J Thromb Haemost 104:780–787
Peter V, Martine J, Godelieve G, Devis J, Maleux G, Stas M (2009) A pilot trial of microplasmin in patients with long-term venous access catheter thrombosis. J Thromb Haemost 28:477–481
Fu J, Ren J, Zou L, Bian G, Li R, Lu Q (2008) The thrombolytic effect of miniplasmin in a canine model of femoral artery thrombosis. Thromb Res 122:683–690
Varma R, Haller JA, Kaiser PK (2015) Improvement in patient-reported visual function after ocriplasmin for vitreomacular adhesion: results of the microplasmin for intravitreous injection–traction release without surgical treatment (mivi-trust) trials. JAMA Ophthalmol 133:97–100
Shi GY, Wu HL (1988) Isolation and characterization of microplasminogen. A low molecular weight form of plasminogen. J Biol Chem 263:17071–17075
Wu HL, Shi GY, Bender ML (1988) Preparation and purification of microplasmin. Proc Natl Acad Sci USA 84(23):8292–8295
Liu R, Bing Z, Zhang Y, Gu J, Yu M, Song H, Yu M, Mo W (2015) High-level expression, purification, and enzymatic characterization of truncated human plasminogen (Lys531-Asn791) in the methylotrophic yeast Pichia pastoris. BMC Biotechnol 15:50. doi:10.1186/s12896-015-0179-z
Astrup T, Mullertz S (1952) The fibrin plate method for estimating fibrinolytic activity. Arch Biochem Biophys 40:346–351
Walkowiak B, Kralisz U, Michalec L, Majewska E, Koziolkiewicz W, Ligocka A, Cierniewski CS (2000) Comparison of platelet aggregability and P-selectin surface expression on platelets isolated by different methods. Thromb Res 99:495–502
Wang X, Terzyan S, Tang J, Loy JA, Lin X, Zhang XC (2000) Human plasminogen catalytic domain undergoes an unusual conformational change upon activation. J Mol Biol 295:903–914
Spraggon G, Phillips C, Nowak UK, Ponting CP, Saunders D, Dobson CM, Stuart DI, Jones EY (1995) The crystal structure of the catalytic domain of human urokinase-type plasminogen activator. Structure 3:681–691
Ewald GA, Eisenberg PR (1995) Plasmin-mediated activation of contact system in response to pharmacological thrombolysis. Circulation 91:28–36
Chang WC, Shi GY, Chow YH, Chang LC, Hau JS, Lin MT, Jen CJ, Wing LY, Wu HL (1993) Human plasmin induces a receptor- mediated arachidonate release coupled with G proteins in endothelial cells. Am J Physiol 264:C271–C281
Kimura M, Andersen TT, Fenton JW 2nd, Bahou WF, Aviv A (1996) Plasmin-platelet interaction involves cleavage of functional thrombin receptor. Am J Physiol 271:C54–C60
Ogiwara K, Nogami KK, Shima M (2010) Plasmin-induced procoagulant effects in the blood coagulation: a crucial role of coagulation factors V and VIII. Blood Coagul Fibrinolysis 21:568–576
Christensen U, Bangert K, Thorsen S (1996) Reaction of human alpha2- antiplasmin and plasmin stopped-flow fluorescence kinetics. FEBS Lett 387:58–62
Nagai N, Demarsin E, Van Hoef B, Wouters S, Cingolani D, Laroche Y, Collen D (2003) Recombinant human microplasmin: production and potential therapeutic properties. J Thromb Haemost 1:307–313
Suzuki Y, Nagai N, Collen D (2004) Comparative effects of microplasmin and tissue-type plasminogen activator (tPA) on cerebral hemorrhage in a middle cerebral artery occlusion model in mice. J Thromb Haemost 2:1617–1621
Acknowledgments
This work was supported by Grants B2013105 from Chinese Hubei Provincial Department of Education and grants 81102502 from Chinese National Natural Science Funds. We are grateful to Shuang Zhu for providing the electroporator and Yun Xiao for anti-platelet aggregation analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Rights and permissions
About this article

Cite this article
Chen, W., Li, Y., Chen, P. et al. In vitro fibrinolysis and antithrombosis characterizations of novel recombinant microplasminogen with RGD and GPRP peptides. J Thromb Thrombolysis 42, 118–126 (2016). https://doi.org/10.1007/s11239-016-1334-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-016-1334-7