
Abstract
Following our quest for N-heterocyclic Hammick silylenes, we have to probe the electronegative and electropositive substitutions on the singlet (s) and triplet (t) silapyridine-4-ylidene fused by two furan rings compared to the synthesized silylenes by West (I-s), Denk (II-s), and Kira (III-s). In all cases, s silapyridine-4-ylidenes emerge as ground state, revealing more stability than their corresponding t analogous. All species seem to have a minimum on their energy surfaces, demonstrating the positive force constant and the positive frequency. The optimized s silylenes show bond length, divalent and dihedral bond angles, somewhat similar to their corresponding t congeners. Irrespective of how substituent groups are arranged in either the “W (ortho)” or “chair (para)” positions of the silylenic center, the most stability is verified by the substitution of more electronegative NH and O groups (as electron withdrawing groups; EWGs) in the corresponding furan rings, while the least stability is respected by III-s. In contradiction to previous reports on the N-heterocyclic Hammick carbenes, silylenes, and germylenes (NHCs, NHSis, and NHGes) that size, type, and orientation, in addition to the number of fused rings, formulated influence on ΔΕs-t, ΔΕHOMO-LUMO, and reactivity of the corresponding divalent species, now PH, AsH, S, and Se (as σ and π- electron donating groups; EDGs) similar to EWGs stabilize their silylenic derivatives. In going from second row to third row and from third row to forth row of every group in the periodic table, stability is decreased. As a result, the stability and electronic properties of s and t NHSis are considerably dependent on the electronegativity and radius of the substituted dopants.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The chemistry of divalent compounds such as carbenes, organosilicon, etc. has fascinated a considerable attention in recent years [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. Divalent silylenes are congeners of carbenes, but mostly seem to have the s configuration as the ground state [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. The larger size of the valence orbitals of silicon reduces the electron–electron repulsion of the lone pair on the Si atom accordingly enlarges the split energy of electrons. Thus, a silylene energetically favors the s ground state as a stable configuration [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. These intermediates are employed in light-emitting diode, electroluminescence, Si chemical vapor deposition procedures, optics, electronics, and semiconductors [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. The simplest, acyclic, linear, and unsaturated silylene with H2C = Si molecular structure has been of great interest as a possible divalent intermediate in numerous organosilicon reactions. It may be established in the interstellar medium [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. Silylidenes have been the subject of various theoretical investigations and experimental surveys [16,17,18,19,20]. The first unsaturated silylene was searched by Murrell et al. in 1977 at theoretical methods and levels [21]. The experimental findings recognized silylidene via its electronic absorption spectrum, in 1979 [22] and later its microwave spectrum [23]. Optical properties, structural parameters, rotational constants, force constants, and vibrational frequencies of silavinylidene derivatives were estimated using computational chemistry [24,25,26,27]. In 1997, laser-induced fluorescence spectroscopy and theoretical expectations were used in order to establish the ground state of this divalent species [28]. The influences of the substituted polar groups on thermodynamic stability (ΔΕs-t) were first described by Hopkinson et al. [29]. In spite of many investigations, achieving at s (ΔΕs-t > 0) vs. t ground state (ΔΕs-t < 0) for saturated and unsaturated NHCs, NHSis, and NHGes seems as a challenging issue. If the promotion energy is increased, the s ground state will be reached. If the promotion energy is decreased, the t ground state will be reached. To this end, electronic, inductive, mesomeric, and steric influences are applied to change the multiplicity of NHCs, NHSis, and NHGes [29,30,31,32,33,34]. For instance, the substituted EDGs increase the 3p-character of Si valence orbitals which leads to t configuration. Gordon identified the first t ground state silylenes [17, 18, 30]. Apeloig researched the effects of EDGs on the multiplicity of silylenes [19, 20, 31]. Considering applications of unsaturated silylenes and increasing demand for stable NHSis [34], here we have studied substituent effects of EWGs and EDGs on two “W” and “chair” orientations with silylenic centers of 1-s, 2x-s, 3x-s, and 1-t, 2x-t, and 3x-t Hammick silylenes (x = NH, PH, AsH, O, S, and Se), at DFT (Fig. 1) [35,36,37,38,39].


The optimized structures of the studied NHSis in this work
Computational methods
Geometry optimizations are carried out without any symmetry constraint, operating the GAMESS [40, 41] program at the (U)B3LYP [42,43,44,45] and (U)M06-2X methods [46, 47] together with 6–311 + + G** and AUG-cc-pVTZ basis sets [48,49,50,51,52,53,54,55]. All optimized structures turn out to be minima on their energy surfaces for showing no imaginary frequency. The s-t energy gap (ΔEs-t), energies of the frontier molecular orbital (FMO), band gap (ΔΕHOMO-LUMO and ΔΕSOMO-SOMO+1 for s and t species, respectively), ionization potential (IP), and electron affinity (EA) are calculated at 298.15 K and 1.00 atm [56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79]. The natural bond orbital (NBO) charges [56,57,58,59], the global reactivity descriptors [80,81,82,83], and the condensed Fukui function (CFF) are provided at the same level of theory [84, 85]. The nucleophilicity index (N) is acquired from the energy difference between EHOMO of s NHSi (or ESOMO of t NHSi) and tetracyanoethylene [80,81,82,83]. The electrophilicity index (ω = μ2/2η), chemical potential (μ = (EHOMO-ELUMO)/2), and the maximum amount of electronic charge index (ΔNmax = − μ/\(\eta\))are obtained as well [80,81,82,83]. The CFF descriptors are calculated using the Multiwfn program via f + A = ρAN + 1 − ρAN (ρA is the electron population number of A atom and N refers to the number of electrons stable states), f + A = qAN + 1 − qAN (nucleophilic attack), f − A = qAN − qAN − 1 (electrophilic attack), and DfA = f + A − f − A (condensed dual descriptor) [84, 85]. The site is favored for a nucleophilic attack if DfA > 0 and is favored for an electrophilic attack if DfA < 0.
Results and discussion
Following the previous research works on divalent compounds [34,35,36,37,38,39], in this research the substitution effects were studied on the singlet (s) and triplet (t) silapyridine-4-ylidene fused by two furan rings. The physical prarmeters have been obtained using DFT calculations according to the previous reports [60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79].The present work covers thermodynamic, kinetic, and electronic effects of EWGs and EDGs on the scrutinized s and t NHSis vs. the first synthesized silylenes at DFT (Schemes 1) [86,87,88,89].

The synthesized silylene by West (I-s), Denk (II-s) as well as Kira (III-s)
Recently, different theoretical studies has been carried out on fusion of the different rings of pyridine-4-ylidene and heavier derivatives (Schemes 2).

The reported newly divalent species in the computational chemistry
The resulted reports reveal that heteroatom type, size of the fused ring, number of fused rings, and orientation of the substituted dopants change stability and other properties of these divalent species. Our s and t NHSis turn out to be minima for showing no negative force constant. Structural parameters including bond length (R; C—Si and Si—C) in angstrom, divalent and dihedral bond angles in degree (\(\widehat{\mathrm{A}}\) (C − Si − C) and \(\widehat{\mathrm{D}}\) (C − Si − C − X), respectively), and symmetry of silylenes are considered. Except for 3AsH-s and 3PH-s with C1 symmetry, structural optimizations of other silylenes show Cs symmetry for all s and t states. All silylenes show \(\widehat{\mathrm{D}}\cong\) 180°. In each of the three series, 1-s, 2x-s, and 3x-s silylenes show more R (about 0.020–0.040 Å) and less \(\widehat{\mathrm{A}}\) (about 10°) than their 1-t, 2x-t, and 3x-t analogous. For instance, 2O-s and 3AsH-s show R of 1.868, 1.890, 1.853, and 1.846 Å, also \(\widehat{\mathrm{A}}\) of 92.461° and 94.434°, respectively. Likewise, 2O-t and 3AsH-t show R of 1.878, 1.876, 1.864, and 1.836 Å, also \(\widehat{\mathrm{A}}\) of 102.021° and 104.150°, respectively. Our silylenes turn out as singlet ground state for showing positive ΔEs-t values [33]. In series 1, while fusion of two benzene rings thermodynamically stabilizes the corresponding silylene (ΔEs-t = 185.30 kJ/mol), in series 2 and 3, by decreasing the electronegativity and increasing the atomic radius, the ΔEs-t diminishes from 215.10 kJ/mol for EWGs to 193.66 kJ/mol for EDGs (Table 1).
All silylenes show more ΔEs-t than III (138.48 kJ/mol). This is because of the higher electronegativity of EWGs which makes them a stronger π-acceptor and hence prefers the singlet over the triplet state which causes a higher ΔEs-t. In going from the most electronegative heteroatom to the least electronegative one, the ΔEs-t is decreased. More π-electron delocalization of the substituted dopantʼs free electron pair in two furan rings and a silapyridine ring makes possible more polarity and less polarizability of the s NHSis with regard to the t ones. Then again, substitution of either EWGs or EDGs groups in a zigzag (W) arrangement to the silylenic center produces less polarity. In accordance with Hoffmann et al.’s statement [89], a molecule could be known as stable if its smallest vibrational frequency is at least 100 cm−1 and it reveals an enormous FMO energy difference (here ΔEHOMO-LUMO for s states and ΔΕSOMO-SOMO+1 for t ones) [86,87,88]. Now, vibrational analysis demonstrates that the scrutinized NHSis including 1-s, 2x-s, 3x-s, 1-t, 2x-t, and 3x-t, where x = NH, PH, AsH, O, S, and Se, are real minimums revealing positive frequency, and hence positive force constant (Table 2).
While fusion of two benzene rings kinetically stabilizes the s and t states of the corresponding silylenes, 1-s (ΔEHOMO-LUMO = 364.96 kJ/mol) and 1-t (ΔΕSOMO-SOMO+1 = 290.09 kJ/mol), in two other series by decreasing the electronegativity of x dopants and increasing their atomic radius, the kinetic stability is diminished from 393.46 for EWGs to 191.61 kJ/mol for EDGs (Table 2). Hence, amid s NHSis, the most kinetic stable structures are 2NH-s and 2O-s (393.38 and 393.46 kJ/mol), and the least stable ones are 3PH-s and 3AsH-s (378.04 and 379.50 kJ/mol), correspondingly, which they are stabilized considerably more than IIIs (351.33 kJ/mol). Between t NHSis, 2NH-t and 2O-t species to some extent accomplish the abovementioned conditions by Hoffmann et al., indicating the smallest vibrational frequency of 73.42 and 73.57 cm−1 besides ΔΕSOMO-SOMO+1 of 299.92 and 277.68 kJ/mol respectively. Between t NHSis, 3AsH-t species contravene the above conditions, emerging lower vibrational frequency of 66.96 cm−1 and a lower ΔΕSOMO-SOMO+1 (191.61 kJ/mol) than that of the distinguished by Hoffmann and co-workers. The band gap of all s and t NHSis is more than III-s (ΔEHOMO-LUMO = 351.33 kJ/mol) and III-t (ΔΕSOMO-SOMO+1 = 138.32 kJ/mol). Among these synthesized silylenes, the silylenic centers of I-s and II-s molecules completely enjoy from π-conjugation of two nitrogen groups adjacent to their silylenic centers, while III-s benefits only from the hyper-conjugation effect of four SiMe3 groups. This difference significantly is influenced on stability of them. Here, the optimized NHSis similar to III silylene suffer from lack of π-donating of the neighboring nitrogen groups to silylenic centers. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of s NHSis is considered dissimilar to the semi occupied molecular orbital (SOMO, SOMO + 1) of t NHSis (for instance, 2O-s and 2O-t silylenes, Fig. 2).

The FMO shapes of the selected 2O-s and 2O-t silylenes
Replacement of two EWGs in chair organizations to the silylenic center of 2O-s progresses unbroken π-conjugation by employing 3σ2 or sp2—lone pair of the divalent center and reduces HOMO energy of 1-s from −520.13 to −581.25 kJ/mol in 2O-s and hence increases stability of HOMO of 2O-s structure. Undoubtedly, conjugation of the free electron pairs on EWGs with π-electrons of the fused double bond (C = C) in the silapyridine ring of 2O-s structure, decreases LUMO energy of 1-s from −155.28 to −187.80 kJ/mol in 2O-s (Fig. 2). Moreover, conjugation of the unpaired lone pairs on EWGs with the semi-filled 3pπ-orbital of the silylenic center of 2O-t diminishes the SOMO energy of 1-t from −322.10 to −354.89 kJ/mol in 2O-t and henceforth increases the stability of SOMO in 2O-t. Furthermore, replacement of two EWGs in chair positions to the silylenic center of the 2O-t structure adjusts π-conjugation by employing the half-captured s1p1 orbital of the silylenic center and free electrons of furanʼs oxygen and hence reduces SOMO + 1 energy of 1-t from −32.00 to −77.11 kJ/mol then increases stability of SOMO + 1 in 2O-t (Fig. 2). Delocalization of free electron pairs on EDGs and oxygen heteroatoms in the silapyridine ring not only leads to low leveling of HOMO and SOMO (e.g., from −520.13 in 1-s to −538.49 kJ/mol in 3AsH-s and from −322.10 in 1-t to −330.75 kJ/mol in 3AsH-t) but also delocalization in the fused furan rings changes LUMO and SOMO + 1 energy (e.g., from −155.28 in 1-s to −158.95 kJ/mol in 3AsH-s and from −32.00 in 1-t to −139.02 kJ/mol in 3AsH-t, Fig. 3).

The FMO shapes of the selected 3AsH-s and 3AsH-t silylenes
Interestingly, the presence of two EWGs in the fused furan rings either in the “W” or “chair” positions of the silylenic center of the s and t NHSis engages π-cross conjugation (β-SOMO + 1 in 2O-t, Fig. 2) and the resulted ring current of the silapyridine ring has more than two EDGs. The polarity and polarizability of s NHSis with the substituted EWGs are different from those of s NHSis with the substituted EDGs and even the corresponding t ones. This observation implies that besides the inductive effect of more electronegative elements, by involving lone pairs of the substituted groups and silylenic centers in mesomeric effect, and π-conjugation polarity is decreased due to the increment of ring current with respect to chair arrangement to silylenic center (Table 2). Consistent with the DOS plots, every s NHSi displays more Eg value than its corresponding t NHSi so that the most value is estimated for the substituted EWG—singlet silylenes, and the least value is calculated for the substituted EDG—triplet silylenes. For example, 4.00 eV for 2O-s vs. 2.56 eV for 2O-t (Fig. 4), also 3.94 eV for 3AsH-s vs. 1.99 eV for 3AsH-t (Fig. 5).

The density of state (DOS) plots of the selected 2O-s and 2O-t silylenes

The DOS plots of the selected 3AsH-s and 3AsH-t silylenes
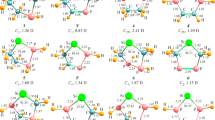
Dissimilar to nonpolar and symmetric (achiral) molecules, the NBO charge is not commonly dispersed over the fused furan and silapyridine rings, revealing the inductive effect along with the mesomeric effect of the substituted EWGs and EDGs, respectively. For instance, we are shown the calculated NBO charges for the 2O-s, 2O-t, 3AsH-s, and 3AsH-t silylenes (Figs. 6 and 7).

The NBO charges calculated for the selected 2O-s and 2O-t silylenes

The NBO charges calculated for the selected 3AsH-s and 3AsH-t silylenes
Regardless of the W (zigzag) and/or chair arrangement, the positive NBO charge is dispersed over hydrogen, carbon, silylenic center, and EDGs, while the negative NBO charge is dispersed over silapyridinicʼs nitrogen, carbon, and EWGs. The dispersed NBO charge qualitatively confirms that the substituted EWGs and EDGs in the difuranosilapyridine structure either in the “W” or “chair” positions of the silylenic center stabilize not only the s NHSis but also the t ones. The negative and positive NBO charges of the s and t NHSis indicate that these sites can be attacked more easily by electrophilic and nucleophilic regents, correspondingly. The electrostatic potential (ESP) maps qualitatively confirm blue color for positive charge, red color for negative charge, the electron cloud in the middle of two rings, and the dependency of NBO charge on electronegativity of EWGs and EDGs (Fig. 8).

The ESP maps and contours of the selected 2O-t and 3AsH-t silylenes. The red and blue regions on the scale bar indicate the lowest and the highest ESP values, respectively
For enquiry of the substituent effects of EWGs and EDGs on global reactivity of their corresponding s and t NHSis, we are calculated N, ω, μ, η, S, and ΔNmax (Table 3).
A commonly found finding is the lower N, higher ω, higher absolute value of μ, higher η, and lower S of every s NHSis than every t congener. The positive ΔNmax index exhibits the positive charge capacity of every s and t NHSi, and here all species are expected electron acceptor in organic chemistry reactions or catalysts. Certainly, the five-membered furan ring contains lower basicity than the six-membered pyridine ring and other amines. The reduced basicity is strengthened by π-delocalization of the free electron on the oxygen atom of the furan ring. In fact, the smaller bond angle of the furan ring (about 12°) than the pyridine ring enlarges the p character of the bonding sp2 orbital and the s character of the nonbonding σ orbital. Here, the higher s character of the nonbonding σ orbital of either the doped EWGs or EDGs in either W or chair positions leads to lower N and higher ω of their s and t NHSis than in 1-s and 1-t structures. The Hirshfield charges, CFFs, atomic, and global indices are calculated to predict the chemical system’s reactive sites and character. Here, the Hirshfield charges, CFFs, and Dfs for silylenic centers of the selected singlet NHSis display dissimilar trends dependent on the electronegativity, size, and topology of the substituted heteroatoms of NH, PH, AsH, O, S, and Se, either in the “W (ortho)” or “chair (para)” position of the silylenic center, in the fused rings (Table 4).
For example, the silylenic center of the 2NH-s species shows the most nucleophilicity via the highest f − (−0.3889 e), the lowest f + (−0.1241 e), and the most positive value of Df (+ 0.2649 e) between NHSis, while the silylenic center of the 3AsH-s species shows the most electrophilicity via the lowest f − (−0.1611 e), the highest f + (−0.5044 e), and the most negative value of Df (-0.3433 e) between NHSis. Comparatively, at the level of computation, the IP and EA for benzene (a reference molecule) have been reported to be 889.69 and −66.51 kJ/mol, respectively [90]. A closed look at 1-s and the substituted NHSis (2x-s and 3x-s) shows that the electrons are more and less binded than those of benzene, respectively (Table 5).
Interestingly, the 1-s species shows the most positive value of IP and EA (691.44 and 303.75 kJ/mol), then the 2NH-s and 3NH-s isomers show the most positive value of IP and EA (361.00 and 50.49 kJ/mol, respectively), while the 3AsH-s species shows the most negative value of EA (−170.42 kJ/mol) between NHSis. Hence, substituent effects compete with the aromaticity of NHSis because substituentsʼ interaction with the π-system decreases degree the of π-electron delocalization.
Conclusion
To reach for novel s and t NHSis, we have studied electronic effects on structural, thermodynamic, and kinetic factors of the fused dibenzo and difuranosilapyridine-4-ylidenes. To this end, three series structures (from 1-s to 3x-t; x = NH, PH, AsH, O, S, and Se) are compared and contrasted with some of the synthesized silylenes using DFT. Every s NHSi exhibits more polarity, lower polarizability, more positive frequency, and higher ΔΕHOMO-LUMO than its corresponding t NHSi; yet ΔΕHOMO-LUMO of them is more than III-s and III-t. In other words, all silylenes substituted by EWGs and EDGs display more stability than III, so that the ΔEs-t and ΔΕHOMO-LUMO ranges are changed from 193.66 and 379.50 kJ/mol for 3AsH to 215.10 and 393.46 kJ/mol for 2O-s vs. 138.48 and 351.33 kJ/mol for III-s, respectively. The ESP contour maps qualitatively confirm the distributed NBO atomic charge and determine nucleophilic and electrophilic sites. Moreover, s silylenes show lower nucleophilicity (N), higher electrophilicity (ω), chemical potential (μ), and global hardness (η) than their related t congeners. The fused furan NHSis (either s or t ones) reveal lower N and higher ω than 1-s and 1-t structures on account of the inductive effect and mesomeric effect of the EWGs and EDGs on the completed σ2 orbital and 3pπ2 orbital of their silylenic centers. Indeed, the stability and electronic properties of s and t NHSis are considerably dependent on the electronegativity and radius of the substituted groups. We hope for experimental investigations that substitute the different heteroatoms in two fused furan silapyridines either the “W” or “chair” position of the silylenic center with the purpose of stabilizing not only the s silylenes but also the t congeners. Based on the Hirshfield charge, CFF, and Df results, the silylenic center of the 2NH-s species shows the highest nucleophilicity, whereas the silylenic center of the 3AsH-s species shows the highest electrophilicity. A close look at 1-s and the substituted NHSis (2x-s and 3x-s) shows that the electrons are more and less binded than benzene.
Data availability
Not applicable.
References
Koohi M (2019) Cyclonona-3,5,7-trienylidene and its Si, Ge, Sn, and Pb analogs versus their α-halogenated derivatives at B3LYP and MP2 methods. J Phys Org Chem 32:e4013
Koohi M (2020) Estimating the stability and reactivity of cyclic tetrahalo substituted germylenes: a density functional theory investigation. J Phys Org Chem 33:e4032
Koohi M, Bastami H (2020) A quest for stable 2,2,9,9-tetrahaloplumbacyclonona-3,5,7-trienylidenes at density functional theory. Struct Chem 31:877
Koohi M, Bastami H (2020) Substituent effects on stability, MEP, NBO analysis, and reactivity of 2,2,9,9-tetrahalosilacyclonona-3,5,7-trienylidenes, at density functional theory. Monatshefte für Chemie - Chemical Monthly 151:11
Koohi M, Bastami H (2020) A density functional theory perspective on 2,2,9,9-tetrahalostannacyclonona-3,5,7-trienylidenes. J Phys Org Chem 33:e4031
Koohi M, Bastami H (2020) Substituted Hammick carbenes: the effects of fused rings and hetero atoms through DFT calculations. J Phys Org Chem 33:e4023
Kassaee MZ, Koohi M (2013) Breathing viability into cyclonona-3,5,7-trienylidenes via a-dimethyl and ά-moieties at DFT. J Phys Org Chem 26:540
Kassaee MZ, Koohi M, Mohammadi R, Ghavami M (2013) 2,2,9,9-Tetramethylcyclonona-3,5,7-trienylidene vs. its heterocyclic analogues: a quest for stable carbenes at DFT. J Phys Org Chem 26:908
Koohi M, Kassaee MZ, Haerizade BN, Ghavami M, Ashenagar S (2015) Substituent effects on cyclonona-3,5,7-trienylidenes: a quest for stable carbenes at density functional theory level. J Phys Org Chem 28:514
Gehrhus B, Lappert MF, Heinicke J, Boese R, Bläser D (1995) Synthesis, structures and reactions of new thermally stable silylenes. J Chem Soc Chem Commun 0:1931
Heinicke J, Oprea A, Kindermann MK, Karpati T, Nyulaszi L, Veszpremi T, Hitchcock PB, Lappert MF, Maciejewski H (1998) Silylenenickel(0) or silyl(silylene)platinum(II) complexes by reaction of Si[(NCH2But)2C6H4–1,2] with [NiCl2(PPh3)2], [Ni(cod)2], or [PtCl2(PPh3)2]. Organomet 17:5599
Becerra R, Walsh R (2010) Dalt Trans 39:9217
Kassaee MZ, Zandi H, Haerizade BN, Ghambarian M (2012) Effects of α-mono heteroatoms (N vs. P), and β-conjugation on cyclic silylenes. Comput Theor Chem 1001:39
Ayoubi‐Chianeh M, Kassaee MZ, Ashenagar S, Cummings PT (2019) Nucleophilicity of cyclic conjugated silylenes using DFT method. J Phys Org Chem 32:ee3956
Cote DR, Van Nguyen S, Stamper AK, Armbrust DS, Tobben D, Conti RA, Lee GY (1999) Plasma-assisted chemical vapor deposition of dielectric thin films for ULSI semiconductor circuits. IBM J Res Dev 43(1.2):5
Hopkinson AC, Lien MH, Csizmadia LG (1983) Ab initio calculations on the singlet and triplet energy surfaces for CsiH2. Chem Phys Lett 95:232
Gordon MS, Pople JA (1981) Structure and stability of a silicon-carbon triple bond. J Am Chem Soc 103:2945
Gordon MS (1982) Effects of polar substituents on carbon-silicon multiple bonds. J Am Chem Soc 104:4352
Luke BT, Pople JA, Krogh-Jespersen M-B, Apeloig Y, Karni M, Chandrasekhar J, Schleyer PVR (1986) A theoretical survey of unsaturated or multiply bonded and divalent silicon compounds. Comparison with carbon analogs. J Am Chem Soc 108(2):270
Kosa M, Karni M, Apeloig Y (2013) Were reactions of triplet silylenes observed? J Am Chem Soc 135(24):9032
Murrell JN, Kroto HW, Guest MF (1977) Double-bonded divalent silicon: ab-initio calculations on the species HSiN, HNSi, HCSiH, and H2CSi. J Chem Soc Chem Commun 17:619
Leclercq H, Dubois I (1979) The absorption spectrum of the H2CSi radical. J Mol Spectrosc 76(1–3):39
Izuha M, Yamamoto S, Saito S (1996) Microwave spectrum of silylidene H2CSi. J Chem Phys 105:4923
Smith TC, Evans CJ, Clouthier DJ (2003) Discovery of the optically forbidden S1–S0 transition of silylidene (H2C=Si). J Chem Phys 118:1642
Sherrill CD, Schaefer HF III (1995) 1-silavinylidene: the first unsaturated silylene. J Phys Chem 99:1949
Hoffmann MR, Yoshioka Y, Schaefer HF III (1983) Vibrational frequencies for silaacetylene and its silylidene and vinylidene isomers. J Am Chem Soc 105:1084
Sharma M, Sharma MK, Verma UP, Chandra S (2014) Radiative transfer in silylidene molecule. Serbian Astron J 2014:37
Harper WW, Ferrall EA, Hilliard RK, Stogner SM, Grev RS, Clouthier DJ (1997) Laser spectroscopic detection of the simplest unsaturated silylene and germylene. J Am Chem Soc 119(35):8361
Bourissou D, Guerret O, Gabbai FP, Bertrand G (2000) Stable carbenes. Chem Rev 100:39
Gordon MS (1985) Potential-energy surfaces in singlet and triplet silylene. Chem Phys Lett 114:348
Holthausen MC, Koch W, Apeloig Y (1999) Theory predicts triplet ground-state organic silylenes. J Am Chem Soc 121(11):2623
Nemirowski A, Schreiner PR (2007) Electronic stabilization of ground state triplet carbenes. J Org Chem 72(25):9533
Dubois I, Herzberg G, Verma RD (1967) Spectrum of SiH2. J Chem Phys 47(10):4262
Ghaghaei M, Babazadeh M, Behmagham F, Edjlali L, Vessally E (2023) A theoretical evaluation for new fused remote N-heterocyclic silylenes (RNHSis) using density functional theory. J Phys Org Chem 16(7):e4475. https://doi.org/10.1002/poc.4475
Zhao H, Yang D, Zhou Y, Fang Y, Shi M, Vessally E (2020) A computational quest for the effects of fused rings on the stability of Hammick carbenes type remote N-heterocyclic carbenes. J Chin Chem Soc 68:76
Delir Kheirollahi Nezhad P, Youseftabar-Miri L, Ahmadi S, Ebrahimiasl S, Vessally E (2021) A DFT quest for effects of fused rings on the stability of remote N-heterocyclic carbenes. Struct Chem 32:787
Hassanpour A, Poor Heravi MR, Rahmani Z, Ebadi AG, Ahmadi S (2021) Characterization of novel pyridine-derived N-heterocyclic silylenes via density functional theory perspective. J Chin Chem Soc 68:1405
Söğütlü İ, Soltanzadeh M, Mert H, Mert N, Vessally E (2021) Substituent effects on the stability of cyclic - unsaturated remote N-heterocyclic Hammick carbenes using density functional theory. J Mol Struct 1230:129821
Zhao K, Zhang Y, Ma Y, Jin Z, Rashid Sheykhahmad F (2020) Stabilization of novel N-heterocyclic Germylenes (NHGes): a computational perspective. J Chin Chem Soc 68:274
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14(11):1347
Sobolewski AL, Domcke W (2002) Ab initio investigation of the structure and spectroscopy of hydronium−water clusters. J Phys Chem A 106:4158
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098
Becke AD (1993) Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648
Becke AD (1996) Density‐functional thermochemistry. IV. A new dynamical correlation functional and implications for exact‐exchange mixing. J Chem Phys 104:1040
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41(2):157
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120:215
Hariharan PC, Pople JA (1974) Accuracy of AH, equilibrium geometries by single determinant molecular orbital theory. J Mod Phys 27:209
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second row elements. J Chem Phys 77:3654
Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PVR (1983) Efficient diffuse function-augmented basis sets for anion calculations. III. The 3–21+G set for first-row elements, Li-F. J Comput Chem 4:294
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25: supplementary functions for Gaussian basis sets. J Chem Phys 80:3265
Hehre WJ, Radom L, PvR S, Pople JA (1986) Ab initio molecular orbital theory. John Wiley & Sons, New York
Foresman JB, Frisch A (1996) Exploring chemistry with electronic structure methods, Gaussian, Inc., Pittsburgh. PA
Kendall RA, Dunning TH Jr, Harrison RJ (1992) Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796
Krishna R, Frisch MJ, Pople JA (1980) Contribution of triple substitutions to the electron correlation energy in fourth order perturbation theory. J Chem Phys 72:4244
Weinhold F, Glendening ED (2001) NBO 7.0 Program Manual Natural Bond Orbital Analysis Programs
Weinhold F (2012) Natural Bond Orbital Analysis: A Critical Overview of Relationships to Alternative Bonding Perspectives. J Comput Chem 33:2363
Glendening ED, Landis CR, Weinhold F (2012) Natural bond orbital methods. Wiley Interdiscip Rev Comput Mol Sci 2:1
Zhang G, Musgrave CB (2007) Comparison of DFT Methods for Molecular Orbital Eigenvalue Calculations. J Phys Chem A 111:1554
Gharibzadeh F, Vessally E, Edjlali L, Es’haghi M, Mohammadi R (2020) A DFT study on sumanene, corannulene and nanosheet as the anodes in Li−ion batteries. Iran J Chem Chem Eng 39:51
Afshar M, Khojasteh RR, Ahmadi R, Nakhaei Moghaddam M (2021) In silico adsorption of lomustin anticancer drug on the surface of boron nitride nanotube. Chem Rev Lett 4:178
Vessally E, Hosseinian A (2021) A computational study on the some small graphene-like nanostructures as the anodes in Na−ion batteries. Iran J Chem Chem Eng 40:691
Hashemzadeh B, Edjlali L, Delir Kheirollahi Nezhad P, Vessally E (2021) A DFT studies on a potential anode compound for Li-ion batteries: hexa-cata-hexabenzocoronene nanographen. Chem Rev Lett 4:232
Vessally E, Farajzadeh P, Najafi E (2021) Possible sensing ability of boron nitride nanosheet and its Al– and Si–doped derivatives for methimazole drug by computational study. Iran J Chem Chem Eng 40:1001
Cao Y, Rostami Z, Ahmadi R, Azimi SB, Nayini MMR, Ansari MJ (2022) Study the role of MgONTs on adsorption and detection of carbon dioxide: First-principles density calculations. Comput Theor Chem 1208:113572
Salehi N, Vessally E, Edjlali L, Alkorta I, Eshaghi M (2020) Nan@Tetracyanoethylene (n=1-4) systems: sodium salt vs sodium electride. Chem Rev Lett 3:207
Soleimani-Amiri S, Asadbeigi N, Badragheh S (2020) A theoretical approach to new triplet and quintet (nitrenoethynyl) alkylmethylenes,(nitrenoethynyl) alkylsilylenes, (nitrenoethynyl) alkylgermylenes. Iran J Chem Chem Eng (IJCCE) 39(4):39
Arshadi S, Abdolahzadeh F, Vessally E (2023) Butadiyne-linked porphyrin nanoring as a highly selective O2 gas sensor: A fast response hybrid sensor. J Mol Graph Model 119:108371
Khalif M, Daneshmehr S, Arshadi S, Söğütlü İ, Mahmood EA, Abbasi V, Vessally E (2023) Adsorption of O2 molecule on the transition metals (TM (II)= Sc2+, Ti2+, V2+, Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) porphyrins induced carbon nanocone (TM (II) PCNC). J Mol Graph Model 119:108362.
Kamel M, Mohammadifard M (2021) Thermodynamic and reactivity descriptors Studies on the interaction of flutamide anticancer drug with nucleobases: a computational view. Chem Rev Lett 4:54
Vessally E, Musavi M, Poor Heravi MR (2021) A density functional theory study of adsorption ethionamide on the surface of the pristine, Si and Ga and Al-doped graphene. Iran J Chem Chem Eng (IJCCE) 40:1720
Vakili M, Bahramzadeh V, Vakili M (2020) A Comparative study of SCN- adsorption on the Al12N12, Al12P12, and Si and Ge -doped Al12N12 nano-cages to remove from the environment. J Chem Lett 1:172
SÖĞÜTLÜ İ, Arshadi S, Mahmood EA, Abbasi V, Kamalinahad S, Vessally E (2023) In silico investigation of metalophthalocyanine substituted in carbon nanocones (TM-PhCCNC, TM= Sc2+, Cr2+, Fe2+ and Zn2+) as a promising sensor for detecting N2O gas involved. J Mol Struc 135263. https://doi.org/10.1016/j.molstruc.2023.135263
Mosavi M (2020) Adsorption behavior of mephentermine on the pristine and Si, Al, Ga- doped boron nitride nanosheets: DFT studies. J Chem Lett 1:164
Vessally E, Siadati SA, Hosseinian A, Edjlali L (2017) Selective sensing of ozone and the chemically active gaseous species of the troposphere by using the C20 fullerene and graphene segment. Talanta 162:505
Kadhim MM, Mahmood EA, Abbasi V, Heravi MRP, Habibzadeh S, Mohammadi-Aghdam S, Soleimani-Amiri S (2023) Investigation of the substituted—Titanium nanocages using computational chemistry. J Mol Graph Model 118:108317
Jalali Sarvestani MR (2022) Venlafaxine Interaction with Fullerene (C20): DFT Studies. J Chem Lett 3:169
Siadati SA, Vessally E, Hosseinian A, Edjlali L (2016) Possibility of sensing, adsorbing, and destructing the Tabun-2D-skeletal (Tabun nerve agent) by C20 fullerene and its boron and nitrogen doped derivatives. Synth Met 220:606
Kadhim MM, Mahmood EA, Abbasi V, Heravi MRP, Habibzadeh S, Mohammadi-Aghdam S, Shoaei SM (2022) Theoretical investigation of the titanium—nitrogen heterofullerenes evolved from the smallest fullerene. J Mol Graph Model 117:108269
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study J Org Chem 73:4615
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, New York
Parr RG, Szentpaly L, Liu S (1999) Electrophilicity Index. J Am Chem Soc 121:1922
Wang B, Rong C, Chattaraj PK, Liu S (2019) A comparative study to predict regioselectivity, electrophilicity and nucleophilicity with Fukui function and Hirshfeld charge. Theor Chem Account 138(12):124
Agwupuye JA, Louis H, Enudi OC, Unimuke TO, Edim MM (2022) Theoretical insight into electronic and molecular properties of halogenated (F, Cl, Br) and hetero-atom (N, O, S) doped cyclooctane. Mater Chem Phys 275:125239
West R, Denk M (1996) Stable silylenes: synthesis, structure, reactions. Pure Appl Chem 68(4):785
Driess M, Yao S, Brym M, Cv W, Lentz D (2006) A new type of N-heterocyclic silylene with ambivalent reactivity. J Am Chem Soc 128(30):9628
Kira M, Ishida S, Iwamoto T, Kabuto C (1999) The first isolable Dialkylsilylene. J Am Chem Soc 121:9722
Hoffmann R, PvR S, Schaefer HF (2008) Predicting molecules—more realism, please! Angew Chem Int Ed 47:7164
Ebbesen TW (2016) Hybrid light–matter states in a molecular and material science perspective. Acc Chem Res 49(11):2403
Author information
Authors and Affiliations
Contributions
Marziyeh Mohammadi: Writing—review, editing, writing—original draft, project administration, investigation, visualization, methodology, formal analysis, data decoration, conceptualization, validation, software, resources, and project administration.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohammadi, M. Electronic effects of the substituted dopants on stability and reactivity of difuranosilapyridine-4-ylidenes: DFT approach. Struct Chem 34, 2347–2364 (2023). https://doi.org/10.1007/s11224-023-02151-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02151-x