
Abstract
The classification of central nervous system tumours has more recently been shaped by a focus on molecular pathology rather than histopathology. We re-classified 82 glial tumours according to the molecular-genetic criteria of the 2016 revision of the World Health Organization (WHO) Classification of Tumours of the Central Nervous System. Initial diagnoses and grading were based on the morphological criteria of the 2007 WHO scheme. Because of the impression of an oligodendroglial component on initial histological assessment, each tumour was tested for co-deletion of chromosomes 1p and 19q and mutations of isocitrate dehydrogenase (IDH-1 and 2) genes. Additionally, expression of proteins encoded by alpha-thalassemia X-linked mental retardation (ATRX) and TP53 genes was assessed by immunohistochemistry. We found that all but two tumours could be assigned to a specific category in the 2016 revision. The most common change in diagnosis was from oligoastrocytoma to specifically astrocytoma or oligodendroglioma. Analysis of progression free survival (PFS) for WHO grade II and III tumours showed that the objective criteria of the 2016 revision separated diffuse gliomas into three distinct molecular categories: chromosome 1p/19q co-deleted/IDH mutant, intact 1p/19q/IDH mutant and IDH wild type. No significant difference in PFS was found when comparing IDH mutant grade II and III tumours suggesting that IDH status is more informative than tumour grade. The segregation into distinct molecular sub-types that is achieved by the 2016 revision provides an objective evidence base for managing patients with grade II and III diffuse gliomas based on prognosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The 2016 revision of the World Health Organization (WHO) Classification of Tumours of the Central Nervous System [1] mandates the inclusion of molecular-genetic alterations in the diagnosis of diffuse glial tumours and, as such, is a significant departure from the morphology-based 2007 classification [2]. Molecular data hold sway over histology in all instances of morphological ambiguity. The trend towards molecular classification has implications not only for survival prediction but also the tailoring of post-surgical management and patient selection in the evaluation of newer molecular targeting treatments as they make their way into clinical practice [3, 4].
In the 2016 revision, astrocytomas and oligodendrogliomas are considered as a single category—diffuse glioma. Astrocytic tumours, that is, diffuse astrocytoma, anaplastic astrocytoma and glioblastoma, are sub-typed by the presence or absence of mutations of the isocitrate dehydrogenase genes (IDH-1&2) i.e. IDH mutated or wild type. Tumours can be assigned to a “Not Otherwise Specified” (NOS) category if molecular-genetic testing is not available or if the data are incomplete or inconclusive. The diagnosis of oligodendroglioma is wholly based on the demonstration of IDH mutation and deletion of both the short arm of chromosome 1 (1p) and the long arm of chromosome 19 (19q). The revision discourages the diagnoses of oligoastrocytoma and glioblastoma with an oligodendroglioma component (GBMO) and takes the position that, in most instances, morphologically mixed tumours will be shown to have the molecular profile of either astrocytoma or oligodendroglioma.
In this retrospective cohort study, we used data from a single institution in Melbourne, Australia, to compare the 2007 and 2016 classifications systems in patients who had undergone 1p/19q co-deletion testing around the time of original diagnosis. We compared the survival outcomes based on WHO grading and molecular and immunohistochemical characteristics. We hypothesized that molecular classification may provide more robust prognostic information than WHO grading alone.
Materials and methods
Case selection
The study cohort comprised 82 patients with diffuse glial tumours managed at The Royal Melbourne Hospital, between August 2010 and June 2016. All were tested for chromosome 1p/19q co-deletion at initial histological diagnosis because of the suspicion of an oligodendroglial component. To provide additional prognostic information, immunohistochemistry was undertaken to detect the most common IDH mutation (IDH R132H) and expression of proteins encoded by ATRX and TP53 genes. Original histological diagnoses and WHO grades were determined for each tumour according to criteria of the 2007 WHO Classification of Central Nervous System Tumours [2]. There were 78 grade II/III tumours and 4 grade IV tumours. These were re-classified to tumour sub-types according to the 2016 revision on the basis of molecular data provided by fluorescence in-situ hybridization (FISH), immunohistochemistry and DNA sequencing. The WHO grade IV tumours were subsequently excluded from the survival analysis as they represented only a very small proportion of all grade IV tumours at the institution within the study period.
Molecular testing and immunohistochemistry
For each tumour, the presence of IDH-1 R132H mutation and ATRX and TP53 expression were determined by immunohistochemistry. The following antibodies were used on a Leica Bond III immunohistochemistry platform—IDH1 (R132H) (H09 clone monoclonal antibody, Dianova, Hamburg, Germany; 1:50 dilution), ATRX (rabbit polyclonal antibody, Sigma-Aldrich, Castle Hill, Australia; 1:300 dilution) and TP53 (mouse monoclonal antibody, Novacastra, Newcastle upon Tyne, UK; 1:50 dilution). These have been shown to be reliable surrogate immunohistochemical markers for mutations of the respective genes [5,6,7,8]. Although the association is imperfect, loss of nuclear staining generally indicates ATRX mutation. Although some mutations may still be present, preserved nuclear staining usually indicates ATRX wild type. Conversely, for TP53, overexpression in tumour cell nuclei, frequently, but not always, indicates mutation while absent or weak nuclear staining usually indicates wild type.
FISH for determining whole-of-arm deletions of chromosomes 1p and 19q used Vysis (Abbott Molecular, Des Plaines, IL) 1p36 and 19q13.3 Spectrum Orange test probes and 1q25 and 19p13 Spectrum Green reference probes in a previously published protocol [9]. Tumours that were immunonegative for IDH1 (R132H) were analysed for IDH-1&2 mutations by pyrosequencing as previously described [10]. IDH mutation status has been shown to be preserved throughout progression of tumours from low to high grade [11, 12]. As this was a retrospective study, matched peripheral blood samples were not available and so LOH analysis to detect small interstitial deletions of chromosomes 1p and 19q could not be performed.
Statistical analysis
Progression-free survival (PFS) was measured from date of diagnosis to date of progression or death, whichever occurred first. Progression was defined as the date at which a decision was made to change management based on radiological or clinical data. Overall survival was not analysed as only 12 patients (15% of the cohort) had died at the time of analysis.
For each WHO classification (2007 and, separately, 2016) a Cox proportional hazards model was fitted and the C-index was extracted. This gives an idea of the ‘goodness-of-fit’ of the model; the C-index is a measure of a model’s discriminability as it quantifies the level of concordance between predicted probabilities and the actual chance of having an event. As the numbers of grade IV tumours were small, these were not included in the survival analyses.
Kaplan–Meier (KM) plots were generated to determine interactions between 1p19q and IDH status; 2016 WHO grade and IDH status; IDH status in patients with no 1p19q deletion, loss of nuclear ATRX expression versus preserved expression and TP53 overexpression versus weak or absent expression.
All statistical analyses were conducted using Stata Version 14 (StataCorp LLC, College Station, TX).
Results
Molecular testing and immunohistochemistry
The results of FISH for chromosome 1p/19q deletion, immunohistochemistry and DNA sequencing for IDH mutations, and immunohistochemistry for ATRX and TP53 expression are summarized in Table 1. A total of 34 tumours (41%) were co-deleted for chromosomes 1p and 19q. Of these, 32 (94%) were IDH1 (R132H) mutated and 2 were IDH wild type by immunohistochemistry and pyrosequencing and were assigned to “oligodendroglioma NOS”. As we were unable to undertake LOH testing, we cannot exclude small interstitial deletions in these two tumours, particularly involving chromosome 1p. These are associated with aggressive behaviour in glial tumours with oligodendroglial morphology [13]. All 34 co-deleted tumours showed preserved nuclear immunostaining for ATRX and lacked TP53 expression. This pattern was mutually exclusive of 1p/19q co-deletion. Thirty-nine tumours (48%) were chromosome 1p/19q intact. Of these, 26 (64%) were IDH mutated (25 R132 H; 1 R132S) and 13 (33%) were IDH wild type by immunohistochemistry and pyrosequencing. Of the 39 1p/19q intact tumours, 25 (64%) showed loss of nuclear immunostaining for ATRX and 33 (85%) showed nuclear overexpression of TP53. A single tumour with 1p deletion only was IDH1 (R132H) mutated with loss of ATRX expression and TP53 overexpression, a profile most consistent with molecular astrocytoma. Chromosome 19q only deletion was seen in 8 tumours; all 8 were IDH1 (R132H) mutated, 7 showed loss of ATRX expression and 7 overexpressed TP53, suggesting that 19q only deletion is also more likely to be associated with molecular astrocytoma.
Re-assignment of 2007 WHO tumours to the 2016 revised classification
Re-assignment of the 82 tumours to categories in the 2016 revision is summarised in Fig. 1.

Summary of re-assignment of 82 tumours from 2007 WHO classification to the 2016 revised classification: a re-assignment of WHO 2007 oligoastrocytomas grades II and III (n = 41); b re-assignment of WHO 2007 oligodendrogliomas grades II and III (n = 20); c re-assignment of WHO 2007 diffuse astrocytomas grade II (n = 12); d re-assignment of WHO 2007 anaplastic/high grade gliomas grade III (n = 5), glioblastomas (n = 3) and glioblastoma with an oligodendroglioma component (GBMO) (n = 1). All but 2 tumours could be assigned to a specific category within the 2016 revision
WHO 2007 oligoastrocytoma
Forty one tumours were initially diagnosed as oligoastrocytoma, grade II or III. Of these, 16 (39%) were re-assigned to oligodendroglioma, IDH mutant 1p/19q co-deleted, WHO grade II. Of the remaining 25 (61%) with intact 1p/19q, 18 were re-assigned to diffuse astrocytoma grade II, 4 to anaplastic astrocytoma grade III and 3 to glioblastoma IDH mutant. Seventeen of these 25 1p/19q intact tumours were IDH mutant and 8 IDH wild type. Loss of ATRX expression was seen in 20, TP53 overexpression in 22 and absent TP53 expression in 3.
WHO 2007 oligodendroglioma
Twenty tumours were initially diagnosed as oligodendroglioma, grade II or III. Of these, 13 (65%) were 1p/19q co-deleted and IDH mutated and retained the same diagnosis of oligodendroglioma grade II or III. One tumour was 1p/19q co-deleted and IDH wild type by both immunohistochemistry and DNA sequencing and was re-assigned to oligodendroglioma NOS grade II. Of the remaining 6 tumours with intact 1p/19q, 2 were re-assigned to diffuse astrocytoma (1 IDH mutant, 1 IDH wild type), 2 to anaplastic astrocytoma (both IDH mutant) and 2 to glioblastoma IDH mutant. IDH mutation was found to strongly co-segregate with 1p/19q co-deletion.
WHO 2007 diffuse astrocytoma grade II
Twelve tumours were initially diagnosed as diffuse astrocytoma grade II. Two of these were 1p/19q co-deleted and IDH mutated and were re-assigned to oligodendroglioma. The remaining 10 tumours with intact 1p/19q retained the same diagnosis: diffuse astrocytoma grade II. Nine were IDH mutant, 1 IDH wild type, 5 showed loss of nuclear ATRX expression, 5 had preserved nuclear ATRX expression, 8 overexpressed TP53 and 2 had weak/absent TP53 expression. Five tumours showed both loss of ATRX expression and TP53 overexpression.
WHO 2007 anaplastic/high grade glioma grade III, glioblastoma and glioblastoma with an oligodendroglioma component (GBMO)
Of 5 WHO 2007 anaplastic/high grade gliomas Grade III, one was 1p/19q co-deleted and IDH mutated and was re-assigned to oligodendroglioma. The remaining 4 tumours with intact 1p/19q retained the same diagnosis of anaplastic astrocytoma grade III. Three were IDH mutant, 1 IDH wild type, 2 showed loss of ATRX expression and 2 had preserved ATRX expression. All 4 tumours overexpressed TP53.
Three tumours satisfied 2007 morphological criteria for glioblastoma. Two were re-assigned to glioblastoma IDH wild type. The third tumour was 1p/19q co-deleted and IDH wild type and was re-assigned to oligodendroglioma NOS grade III. Loss of nuclear ATRX expression and TP53 overexpression were seen in all three tumours.
The single tumour originally diagnosed as GBMO was found to be chromosome 1p/19q intact and IDH1 (R132H) mutated and was re-classified as glioblastoma IDH mutant.
PFS, WHO grade, and molecular markers
Progression-free survival data were available for 79 (96%) of patients.
WHO grading (Table 2)
According to the 2007 WHO classification scheme, 56 patients were grade II, 22 were grade III and 4 were grade IV. Applying the 2016 revision, 6 patients were re-classified into a different grade; 1 from grade IV to grade III and 5 from grade III to grade IV. C-statistics (Table 2) suggest better discriminability with grading according to the 2016 revision. The KM survival curves for PFS, based on tumour grade in the 2007 and 2016 schemata are shown in Fig. 2a, b respectively. The curves show that there was no significant association between WHO grade and PFS (log-rank test: p = 0.83 and p = 0.66 respectively).

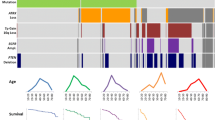
Kaplan-Meir survival curves of progression free survival (PFS) by WHO grade and molecular and immunohistochemistry characteristics. a No difference in PFS by 2007 WHO grade (p = 0.83), b No difference in PFS by 2016 WHO grade (p = 0.66). c IDH mutation is associated with improved PFS regardless of WHO grade (p < 0.001), d IDH mutation is associated with improved PFS regardless of 1p/19q status (p < 0.001). Non co-deleted tumours (n = 46) included those with either 1p or 19q deletion. e loss of nuclear ATRX expression in 1p/19q intact tumours does not affect PFS (p = 0.63), and f TP53 expression is associated with poorer PFS (p = 0.02)
IDH status and WHO grade
Figure 2c shows the KM survival curves for IDH status by 2016 WHO grade. The curves suggest that patients with IDH mutated tumours have a better prognosis regardless of WHO grade (log rank p < 0.001).
IDH mutation and 1p/19q status
The KM plot in Fig. 2d shows the survival curves for IDH and 1p/19q status irrespective of tumour grade. There was a significant difference between survival curves based on a log rank test (p < 0.001). The curves suggest that patients whose tumours were IDH mutated had a better prognosis (IDH wt vs. IDH mut: median PFS 3.97 vs. 1.24 years: HR 4.12, 95% CI: [1.70–9.98], p = 0.002) and even more so in those with intact 1p/19q.
Loss of ATRX expression and TP 53 overexpression
Figure 2e, f show the KM survival curves for loss of ATRX expression and TP53 overexpression respectively. There was no significant difference between tumours with absent and those with preserved nuclear ATRX expression in the subgroup of patients (n = 46) with intact 1p/19q tumours (median PFS: 2.66 and 3.92 years respectively, HR: 0.81, 95% CI: [0.34–1.92], p = 0.63). There was a significant association between TP53 overexpression and shorter progression-free survival (median PFS: overexpressed 3.92 vs. 5.35 years or weak/absent expression, HR: 0.46, 95% CI: [0.23–0.89], p = 0.02).
Discussion
The 2016 revision of the WHO Classification of Tumours of the Central Nervous System broke new ground by utilizing molecular-genetic data to separate tumour sub-types. This alteration has particularly affected the classification of glial tumours. The expectation was that objective molecular-genetic criteria would provide a clearer indication of likely tumour behaviour than had been achieved by previous grading schemes founded on morphological features alone.
Since the release of the revised classification in July 2016, several studies have been published that have re-assigned diffuse glial tumours from 2007 WHO categories to those in the 2016 revision. The largest of these, the French nationwide POLA cohort, re-assigned 1041 tumours and found that classification by the 2016 revision criteria provided clearer prognostic discrimination between diffuse glioma sub-types than grading based on morphology [14]. The analysis of 645 diffuse gliomas in The Cancer Genome Atlas Consortium database by Cimino and co-workers [15] showed that the revised 2016 criteria segregated these tumours into three distinct molecular sub-groups: chromosome 1p/19q co-deleted IDH mutant with oligodendroglial morphology, IDH mutant with astrocytic morphology and IDH wild type with astrocytic morphology. They also reported that grading based on morphology was non-discriminatory. In the study by Pekmezci et al., the 2016 revision criteria were applied retrospectively to 1206 diffuse glial tumours [16]. Based on overall survival analysis, there was clear separation of four molecular sub-types: oligodendroglioma 1p/19q co-deleted/IDH mutant, IDH mutant astrocytoma, IDH wild astrocytoma and IDH wild type glioblastoma. There was no clear difference in overall survival for IDH wild type astrocytoma compared with a fifth sub-type, IDH mutant glioblastoma. In addition, they assessed the influence of telomerase reverse transcriptase promoter (TERTp) and ATRX mutations, deduced from loss of nuclear ATRX immunostaining, on overall survival. Oligodendrogliomas IDH mutant and 1p/19q co-deleted with TERTp mutations had a more favourable outcome compared with TERTp wild type tumours. Among IDH wild type astrocytomas, those that were TERTp wild type had significantly longer survivals compared with TERTp mutated tumours. ATRX mutations were associated with more favourable outcome in IDH wild type glioblastomas compared with ATRX wild type tumours. In a fourth study, by Mellai et al., 204 of 206 diffuse gliomas could be re-assigned to specific categories within the 2016 revision [17]. Survival data were not analysed. This study also showed close association between loss of ATRX protein expression by immunohistochemistry and IDH mutations, particularly IDH1 (R132H). Loss of ATRX protein expression was mutually exclusive of 1p/19q co-deletion.
The data generated in our study corroborate these previous investigations. We found that the molecular criteria in the 2016 revision for the classification of diffuse gliomas are straightforward in their application and segregate diffuse gliomas into three distinct molecular sub-types: 1p/19q co-deleted/IDH mutant, 1p/19q intact/IDH mutant and 1p/19q intact/IDH wild type. The first sub-type is associated with oligodendroglial morphology whereas tumours in the other two categories have astrocytic morphology. As in other studies (5, 11–15) we found that the commonest IDH mutation, IDH-1 R132H [18], is reliably detected by immunohistochemistry. A single immunonegative tumour was found to have an R132S mutation of IDH-1. All remaining 14 immunonegative tumours were found to be IDH wild type by pyrosequencing.
Analysis of PFS showed substantial separation of these three molecular categories. Discrepancies between grade and expected survival have been previously reported [19, 20], and similarly we were unable to show any significant survival difference between WHO grade II and III tumours, indicating that IDH mutation status is more informative than tumour grade. The influence of other factors, such as tumour location, on IDH mutation status and prognosis continues to be investigated [21]. There is currently no consensus on how patients with grade II and III diffuse gliomas should be managed and treatment protocols have previously been guided predominantly by tumour grade [22,23,24,25,26,27,28,29,30]. The clear separation of these tumours into distinct molecular sub-types that is achieved by the 2016 revision provides an objective evidence base for tailoring personalized treatment and guidelines are now being updated [31].
In agreement with other studies [32,33,34], we found a close association between loss of nuclear immunostaining for ATRX and IDH mutations and that no 1p/19q co-deleted tumour showed loss of nuclear ATRX staining. This finding supports the proposal that testing for 1p/19q co-deletion may not be indicated in an ATRX immunonegative tumour [32]. We also found a strong positive association between IDH mutation and TP53 overexpression and a strong negative association between TP53 overexpression and chromosome 1p/19q co-deletion.
The grading system that has been incorporated into previous WHO classifications has largely been determined by histopathological features. Increasing grade has been associated with cumulative anaplastic features; in particular, mitotic activity, endothelial cell hyperplasia and necrosis. The segregation of diffuse astrocytoma into distinct molecular sub-types, as shown in our and other studies, suggests that a molecular grading scheme could now be developed [34,35,36].
We conclude that the objective molecular-genetic criteria of the 2016 revision provide for clearer separation of diffuse glioma sub-types than was achieved by the morphological criteria of the 2007 scheme. In its current form however, the revision is confined to 1p/19q and IDH alterations for the classification of diffuse gliomas. Our and other studies suggest that alterations in ATRX and TP53 genes give additional information that is useful in separating oligodendroglial from astrocytic tumours and also provide prognostic data. As well, TERTp alterations appear to provide further segregation of 1p/19q co-deleted IDH mutated tumours and those that are found to be IDH wild type with intact 1p/19q [15]. Future iterations of the WHO Classification of CNS tumours may incorporate these additional molecular features into both classification and “molecular grading” as the capacity of laboratories to perform molecular testing expands.
References
von Deimling A (eds) (2016) WHO classification of tumours of the central nervous system. IARC, Lyon
Louis DN, Ohgaki H, Wiestler OD, Cavanee WK (eds) (2007) WHO classification of tumours of the central nervous system. IARC, Lyon
Buckner J, Giannini C, Eckel-Passow J, Lachance D, Parney I, Laack N, Jenkins R (2017) Management of diffuse low-grade gliomas in adults: use of molecular diagnostics. Nat Rev Neurol 13:340–351. https://doi.org/10.1038/nrneurol.2017.54
Wang H, Xu T, Jiang Y, Xu H, Yan Y, Fu D, Chen J (2015) The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia 17:239–255. https://doi.org/10.1016/j.neo.2015.02.002
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601. https://doi.org/10.1007/s00401-009-0595-z
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, Diaz LA Jr, Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos N (2011) DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331:1199–1203. https://doi.org/10.1126/science.1200609
Pardo FS, Hsu DW, Zeheb R, Efird JT, Okunieff PG, Malkin DM (2004) Mutant, wild type, or overall p53 expression: freedom from clinical progression in tumours of astrocytic lineage. Br J Cancer 91:1678–1686. https://doi.org/10.1038/sj.bjc.6602161
Takami H, Yoshida A, Fukushima S, Arita H, Matsushita Y, Nakamura T, Ohno M, Miyakita Y, Shibui S, Narita Y, Ichimura K (2015) Revisiting TP53 mutations and immunohistochemistry: a comparative study in 157 diffuse gliomas. Brain Pathol 25:256–265. https://doi.org/10.1111/bpa.12173
Gonzales M, Dale S, Susman M, Mills J (2006) Quantitation of chromosome 1p and 19q deletions in glial tumours by interphase FISH on formalin-fixed paraffin-embedded tissue. J Clin Neurosci 13:96–101. https://doi.org/10.1016/j.jocn.2005.06.004
Gupta R, Flanagan S, Li CC, Lee M, Shivalingham B, Maleki S, Wheeler HR, Buckland ME (2013) Expanding the spectrum of IDH1 mutations in gliomas. Mod Pathol 26:619–625. https://doi.org/10.1038/modpathol.2012.210
Cai J, Zhang C, Zhang W, Wang G, Yao K, Wang Z, Li G, Qian Z, Li Y, Jiang T, Jiang C (2016) ATRX, IDH1-R132H and Ki-67 immunohistochemistry as a classification scheme for astrocytic tumors. Oncoscience 3:258–265
Juratli TA, Kirsch M, Robel K, Soucek S, Geiger K, von Kummer R, Schackert G, Krex D (2012) IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol 108:403–410. https://doi.org/10.1007/s11060-012-0844-1
Iuchi T, Namba H, Iwadate Y, Shishikura T, Kageyama H, Nakamura Y, Ohira M, Yamaura A, Osato K, Sakiyama S, Nakagawara A (2002) Identification of the small interstitial deletion at chromosome band 1p34-p35 and its association with poor outcome in oligodendroglial tumors. Genes Chromosomes Cancer 35:170–175. https://doi.org/10.1002/gcc.10080
Tabouret E, Nguyen AT, Dehais C, Carpentier C, Ducray F, Idbaih A, Mokhtari K, Jouvet A, Uro-Coste E, Colin C, Chinot O, Loiseau H, Moyal E, Maurage CA, Polivka M, Lechapt-Zalcman E, Desenclos C, Meyronet D, Delattre JY, Figarella-Branger D, For Pola Network (2016) Prognostic impact of the 2016 WHO classification of diffuse gliomas in the French POLA cohort. Acta Neuropathol 132:625–634. https://doi.org/10.1007/s00401-016-1611-8
Cimino PJ, Zager M, McFerrin L, Wirsching HG, Bolouri H, Hentschel B, von Deimling A, Jones D, Reifenberger G, Weller M, Holland EC (2017) Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun 5:39. https://doi.org/10.1186/s40478-017-0443-7
Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H, Sicotte H, Kollmeyer TM, McCoy LS, Sarkar G, Perry A, Giannini C, Tihan T, Berger MS, Wiemels JL, Bracci PM, Eckel-Passow JE, Lachance DH, Clarke J, Taylor JW, Luks T, Wiencke JK, Jenkins RB, Wrensch MR (2017) Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol 133:1001–1016. https://doi.org/10.1007/s00401-017-1690-1
Mellai M, Annovazzi L, Senetta R, Dell’Aglio C, Mazzucco M, Cassoni P, Schiffer D (2017) Diagnostic revision of 206 adult gliomas (including 40 oligoastrocytomas) based on ATRX, IDH1/2 and 1p/19q status. J Neurooncol 131:213–222. https://doi.org/10.1007/s11060-016-2296-5
Waitkus MS, Diplas BH, Yan H (2016) Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncology 18(1):16–26. https://doi.org/10.1093/neuonc/nov136
Reuss DE, Mamatjan Y, Schrimpf D, Capper D, Hovestadt V, Kratz A, Sahm F, Koelsche C, Korshunov A, Olar A, Hartmann C, Reijneveld JC, Wesseling P, Unterberg A, Platten M, Wick W, Herold-Mende C, Aldape K, von Deimling A (2015) IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol 129:867–873. https://doi.org/10.1007/s00401-015-1438-8
Lasocki A, Tsui A, Tacey MA, Drummond KJ, Field KM, Gaillard F (2014) MRI grading versus histology: predicting survival of World Health Organization grade II–IV astrocytomas. Am J Neuroradiol 36:77–83. https://doi.org/10.3174/ajnr.A4077
Paldor I, Drummond KJ, Kaye AH (2016) IDH1 mutation may not be prognostically favorable in glioblastoma when controlled for tumor location: a case-control study. J Clin Neurosci 34:117–120. https://doi.org/10.1016/j.jocn.2016.05.016
Dong X, Noorbakhsh A, Hirshman BR, Zhou T, Tang JA, Chang DC, Carter BS, Chen CC (2015) Survival trends of grade I, II, and III astrocytoma patients and associated clinical practice patterns between 1999 and 2010: a SEER-based analysis. Neuro-Oncol Pract. https://doi.org/10.1093/nop/npv016
Oberheim Bush NA, Chang S (2016) Treatment strategies for low-grade glioma in adults. J Oncol Pract 12:1235–1241. https://doi.org/10.1200/JOP.2016.018622
Soffietti R, Baumert BG, Bello L, von Deimling A, Duffau H, Frenay M, Grisold W, Grant R, Graus F, Hoang-Xuan K, Klein M, Melin B, Rees J, Siegal T, Smits A, Stupp R, Wick W, European Federation of Neurological (2010) Guidelines on management of low-grade gliomas: report of an EFNS-EANO Task Force. Eur J Neurol 17:1124–1133
van den Bent MJ, Snijders TJ, Bromberg JE (2012) Current treatment of low grade gliomas. Memo 5:223–227. https://doi.org/10.1007/s12254-012-0014-3
Wahl M, Phillips JJ, Molinaro AM, Lin Y, Perry A, Haas-Kogan DA, Costello JF, Dayal M, Butowski N, Clarke JL, Prados M, Nelson S, Berger MS, Chang SM (2017) Chemotherapy for adult low-grade gliomas: clinical outcomes by molecular subtype in a phase II study of adjuvant temozolomide. Neuro Oncol 19:242–251. https://doi.org/10.1093/neuonc/now176
Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, Cohen-Jonathan-Moyal E, Frappaz D, Henriksson R, Balana C, Chinot O, Ram Z, Reifenberger G, Soffietti R, Wick W (2014) EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol 15:e395–e403. https://doi.org/10.1016/s1470-2045(14)70011-7
Zadeh G, Khan OH, Vogelbaum M, Schiff D (2015) Much debated controversies of diffuse low-grade gliomas. Neuro Oncol 17:323–326. https://doi.org/10.1093/neuonc/nou368
Rosenthal MA, Drummond KJ, Dally M, Murphy M, Cher L, Ashley D, Thursfield V, Giles GG (2006) Management of glioma in Victoria (1998–2000): retrospective cohort study. Med J Aust 184:270–273
Gan HK, Rosenthal MA, Cher L, Dally M, Drummond K, Murphy M, Thursfield V (2015) Management of glioblastoma in Victoria, Australia (2006–2008). J Clin Neurosci 22:1462–1466. https://doi.org/10.1016/j.jocn.2015.03.029
Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, Henriksson R, Rhun EL, Balana C, Chinot O, Bendszus M, Reijneveld JC, Dhermain F, French P, Marosi C, Watts C, Oberg I, Pilkington G, Baumert BG, Taphoorn MJB, Hegi M, Westphal M, Reifenberger G, Soffietti R, Wick W (2017) European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol 18:e315–e329. https://doi.org/10.1016/s1470-2045(17)30194-8
Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Muhleisen H, Eckert F, Tabatabai G, Schittenhelm J (2016) ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun 4:60. https://doi.org/10.1186/s40478-016-0331-6
Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, Schweizer L, Korshunov A, Jones DT, Hovestadt V, Mittelbronn M, Schittenhelm J, Herold-Mende C, Unterberg A, Platten M, Weller M, Wick W, Pfister SM, von Deimling A (2015) ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129:133–146. https://doi.org/10.1007/s00401-014-1370-3
Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, Platten M, Weller M, Wick W (2013) ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126:443–451. https://doi.org/10.1007/s00401-013-1156-z
Dubbink HJ, Atmodimedjo PN, Kros JM, French PJ, Sanson M, Idbaih A, Wesseling P, Enting R, Spliet W, Tijssen C, Dinjens WN, Gorlia T, van den Bent MJ (2016) Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: a report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro Oncol 18:388–400. https://doi.org/10.1093/neuonc/nov182
Kim Y-H, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Sure U, Wrede K, Nakazato Y, Vital A, Mariani L, Stawski R, Watanabe T, De Girolami U, Kleihues P, Ohgaki H (2010) Molecular classification of low-grade diffuse gliomas. Am J Pathol 177(6):2708–2714. https://doi.org/10.2353/ajpath.2010.100680
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rogers, T.W., Toor, G., Drummond, K. et al. The 2016 revision of the WHO Classification of Central Nervous System Tumours: retrospective application to a cohort of diffuse gliomas. J Neurooncol 137, 181–189 (2018). https://doi.org/10.1007/s11060-017-2710-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-017-2710-7