
Abstract
Purpose
Chitosan-g-PEG/heparin polyelectrolyte complex micelles were prepared for inducing apoptotic death of cancer cells.
Materials and methods
The cytotoxicity of polyelectrolyte complex micelles was evaluated by examining the growth inhibition of mouse melanoma B16F10 cells. Cellular uptake and apoptosis-inducing effect were investigated by confocal laser scanning microscopy and flow cytometric analysis, respectively.
Results
The prepared polyelectrolyte complex micelles had a spherical shape with an average diameter of 162.8 ± 18.9 nm. They were highly stable and well dispersed even in the presence of serum due to the presence of hydrophilic PEG shell layer surrounding the micelles. Moreover, they exhibited significantly higher cytotoxic activity against B16F10 cells compared to heparin or chitosan-g-PEG at the same concentration. The polyelectrolyte complex micelles were internalized by cancer cells to a greater extent than free heparin alone, indicating that the dramatic cell death was attributed to the increased cellular uptake of heparin. The internalized heparin was shown to induce apoptotic death of the cancer cells via a caspase-dependent pathway.
Conclusions
Nanosized and stable chitosan-g-PEG/heparin polyelectrolyte complex micelles were produced by a self-assembly process. The polyelectrolyte complex micelles facilitated the intracellular delivery of heparin, triggered the caspase activation, and consequently promoted apoptotic death of cancer cells.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past decade, nanoscale drug delivery systems based on polymeric micelles have drawn significant attention due to their unique properties for therapeutic and diagnostic applications. Particularly, polymeric micelles formed by self-assembly of amphiphilic copolymers in aqueous solution have been widely explored for effective cancer therapy because they can encapsulate diverse water-insoluble anti-cancer drugs such as doxorubicin and paclitaxel in their hydrophobic core (1–3). Polyelectrolyte complex micelles have recently emerged as an alternative class of polymeric micelles for efficient delivery of charged therapeutic agents. They are usually produced by self-assembly of di-block copolymers composed of a cationic segment and a hydrophilic segment, and anionic macromolecules. Electrostatic interactions between oppositely charged molecules generate a charge neutralized core surrounded by a layer of the hydrophilic polymer (4,5). These polyelectrolyte complex micelles have distinctive advantages for intravenous and intracellular drug delivery. For example, the nanoscale size and hydrophilic shell layer render the micelles to escape from degradation by enzymes and avoid nonspecific uptake by macrophages distributed in body organs (6,7). Furthermore, the polyelectrolyte complex micelles can be extensively applied for delivery of various bioactive macromolecules including nucleic acids and proteins.
Heparin, a highly sulfated natural glycosaminoglycan, is clinically used as an injectable anticoagulant. Heparin is known to interact with a diverse group of proteins having heparin-binding domains to regulate a variety of physiological processes including cell proliferation, differentiation, and inflammation (8). Recent studies have revealed that heparin exhibits various anti-cancer activities in the processes of tumor progression and metastasis. For instance, it has been found that heparin strongly binds to various growth factors such as vascular endothelial growth factor (VEGF), and consequently inhibits angiogenesis critical for tumor progression (9,10). In addition, heparin is able to exhibit inhibitory effects on tumor metastasis by blocking selectin-mediated adherence of cancer cells to vascular endothelium or platelets (11,12). More recently, it was discovered that free heparin molecules within cells interfered with activity of various transcription factors that played an important role in cell survival and growth, ultimately leading to apoptotic cell death (13–15). This suggests that intracellular delivery of free heparin can be used as a new class of effective apoptotic agent to kill cancer cells.
We present herein the development of chitosan-g-poly(ethylene glycol)/heparin polyelectrolyte complex micelles for inducing apoptotic death of cancer cells. Chitosan is a natural polysaccharide composed of glucosamine and N-acetyl-d-glucosamine and is receiving considerable attention for biomedical and pharmaceutical applications due to its highly cationic, biocompatible, and biodegradable nature (16). Owing to its unique mucoadhesive property, chitosan has been extensively exploited as promising oral drug delivery vehicles to increase the transport of drugs across intestinal epithelium (17,18). Moreover, chitosan has been reported to interact electrostatically with negatively charged macromolecules such as plasmid DNA and anionic protein drugs (e.g. insulin) to form polyelectrolyte complexes (19,20). In this study, chitosan grafted with polyethylene glycol (chitosan-g-PEG) was synthesized and used to produce self-assembled polyelectrolyte complex micelles by interacting with heparin in aqueous solution. The resultant chitosan-g-PEG/heparin polyelectrolyte complex micelles were expected to facilitate the entry of heparin into cells because the nanoparticulate form was more favorable for the intracellular transport across the cell membrane than free heparin alone. The formation of polyelectrolyte complex micelles was confirmed by dynamic light scattering (DLS) and atomic force microscopy (AFM). The anti-cancer effect of these polyelectrolyte complex micelles was evaluated by investigating their cytotoxicity, intracellular uptake, and apoptosis-inducing activities using mouse melanoma B16F10 cells.
MATERIALS AND METHODS
Materials
Heparin sodium salt extracted from porcine mucosa (M w = 12,000) was obtained from Wako Pure Chemical Industries (Osaka, Japan). Water-soluble chitosan oligosaccharide (M w = 4,000, deacetylation degree = 96%) was supplied by Kittolife Co. (Seoul, Korea). Methoxy poly(ethylene glycol)-succinimidyl succinate (mPEG-SS, M w = 5,000) was a product of Sunbio Co. (Anyang, Korea). Fluorescein-5-maleimide, propidium iodide, and CaspACE colorimetric assay kit were purchased from Promega Corporation (Madison, WI, USA). Cell counting kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Micro-BCA protein assay kit obtained from Pierce Biotechnology (Rockford, IL, USA) was used according to the manufacturer’s instructions. All other chemicals were of analytical grade.
Synthesis of Chitosan-g-Poly(Ethylene Glycol) (Chitosan-g-PEG)
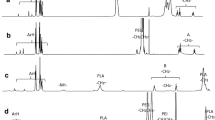
Chitosan-g-PEG copolymer was prepared by conjugating a succinimidyl group of mPEG-SS to amino groups of chitosan. In brief, 125 mg (25 μmol) of mPEG-SS was slowly added to 10 mM phosphate buffer solution (15 mL, pH 8) containing 100 mg (25 μmol) of chitosan. After reaction with stirring for 24 h at room temperature, the solution was dialyzed against deionized water by a Spectra/Por dialysis membrane with a M W cutoff of 5 kDa, and then lyophilized. The synthesis of the product was confirmed by 1H-NMR using Brucker DRX 400 spectrometer operating at 400 MHz. The degree of substitution of PEG to amino groups of chitosan was estimated by comparing the relative peak area of a methylene group (–NH–CO–CH2CH2O–, δ = 2.45 ppm) of PEG and an acetyl group (–COCH3, δ = 1.95 ppm) of the monosaccharide residue in chitosan (19).
Preparation of Chitosan-g-PEG/Heparin Polyelectrolyte Complex Micelles
Chitosan-g-PEG with different amounts (10, 20, 40, and 100 mg/mL) was dissolved in deionized water. A heparin solution of 20 mg/mL was prepared in deionized water, separately. The two solutions with an equal volume were mixed by vortexing vigorously for 3 min, and then incubated for 30 min at room temperature in order to produce chitosan-g-PEG/heparin polyelectrolyte complex micelles. For comparison, chitosan/heparin polyelectrolyte complexes were also prepared with unmodified chitosan at the same concentration. The hydrodynamic diameter of the micelles was analyzed by using a dynamic light scattering instrument (Zeta-Plus, Brookhaven, NY, USA) equipped with a He–Ne laser at a wavelength of 632 nm. Intensity-weighted particle size distribution data were collected at a scattering angle of 90° under physiological temperature (37°C). The ζ potential values of the micelles dispersed in 0.1 M phosphate-buffered saline solution (PBS, pH 7.4) were measured in triplicate. The physical stability in serum was examined by measuring their hydrodynamic diameters continuously every 3 min following the addition of 10% (v/v) fetal bovine serum. Size and shape of the micelles were confirmed by atomic force microscopy (AFM). One hundred microliters of the micelle solution was deposited and dried on a clean mica surface, and then its image was obtained with a PSIA XE-100 AFM system (Santa Clara, CA, USA) in a non-contact mode with a scanned area of 5 × 5 μm. The diameter and height of the micelles were analyzed from AFM images by a PSIA XEI 1.5 software (Santa Clara, CA, USA).
Cell Viability Assay
Mouse melanoma B16F10 cells were seeded in a 24-well plate at a density of 5 × 104 cells per well and grown in DMEM medium supplemented with 10 % (v/v) fetal bovine serum for 24 h at 37°C. The cells were then incubated with the culture medium containing chitosan-g-PEG/heparin polyelectrolyte complex micelles or heparin at varying heparin concentration from 1 to 100 μg/mL for 2 days at 37°C. As a control experiment, chitosan-g-PEG was treated to the cells at the concentration equivalent to the micelles. The number of viable cells was determined by the CCK-8 cell viability assay which depends on the mitochondrial dehydrogenase activity inside the cells. In brief, 20 μL of CCK-8 solution was added to 400 μL of serum-free DMEM medium in each well of the plate. After incubating the plate for 2 h at 37°C, the absorbance at 450 nm was measured using a Bio-Rad microplate reader.
Evaluation of Cellular Uptake of Polyelectrolyte Complex Micelles
To visualize cellular uptake, fluorescein-labeled heparin was prepared by conjugating fluorescein-5-maleimide to thiolated heparin as previously described (15). In brief, 5 mg of thiolated heparin was reacted with 0.1 mg of fluorescein-5-maleimide in 0.1 M PBS solution (pH 7.4). The solution was dialyzed against deionized water (M W cutoff of 6 kDa) and then lyophilized. Using fluorescein-labeled heparin, polyelectrolyte complex micelles were prepared as described above. B16F10 cells were plated over a cover slide on a six-well plate at a density of 2 × 105 cells per well and cultivated for 24 h at 37°C. The cells were then treated with the fluorescein-labeled heparin or polyelectrolyte complex micelles (200 μg/mL in heparin) in serum-free DMEM medium for 2 days at 37°C. The cells were washed with PBS solution and fixed with a 1:1 (v/v) mixture of methanol and acetone. After washing with PBS solution, the cell nuclei were stained with propidium iodide solution (50 μg/mL in PBS solution) for 30 min. The cells were examined by using a LSM510 confocal laser scanning microscope (Carl Zeiss, Germany) equipped with a 488 nm Argon laser and 543 nm HeNe laser.
Flow Cytometry and Caspase-3 Activity Assay
B16F10 cells were seeded in T-75 flasks at a density of 5 × 106 cells per flask and grown in DMEM medium supplemented with 10 % (v/v) fetal bovine serum for 24 h at 37°C. The cells were then treated with heparin or polyelectrolyte complex micelles (600 μg/mL in heparin) in serum-free DMEM medium for 2 days at 37°C. The cells were harvested, washed with PBS solution three times, and then fixed by slowly adding 70 % (v/v) ethanol while gentle vortexing. The fixed cells were stored at 4°C overnight and incubated with hypotonic propidium iodide solution (0.25 mg/mL propidium iodide and 0.1 mg/mL RNase A in PBS solution) at a concentration of 106 cells/mL for 30 min at 37°C. The fluorescence of each nucleus was analyzed by a flow cytometer (FACSCalibur, USA) using a FlowJo software (Tree Star, USA). The enzymatic activity of caspase-3 was measured with a CaspACE colorimetric assay. Briefly, the treated cells were harvested, washed with ice-cold PBS solution, and then resuspended in a cell lysis buffer solution at a concentration of 108 cells/mL. The cell lysate was centrifuged at 15,000 rpm for 30 min at 4°C. The supernatant fraction was collected and incubated for 4 h at 37°C with 0.2 mM caspase-3 substrate (DEVD-pNA), which was labeled with p-nitrophenylaniline (pNA). The yellow color produced by released pNA from the cleaved substrate was monitored at 405 nm. The amount of total protein in each cell lysate was measured with a Micro-BCA protein assay kit. The specific activity of caspase-3 was calculated by dividing the amount of released pNA with the amount of total intracellular protein.
RESULTS AND DISCUSSION
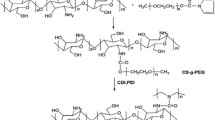
Chitosan-g-poly(ethylene glycol) (chitosan-g-PEG) was prepared by conjugating a succinimidyl group of methoxy PEG-succinimidyl succinate to amino groups of chitosan oligosaccharide. The degree of substitution of PEG to the amino groups of chitosan was approximately 12.8% as determined by 1H-NMR spectroscopy. The generation of a new amide linkage between chitosan and PEG was also observed at δ = 2.45 ppm, indicating that PEG chains were covalently conjugated to the backbone of chitosan oligosaccharide. Figure 1 illustrates the formation of nanosized polyelectrolyte complex micelles through self-assembly process of heparin and chitosan-g-PEG in aqueous solution. A simple mixing of an aqueous heparin solution with a chitosan-g-PEG solution induced spontaneous formation of the polyelectrolyte complex micelles. It was previously reported that PEG grafted cationic copolymers such as poly(l-lysine)-g-PEG and polyethylenimine-g-PEG could form polyelectrolyte complex micelles by electrostatic interactions with anionic macromolecules (e.g. plasmid DNA, oligonucleotides) (7,21,22). In the present study, it was anticipated that the positively charged chitosan backbone in the structure of chitosan-g-PEG ionically interacted with the negatively charged heparin molecules to form a hydrophobic core via charge neutralization, while the grafted PEG chains were exposed outside. The self-assembly process would finally produce spherical micelles having a charge-neutralized chitosan/heparin polyelectrolyte complex core surrounded by a hydrophilic PEG shell layer in aqueous solution.

Synthetic scheme of chitosan-g-PEG/heparin polyelectrolyte complex micelles.
We performed dynamic light scattering (DLS) analysis to examine the effect of various weight ratios between chitosan-g-PEG and heparin on the size and surface charge of chitosan-g-PEG/heparin polyelectrolyte complex micelles in aqueous solution. As presented in Table I, chitosan-g-PEG and heparin self-assembled to form nanoscale polyelectrolyte complexes in the size range of 160–270 nm. When the weight ratio of chitosan-g-PEG/heparin was 0.5, the resultant polyelectrolyte complexes had a negative ζ potential of −10.6 ± 4.3 mV. The ζ potential values gradually increased upon raising the weight ratio of chitosan-g-PEG/heparin. It should be noted that polyelectrolyte complex micelles prepared at a weight ratio of 1:1 had the smallest size (162.8 ± 18.9 nm) and slightly negative surface charge (−4.0 ± 0.6 mV). Since the charge ratio of –NH3 + groups on chitosan to –COO− and –SO3 − groups on heparin was nearly 1:1 at this weight ratio, electrostatic interactions between chitosan and heparin might become strongest, leading to the formation of compact polyelectrolyte complex micelles (20). Based on these results, the polyelectrolyte complex micelles prepared at a weight ratio of 1:1 were selected for the rest of the study.
As shown in Fig. 2A, chitosan-g-PEG/heparin polyelectrolyte complex micelles were produced with a narrow size distribution of 162.8 ± 18.9 nm. However, chitosan/heparin polyelectrolyte complexes prepared with unmodified chitosan formed a binary mixture of inhomogeneous aggregates with an average diameter of 329.3 ± 36.4 nm and 2.79 ± 0.24 μm. It was clearly observed that the chitosan/heparin polyelectrolyte complexes were severely flocculated and finally precipitated out of solution at 24 h after preparation (inset photograph). This phenomenon can be explained by uncontrollable aggregation of charge-neutralized chitosan/heparin polyelectrolyte complexes (16). It was reported previously that stable chitosan/heparin nanoparticles could be produced by polyelectrolyte complexation in aqueous solution (23). Since the formation of these nanoparticles largely depends on electrostatic interactions between chitosan and heparin, their concentration and weight ratio were carefully controlled to produce stable nanoparticles. In the present study, however, chitosan/heparin polyelectrolyte complexes did not exhibit stability at the selected concentration and weight ratio, and especially under serum conditions, resulting in severe aggregation and precipitation. It was noteworthy that the chitosan-g-PEG/heparin polyelectrolyte complex micelles exhibited excellent stability in deionized water, showing a transparent solution without any precipitates. These polyelectrolyte complex micelles had a spherical shape with an average diameter of 202.7 ± 42.2 nm, as visualized by using AFM (Fig. 2B). They were well dispersed and separated each other, suggesting that the hydrophilic PEG shell layer surrounding the micelles effectively prevented the inter-particular aggregation of the charge-neutralized complexes and thus enhanced the dispersion stability in aqueous solution. It has been shown previously that PEG modified polymeric micelles and nanoparticles are highly resistant to nonspecific adsorption of plasma proteins in the blood stream due to the steric stabilization effect of the PEG chains on their surface (24,25). To investigate the effect of PEG on the stability of the polyelectrolyte complex micelles under physiological environment, we measured their hydrodynamic diameter in 0.1 M phosphate-buffered saline solution (pH 7.4) supplemented with serum. As presented in Fig. 2C, the diameter of the chitosan/heparin complexes increased drastically from 330.4 ± 25.7 nm (the main fraction in Fig. 2A) to 2.24 ± 0.25 μm within 9 min following the addition of serum. In contrast, the chitosan-g-PEG/heparin complex micelles didn’t show any significant change in the size and stability for several days. Therefore the above results revealed that the steric shield provided by hydrophilic PEG layer surface played an important role in avoiding nonspecific adsorption of plasma proteins onto the micelles, thereby leading to the stabilization of micelles under physiological environment.

A Hydrodynamic diameter of chitosan-g-PEG/heparin polyelectrolyte complex micelles (prepared at a weight ratio of 1:1) and chitosan/heparin polyelectrolyte complexes. The inset photograph showing chitosan-g-PEG/heparin polyelectrolyte complex micelles (left) and chitosan/heparin polyelectrolyte complexes (right) was taken at 24 h after sample preparation. B AFM image of chitosan-g-PEG/heparin polyelectrolyte complex micelles. C Time course change of hydrodynamic diameter for chitosan-g-PEG/heparin polyelectrolyte complex micelles and chitosan/heparin polyelectrolyte complexes in 0.1 M PBS solution (pH 7.4) upon addition of 10% (v/v) fetal bovine serum (FBS).
We evaluated the cell viability of mouse melanoma B16F10 cells treated with various concentrations of chitosan-g-PEG/heparin polyelectrolyte complex micelles. The entry of native heparin molecules into cancer cells has been shown to promote apoptotic cell death by inhibiting the activity of transcription factors essential for cell proliferation (13–15). It was reported previously that heparin complexed with cationic poly(β-amino ester)s greatly influenced the transcription factor levels in cancer cells. The result suggested that the internalized heparin affected cellular processes associated with transcription factors, and ultimately induced apoptosis of cancer cells. As shown in Fig. 3, the chitosan-g-PEG/heparin polyelectrolyte complex micelles exhibited a remarkable cytotoxic activity against cancer cells as compared with free heparin or chitosan-g-PEG at the same concentration. The treatment of the polyelectrolyte complex micelles at 100 μg/mL induced a 42.4 ± 4.5% reduction in cell viability, whereas free heparin alone decreased cell viability to a negligible extent (3.5 ± 3.7%). It was found that chitosan-g-PEG was non-toxic to the cancer cells, indicating that the cationic polymer itself was not responsible for the cell death. Since heparin is a highly negatively charged macromolecule, the absorptive endocytosis of heparin across the cell membrane would not occur (26). It was most likely that the chitosan-g-PEG/heparin polyelectrolyte complex micelles entered the cell by nonspecific adsorptive endocytosis, inducing the cancer cell death more efficiently than free heparin alone. In addition, the cytotoxic effect of the polyelectrolyte complex micelles significantly increased in a dose-dependent manner, suggesting that heparin molecules were more taken up by cells with increasing the micelle concentration.

Cell viability of mouse melanoma B16F10 cells treated with chitosan-g-PEG, heparin, or chitosan-g-PEG/heparin polyelectrolyte complex micelles (prepared at a weight ratio of 1:1) for 2 days.
The capability of chitosan-g-PEG/heparin polyelectrolyte complex micelles as an efficient carrier for heparin was investigated by confocal laser scanning microscopy. Heparin was fluorescently labeled with fluorescein (green fluorescent dye), in order to visualize its subcellular distribution. Figure 4 shows the confocal microscopic images of B16F10 cells following incubation with fluorescein-labeled heparin or polyelectrolyte complex micelles at an equivalent concentration of heparin. While the cells treated with polyelectrolyte complex micelles displayed strong green fluorescence within the cytoplasm homogeneously, only a marginal fluorescence was observed within the cells treated with free heparin alone, suggesting that the micelles are much more internalized by the cells than free heparin. This revealed that the nanosized polyelectrolyte complex micelles could improve the intracellular transport of heparin via a endocytic process across the cell membrane (27,28). This was likely responsible for the enhanced internalization of the micelles compared to heparin. One interesting point was that only the cells showing intense green fluorescence were round shaped and detached from the surfaces. Since the aberrant detachment of adherent cells is generally considered as a result of cell death (29), it is conceivable that the observed dramatic cytotoxic effect was attributed to the internalized free heparin molecules within cells.

Confocal microscopic images of B16F10 cells following incubation with A chitosan-g-PEG/heparin polyelectrolyte complex micelles (prepared at a weight ratio of 1:1) or B heparin for 2 days. Heparin was fluorescently labeled with fluorescein. Cell nuclei were also stained with propidium iodide. Each pair of images was obtained at the same Z-value.
To examine the extent of apoptotic cell death quantitatively, we conducted flow cytometric analysis after propidium iodide staining of the cell nuclei. Since apoptosis is accompanied by the activation of endogenous nucleases, which cleaves chromosomal DNA into oligonucleosomal fragments, apoptotic cells can be recognized by their diminished DNA content within the nuclei (30,31). The extent of apoptosis was determined by appearance of the cells in a sub-G1 phase (DNA content of less than diploid). As shown in Fig. 5, chitosan-g-PEG/heparin polyelectrolyte complex micelles induced significant accumulation of the sub-G1 cell population (27.13%) compared to heparin (0.65%) or the untreated cells (0.85%). This suggests that they greatly promoted apoptotic death of cancer cells. The molecular mechanisms involved in heparin-induced apoptosis are rarely understood. One plausible explanation is that free heparin internalized within cells likely interrupts the interactions of DNA with various transcription factors, such as Rb, p107, and E2F, which participate in the process of cell proliferation (9,10,13). Afterward, the internalized heparin might trigger apoptosis-inducing stimuli for activation of caspases and subsequent apoptotic cell death. Caspases are a family of cysteine aspartic acid-specific proteases playing an important role in apoptotic processes in mammalian cells. Among them, caspase-3 is an essential protease that degrades other protein substrates inside the cells at an early stage of apoptosis (32,33). The enzymatic activity of caspase-3 was measured by using a colorimetric peptide substrate labeled with p-nitrophenylaniline (DEVD-pNA). This enzymatic technique allows a highly sensitive and quantitative measurement of caspase-3 activity because the colorimetric peptide substrate will release yellow colored pNA product only if it is specifically cleaved by activated caspase-3. As presented in Fig. 5D, the caspase-3 activity within cells was slightly increased from 107.4 ± 16.7 to 208.9 ± 1.5 pmol pNA/μg protein after treatment of free heparin for 2 days. However, the polyelectrolyte complex micelles exhibited far greater cellular activity of caspase-3 (707.7 ± 15.4 pmol pNA/μg protein) than heparin. The increased caspase activity was consistent with the flow cytometry results, implying that the cancer cell death was probably mediated by a caspase-dependent apoptosis pathway. The above results demonstrated that the polyelectrolyte complex micelles significantly enhanced the intracellular delivery extent of heparin with concomitant activation of caspase, thereby triggering the apoptosis of cancer cells. The self-assembled heparin polyelectrolyte complex micelles could be also co-encapsulated with negatively charged apoptosis-related nucleic acid therapeutics, such as plasmid DNA and small interfering RNA, to further promote an apoptotic effect for cancer cells.

Cell cycle analysis of B16F10 cells treated with A PBS solution, B heparin, or C chitosan-g-PEG/heparin polyelectrolyte complex micelles (prepared at a weight ratio of 1:1) for 2 days. D Caspase-3 activity of B16F10 cells after treatment with PBS solution, heparin, or chitosan-g-PEG/heparin polyelectrolyte complex micelles.
CONCLUSIONS
Self-assembled chitosan-g-PEG/heparin polyelectrolyte complex micelles were developed for heparin-induced apoptotic death of cancer cells. The resultant polyelectrolyte complex micelles were highly stable and well dispersed under physiological environment due to the steric stabilization effect of the PEG shell layer. Furthermore, we demonstrated that the internalization of heparin significantly induced the apoptotic death of cancer cells through activation of a caspase-mediated pathway. The current strategy for intracellular delivery of heparin using polyelectrolyte complex micelles has a wide range of potential applications for anti-cancer treatments.
References
J. A. Hubbell. Enhancing drug function. Science. 300:595–596 (2003) doi:10.1126/science.1083625.
R. Duncan. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2:347–360 (2003) doi:10.1038/nrd1088.
M. C. Jones, and J. C. Leroux. Polymeric micelles—a new generation of colloidal drug carriers. Eur. J. Pharm. Biopharm. 48:101–111 (1999) doi:10.1016/S0939-6411(99)00039-9.
Y. Kakizawa, and K. Kataoka. Block copolymer micelles for delivery of gene and related compounds. Adv. Drug Delivery Rev. 54:203–222 (2002) doi:10.1016/S0169-409X(02)00017-0.
T. G. Park, J. H. Jeong, and S. W. Kim. Current status of polymeric gene delivery systems. Adv. Drug Delivery Rev. 58:467–486 (2006) doi:10.1016/j.addr.2006.03.007.
R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin, and R. Langer. Biodegradable long-circulating polymeric nanospheres. Science. 263:1600–1603 (1994) doi:10.1126/science.8128245.
K. Kunath, A. von Harpe, H. Petersen, D. Fischer, K. Voigt, T. Kissel, and U. Bickel. The structure of PEG-modified poly(ethylene imines) influences biodistribution and pharmacokinetics of their complexes with NF-κB decoy in mice. Pharm. Res. 19:810–817 (2002) doi:10.1023/A:1016152831963.
I. Capila, and R. J. Linhardt. Heparin–protein interactions. Angew. Chem. Int. Ed. Engl. 41:390–412 (2002) doi:10.1002/1521-3773(20020201)41:3<390::AID-ANIE390>3.0.CO;2-B.
R. Sasisekharan, Z. Shriver, G. Venkataraman, and U. Narayanasami. Roles of heparansulfate glycosaminoglycans in cancer. Nat. Rev. Cancer. 2:521–528 (2002) doi:10.1038/nrc842.
J. Hasan, S. D. Shnyder, A. R. Clamp, A. T. McGown, R. Bicknell, M. Presta, M. Bibby, J. Double, S. Craig, D. Leeming, K. Stevenson, J. T. Gallagher, and G. C. Jayson. Heparin octasaccharides inhibit angiogenesis in vivo. Clin. Cancer Res. 11:8172–8179 (2005) doi:10.1158/1078-0432.CCR-05-0452.
L. Borsig, R. Wong, J. Feramisco, D. R. Nadeau, N. M. Varki, and A. Varki. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. U.S.A. 98:3352–3357 (2001) doi:10.1073/pnas.061615598.
K. Park, S. K. Lee, D. H. Son, S. A. Park, K. Kim, H. W. Chang, E. J. Jeong, R. W. Park, I. S. Kim, I. C. Kwon, Y. Byun, and S. Y. Kim. The attenuation of experimental lung metastasis by a bile acid acylated-heparin derivative. Biomaterials. 28:2667–2676 (2007) doi:10.1016/j.biomaterials.2007.02.001.
D. Berry, D. M. Lynn, R. Sasisekharan, and R. Langer. Poly(b-amino ester)s promote cellular uptake of heparin and cancer cell death. Chem. Biol. 11:487–498 (2004) doi:10.1016/j.chembiol.2004.03.023.
M. K. Yu, D. Y. Lee, Y. S. Kim, K. Park, S. A. Park, D. H. Son, G. Y. Lee, J. H. Nam, S. Y. Kim, I. S. Kim, R. W. Park, and Y. Byun. Antiangiogenic and apoptotic properties of a novel amphiphilic folate–heparin–lithocholate derivative having cellular internality for cancer therapy. Pharm. Res. 24:705–714 (2007) doi:10.1007/s11095-006-9190-3.
K. H. Bae, H. Mok, and T. G. Park. Synthesis, characterization, and intracellular delivery of reducible heparin nanogels for apoptotic cell death. Biomaterials. 29:3376–3383 (2008) doi:10.1016/j.biomaterials.2008.04.035.
J. Berger, M. Reist, J.M. Mayer, O. Felt, and R. Gurny. Structure and interactions in chitosan hydrogels formed by complexation or aggregation for biomedical applications. Eur. J. Pharm. Biopharm. 57:35–52 (2004) doi:10.1016/S0939-6411(03)00160-7.
M. Thanou, J. C. Verhoef, and H. E. Junginger. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Delivery Rev. 52:117–126 (2001) doi:10.1016/S0169-409X(01)00231-9.
C. Prego, D. Torres, E. Fernandez-Megia, R. Novoa-Carballal, E. Quiñoá, and M.J. Alonso. Chitosan–PEG nanocapsules as new carriers for oral peptide delivery Effect of chitosan pegylation degree. J. Control. Release. 111:299–308 (2006) doi:10.1016/j.jconrel.2005.12.015.
X. Jiang, H. Dai, K. W. Leong, S. H. Goh, H. Q. Mao, and Y. Y. Yang. Chitosan-g-PEG/DNA complexes deliver gene to the rat liver via intrabiliary and intraportal infusions. J. Gene Med. 8:477–487 (2006) doi:10.1002/jgm.868.
Y. H. Lin, F. L. Mi, C. T. Chen, W. C. Chang, S. F. Peng, H. F. Liang, and H. W. Sung. Preparation and characterization of nanoparticles shelled with chitosan for oral insulin delivery. Biomacromolecules. 8:146–152 (2007) doi:10.1021/bm0607776.
Y. H. Choi, F. Liu, J. S. Kim, Y. K. Choi, J. S. Park, and S. W. Kim. Polyethylene glycol-grafted poly-L-lysine as polymeric gene carrier. J. Control. Release. 54:39–48 (1998) doi:10.1016/S0168-3659(97)00174-0.
S. Mao, M. Neu, O. Germershaus, O. Merkel, J. Sitterberg, U. Bakowsky, and T. Kissel. Influence of polyethylene glycol chain length on the physicochemical and biological properties of poly(ethylenimine)-graft-poly(ethylene glycol) block copolymer/siRNA polyplexes. Bioconj. Chem. 17:1209–1218 (2006) doi:10.1021/bc060129j.
Z. Liu, Y. Jiao, F. Liu, and Z. Zhang. Heparin/chitosan nanoparticle carriers prepared by polyelectrolyte complexation. J. Biomed. Mater. Res. 83A:806–812 (2007) doi:10.1002/jbm.a.31407.
J. M. Harris, and R. B. Chess. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2:214–221 (2003) doi:10.1038/nrd1033.
L. E. van Vlerken, T. K. Vyas, and M. M. Amiji. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 24:1405–1414 (2007) doi:10.1007/s11095-007-9284-6.
M. Gohda, T. Magoshi, S. Kato, T. Noguchi, S. Yasuda, H. Nonogi, and T. Matsuda. Terminally alkylated heparin. 2. Potent antiproliferative agent for vascular smooth muscle cells. Biomacromolecules. 2:1178–1183 (2001) doi:10.1021/bm010097x.
A. V. Kabanov, and V. A. Kabanov. DNA complexes with polycations for the delivery of genetic material into cells. Bioconj. Chem. 6:7–20 (1995) doi:10.1021/bc00031a002.
S. A. Agnihotri, N. N. Mallikarjuna, and T. M. Aminabhavi. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release. 100:5–28 (2004) doi:10.1016/j.jconrel.2004.08.010.
J. E. Jr Meredith, B. Fazeli, and M. A. Schwartz. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 4:953–961 (1993).
Z. Daryzynkiewicz, S. Bruno, G. D. Bino, M. A. Hotz, P. Lassota, and F. Traganos. Features of apoptotic cells measured by flow cytometry. Cytometry. 13:795–808 (1992) doi:10.1002/cyto.990130802.
K. C. Cho, J. H. Jeong, H. J. Chung, C. O. Joe, S. W. Kim, and T. G. Park. Folate receptor-mediated intracellular delivery of recombinant caspase-3 for inducing apoptosis. J. Control. Release. 108:121–131 (2005) doi:10.1016/j.jconrel.2005.07.015.
M. O. Hengartner. The biochemistry of apoptosis. Nature. 407:770–776 (2000) doi:10.1038/35037710.
W. C. Earnshaw, L. M. Martins, and S. H. Kaufmann. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383–424 (1999) doi:10.1146/annurev.biochem.68.1.383.
Acknowledgements
This research was supported by the Ministry of Health and Welfare, and the National Research Laboratory program from the Ministry of Science and Technology, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
K. H. Bae and C. W. Moon contributed equally to this work as first authors.
Rights and permissions
About this article
Cite this article
Bae, K.H., Moon, C.W., Lee, Y. et al. Intracellular Delivery of Heparin Complexed with Chitosan-g-Poly(Ethylene Glycol) for Inducing Apoptosis. Pharm Res 26, 93–100 (2009). https://doi.org/10.1007/s11095-008-9713-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9713-1