
Abstract
Background
Pediatric postoperative cognitive dysfunction (POCD) is a prevalent complication following anesthesia and surgery. Hypoxia and propofol are the primary risk factors contributing to pediatric POCD. Our previous in vivo animal research has demonstrated that cognitive dysfunction in immature Sprague-Dawley (SD) rats, induced by hypoxia combined with propofol (HCWP), is closely associated with hippocampal neuron ferroptosis.
Methods and results
In vivo transcriptome sequencing and KEGG functional analysis revealed significant enrichment of the mitophagy pathway. To further elucidate the relationship between mitophagy and ferroptosis, HT22 cells were selected to construct an in vitro HCWP model. Our findings indicate that HCWP activates excessive mitophagy in HT22 cells, leading to decreased mitochondrial membrane potential (ΔΨm), reactive oxygen species (ROS) burst, mitochondrial fragmentation, and the induction of ferroptosis. To explore this causal relationship further, we employed Mdivi-1, a mitophagy inhibitor. Notably, low-dose Mdivi-1 (10 µM) effectively suppressed excessive mitophagy in HT22 cells, improved mitochondrial function and morphology, and mitigated markers associated with ferroptosis. The mechanism by which Mdivi-1 alleviates HCWP-induced ferroptosis in HT22 cells is likely due to its inhibition of excessive mitophagy, thereby promoting mitochondrial homeostasis.
Conclusions
Our study suggests that mitophagy may be an upstream event in HCWP-induced ferroptosis in HT22 cells. Consequently, targeted regulation of mitophagy by Mdivi-1 may represent a promising approach to prevent cognitive dysfunction following HCWP exposure.
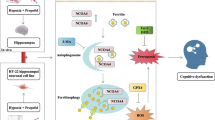
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Postoperative cognitive dysfunction (POCD) is a prevalent complication following anesthesia and surgery, primarily characterized by learning and memory impairment and personality disorders, which severely impact patients’ quality of life and pose a global threat to human health [1, 2]. The pathogenesis of POCD is complex, involving increased brain inflammation, exacerbated oxidative stress, reduced synaptic connections, and neuronal destruction [1, 3, 4]. Children’s brain development is immature, making them more susceptible to brain development damage when exposed to risk factors, resulting in long-term cognitive impairment and even death [5]. However, the specific mechanisms underlying pediatric POCD remain unclear.
Hypoxemia is a common perioperative complication prevalent in pathological conditions such as shock, pulmonary edema, and congenital heart disease shunts in children. Additionally, brain-local hypoxia caused by specific surgical positions or perioperative drugs is often undetected. Research has indicated that nearly 50% of patients in clinical settings experienced varying degrees of hypoxia during the perioperative period [6]. Hypoxia can severely impact cellular function, slow down drug metabolism, and promote cell apoptosis [7]. Propofol is a frequently utilized intravenous general anesthetic, is extensively employed for anesthesia induction and maintenance, as well as for sedation in the pediatric intensive care unit (ICU). Propofol has demonstrated potential neurotoxicity in the developing brain, including promoting the release of neuroinflammatory factors, triggering cell apoptosis, and causing extensive neurodegeneration [8,9,10]. Both hypoxia and propofol are considered risk factors for pediatric POCD [11].
Ferroptosis, an emerging type of regulated cell death (RCD), is characterized by the accumulation of reactive oxygen species (ROS) and iron-dependent lipid peroxidation, posing a substantial threat to cellular homeostasis [12]. This process differs from other types of cell death, such as apoptosis and necrosis, and is increasingly recognized as a significant mediator of neurodegenerative diseases and various brain injuries [13]. Our previous in vivo research has shown that hypoxia combined with propofol (HCWP) leads to cognitive dysfunction in immature SD rats, with the mechanism closely connected to hippocampal neuronal ferroptosis [14]. However, the role of ferroptosis and its upstream pathways and processes in HCWP-induced cognitive impairment remains unknown.
Mitophagy, a type of selective autophagy, maintains mitochondrial function and quantity in cells by eliminating and degrading damaged mitochondria, playing a crucial role in maintaining mitochondrial quality control and cellular redox balance [15]. This degradation can occur through both ubiquitin-dependent and ubiquitin-independent pathways [16]. In the context of ferroptosis, mitophagy has been suggested to be a double-edged sword, exhibiting both protective and detrimental roles depending on the degree of autophagy flux. On the one hand, during cell damage caused by ferroptosis, normal mitophagy may exert a protective effect by clearing dysfunctional mitochondria and reducing their released ROS [17]. On the other hand, excessive mitophagy may ultimately provide additional iron and ROS, thereby amplifying lipid peroxidation and ferroptosis [18]. However, the role of mitophagy in HCWP-induced ferroptosis remains unclear.
Therefore, based on our previous in vivo studies, we selected mouse hippocampal neuron cells (HT22) to further explore the specific molecular mechanisms among HCWP, ferroptosis, and mitophagy in vitro. We aim to provide new insights and intervention targets for the prevention and treatment of pediatric POCD.
Materials and methods
Cell culture
HT22 mouse hippocampal neuronal cells were obtained from the Chinese Academy of Sciences Cell Bank of Type Culture Collection (Shanghai, China) (RRID: CVCL_0321) and cultured in high glucose medium (DMEM, C11995500BT, Gibco, Waltham, U.S.) supplemented with 10% fetal bovine serum (FBS, 10099-141, Gibco, Waltham, U.S.) and 1% penicillin-streptomycin (P0781, Sigma, U.S.). The cells were incubated in a humidified incubator (3131, Thermo Fisher, U.S.) with 5% CO2 at 37 °C and subcultured after reaching 80–90% confluence.
Cell treatment
HT22 cells were divided into 5 groups: CON (blank control group), CA (lipid emulsion solvent + room air group), CH (lipid emulsion solvent + hypoxia group), PA (propofol + room air group), and PH (propofol + hypoxia group). HT22 cells received treatment with 100 µM lipid emulsion solvent (F18020309, Kelun, Sichuan, China) or 100 µM propofol (X21062B, AstraZeneca, Italy), respectively, and then incubated at room air (21% O2) or in a 1% hypoxia incubator (3131, Thermo Fisher, U.S.) for 24 h. In the inhibitor-related experiments, HT22 cells were divided into 4 groups: CON, CON + Mdivi-1, PH, and PH + Mdivi-1. Mdivi-1 (SC8028-10 mM, Beyotime, China) (Lot: 072823231207) stock solution was diluted with DMEM medium to reach a final concentration of 10 µM for use. HT22 cells were pretreated with 10 µM Mdivi-1 for 2 h before modeling.
Cell viability assay
Cell viability was assessed with the Cell Counting Kit-8 (K1018, CCK-8, APExBIO, USA). HT22 cells were planted in 96-well plates (3599, Corning/Costar, U.S.) at a density of 5 × 103 cells per well, with six replicate wells set for each group. When cells growth density reached about 60% confluence, the cells were intervened for 24 h. After incubating HT22 cells with 10% CCK-8 in the dark at 37 °C for 2 h, absorbance at 450 nm was measured by a microplate reader (Cytation 5, BioTek Instruments, U.S.).
Malondialdehyde (MDA) assay
The Cell MDA Assay Kit (A003-4-1, NanjingJiancheng, China) was used to measure the level of MDA. As directed by the manufacturer, the absorbance values were measured at 530 nm using a microplate reader, and then the protein content was determined using the BCA protein assay kit (P0010, Beyotime, China). The MDA content was calculated according to the formula and expressed in nmol/mgprot. MDA content (nmol/mgprot) = [(Measured OD value - Blank OD value)/(Standard OD value - Blank OD value)]×Standard sample concentration/Tested sample protein concentration. Each sample was assayed three times.
Iron content detection
The iron content was measured using a Cell Ferrous Iron Colorimetric Assay Kit (E-BC-K881-M, Elabscience, Wuhan, China). The ferrous ion content standard curve was tested using different concentrations of iron standard samples, as instructed by the manufacturer. The samples to be tested were mixed with the chromogenic solution, incubated for 10 min at 37 °C, and the OD values at 593 nm were measured using the microplate reader. Then the ferrous ion contents in the samples were calculated according to the formula in the instructions.
Intracellular ROS detection
HT22 cells were cultured in a confocal dish (BS-15-GJM, Biosharp, China) with a diameter of 15 mm at a density of 1 × 104 per dish, with intervention based on cell modeling conditions. The ROS levels were determined using the ROS Assay Kit (S0033S, Beyotime, China) in accordance with the manufacturer’s instructions. The DCFH-DA probe was diluted with FBS-free DMEM medium (C11995500BT, Gibco, Waltham, U.S.) to a concentration of 10 µM. After adding 1 mL of solution per well and incubating at 37 °C for 20 min, the dish was rinsed three times, followed by nuclei staining with 1X Hoechst 33,342 (C1028, Beyotime, China) at 37 °C for 10 min. A Nikon C2 confocal microscope (Nikon, Japan) was used to image cells at a magnification of 300 times, and the fluorescence intensity of the FITC channel in the pictures was analyzed using ImageJ software (Version 1.53a).
Mitochondrial membrane potential (ΔΨm) detection
HT22 cells were seeded in 15 mm confocal dishes at a density of 1 × 104 per dish. Cell modeling was performed as previously described. An appropriate amount of JC-1 (200X) (C2003S, Beyotime, China) was taken, and it was diluted with JC-1 buffer to JC-1 working solution (1X), then the cells were washed with DMEM medium once. A mixture of 0.5 mL of JC-1 working solution (1X) and 0.5 mL of DMEM medium was added to each dish. The cells were then cultured at 37 °C in a 5% CO2 incubator for 30 min before being rinsed twice with JC-1 buffer. Cells were imaged at a magnification of 600 times using a Nikon C2 confocal microscope (Nikon, Japan). JC-1 monomer fluorescence was excited using a 488 nm laser and JC-1 aggregate fluorescence was excited using a 561 nm laser. The ImageJ software was used to analyze the fluorescence intensity ratio of FITC and TRITC channels of the image.
Western blotting
Proteins were extracted from the HT22 cells using the Whole-Protein Extraction Kit (KGP250, KeyGEN Biotech, China) and the proteins’ concentration was measured using the BCA Protein Concentration Determination Kit (P0010, Beyotime, China). The PAGE Gel Fast Preparation Kit (PG112/PG113, Epizyme, China) was selected according to the molecular weight of the target protein to prepare the 10% or 12.5% PAGE gel. Equal protein amounts were then separated in a Tris-Glycine buffer system using Bio-Rad (PowerPac™ Universal Power Supply, U.S.) at a constant voltage of 80 V. Then, the proteins were transferred onto a 0.2 μm PVDF membrane (ISEQ00010, Millipore, U.S.) using Bio-Rad at a constant current of 400 mA. Subsequently, the membranes were blocked with 5% skim milk (P0216, Beyotime, China) for 2 h. The membranes were washed three times with 1 × TBST followed by 12 h incubation with appropriate primary antibodies at 4 °C. After the incubation with the primary antibody was completed, the membranes were washed three times with 1 × TBST, for 10 min each time. The membranes were then incubated with appropriate HRP-conjugated secondary antibodies for 2 h at room temperature. The membranes were washed three times with 1 × TBST again. The protein bands were visualized using an ultra-high sensitivity ECL kit (MAO186, meilunbio, China) on a Bio-Rad imager (ChemiDoc, Bio-Rad, U.S.). The expression levels of the target proteins were quantitatively analyzed using ImageLab software (version 6.0, Bio-Rad, USA), with β-actin serving as the reference protein for the whole-lysate proteins. The following primary antibodies were utilized: GPX4 (1:1000, BS90596, Bioworld, China) (Lot: CC02181) (RRID: AB_3105959), FTH1 (1:1000, ET1610-78, HUABIO, China) (Lot: H661963002) (RRID: AB_3069963), ACSL4 (1:1000, BS71431, Bioworld, China) (Lot: CN03212) (RRID: AB_3105960), p62 (1:5000, T55546F, Abmart, China) (Lot: 10,045,859) (RRID: AB_2936967), LC3 (1:2500, 14600-1-AP, Proteintech, China) (Lot: 00107124) (RRID: AB_2137737), PINK1 (1:1000, 23274-1-AP, Proteintech, China) (00140989) (RRID: AB_2879244), Parkin (1:2500, 14060-1-AP, Proteintech, China) (Lot: 00150132) (RRID: AB_2878005), β-actin (1:1500, AM1829B, abcepta, China) (Lot: SG210714Z01) (RRID: AB_10664137). The following HRP-conjugated secondary antibodies were utilized: Goat Anti-Mouse IgG antibody (1:10000, ASP1613, abcepta, China) (Lot: 20,220,621) (RRID: AB_3106369), Goat Anti-Rabbit IgG antibody (1:1000, A0208, Beyotime, China) (Lot: 051022220718) (RRID: AB_2892644).
Double staining observation of mitochondria and lysosomes
HT22 cells were planted in a confocal dish at the above density, and cell modeling was performed as described above. The Mito-Tracker Green (50 nM) (C1048, Beyotime, China) and Lyso-Tracker Red (50 nM) (C1046, Beyotime, China) were added to the dishes. The cells were then cultured at 37 °C in a 5% CO2 incubator for 30 min and washed with 1 × PBS. The 1X Hoechst 33,342 (C1028, Beyotime, China) was used to stain nuclei. Cells were imaged at a magnification of 900 times under oil microscope using a Nikon C2 confocal microscope (Nikon, Japan). The fluorescence mean intensity ratio of lysosomes in TRITC channel was analyzed by ImageJ software.
Mitochondrial morphological analysis
After Mito-tracker Green was used to tack mitochondria, the images of mitochondria were obtained at a magnification of 900 times under the oil microscope of Nikon C2 confocal microscope (Nikon, Japan). The mitochondrial skeleton was analyzed using the Fiji software (Version 2.14.0), and the length of mitochondrial branches was analyzed using the MiNA plugin (Version 3.0.1).
Statistical analysis
Data were analyzed using GraphPad Prism 8.0 software (San Diego, USA) and were presented as means ± standard deviation (SD). Each experiment included at least 3 replicates. The normality of data was assessed using the Shapiro-Wilk test, and all data met normal distribution. Differences among three or more groups were compared using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons tests, with statistical significance defined as p ≤ 0.05.
Results
HCWP triggers ferroptosis in HT22 cells
Initially, we conducted CCK-8 cell viability assays to investigate the effects of propofol concentration, oxygen concentration, and intervention duration. We determined that treatment with 100 µM propofol under 1% oxygen concentration for 24 h was the optimal modeling condition (Fig. 1A and B). Subsequently, under these conditions, we assessed the cell viability of 5 groups (CON, CA, CH, PA and PH). We observed a decrease in cell viability in the propofol groups (PA and PH) compared to the corresponding lipid emulsion solvent groups (CA and CH) (p < 0.05). The PA and PH groups exhibited a statistically significant difference, with the cell viability of the PH group reduced by approximately 40.7% compared to the CON group (Fig. 1C). These data indicated a significant toxic effect of HCWP treatment on HT22 cells.
To determine whether HCWP-induced cytotoxicity is related to ferroptosis, we examined relevant ferroptosis indicators. Malondialdehyde (MDA), a product of membrane lipid peroxidation, and intracellular iron (Fe2+) levels are crucial biomarkers for detecting and assessing ferroptosis [19]. As shown in Fig. 1D and E, MDA and Fe2+ levels in the propofol group (PA and PH) were significantly increased compared to the corresponding lipid emulsion solvent groups (CA and CH) (p < 0.05). Notably, the PH group exhibited the most significant increase compared to the CON group, with MDA and Fe2+ levels increasing by about 2.88 and 4.05 times, respectively. These data indicated that HCWP treatment resulted in excessive lipid peroxidation and iron accumulation in cells. Furthermore, we detected the expression of ferroptosis-related proteins in each group using Western blot analysis. The results demonstrated that, compared to CON, protein expression of glutathione peroxidase 4 (GPX4) and ferritin heavy chain 1 (FTH1) was significantly decreased in the PH group, while protein expression of acyl-CoA synthetase long-chain family member 4 (ACSL4) increased (p < 0.05) (Fig. 1F-K). Collectively, these data suggested that HCWP treatment may trigger ferroptosis in HT22 cells.

HCWP triggers ferroptosis in HT22 cells. (A) The effects of different propofol concentrations and intervention times on the viability of HT22 cells under air condition. (B) The effect of 24 h intervention with different propofol concentrations under different oxygen concentrations on the viability of HT22 cells. (C) The effects of 100 µM propofol and 1% oxygen on the viability of HT22 cells in different groups. (D, E) Malondialdehyde (MDA) and Ferrous Iron (Fe2+) contents in different groups. (F-K) Protein expression of ferroptosis-related markers GPX4, FTH1 and ACSL4, with β-actin as a reference protein (n = 3). Data are presented as mean ± standard deviation; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n.s., not significant
HCWP activates excessive mitophagy in HT22 cells
In our previous HCWP animal model experiment, transcriptome sequencing (RNA-seq) results revealed significant KEGG pathway enrichment of the mitophagy in the PH group compared to the CH group (Fig. 2A). These data led us to hypothesize that mitophagy may also play a crucial role in HCWP-treated cell models. Therefore, we first examined the protein expression levels of autophagy-associated markers LC3 and p62. Western blot (WB) analysis demonstrated an increase in the expression of LC3-II protein in each propofol group (PA and PH) compared to the corresponding lipid emulsion solvent group (CA and CH) (p < 0.05). Additionally, the PH group showed a significantly elevated expression of LC3-II protein compared to the CON group (p < 0.05). Conversely, the expression of p62 protein exhibited an opposite trend (p < 0.05) (Fig. 2B-E), indicating that autophagy was induced following HCWP exposure.
To further investigate the specific types of autophagy, we examined the expression of key proteins PINK1 and Parkin in the ubiquitin-dependent mitophagy pathway in each group. We found that, compared to the corresponding lipid emulsion solvent group (CA and CH), protein expression levels of PINK1 and Parkin were increased in the propofol group (PA and PH) (p < 0.05). Additionally, compared to the CON group, the protein levels of PINK1 and Parkin showed a significant increase in the PH group (p < 0.05) (Fig. 2F-I). Mitophagy typically involves the formation of autophagosomes around damaged mitochondria, which then fuse with lysosomes to facilitate their degradation [20]. Consequently, to further demonstrate the occurrence of mitophagy, we performed double staining targeting mitochondria and lysosomes, followed by confocal microscopy. Initially, we observed abnormal mitochondrial morphology in HT22 cells, characterized by extensive fragmentation, indicating possible mitochondrial damage. Additionally, we observed extensive lysosome aggregation around fragmented mitochondria and noted the engulfment of mitochondria by lysosomes. Further analysis of fluorescence intensity revealed that, compared to the CON group, the mean fluorescence intensity of lysosomes significantly increased in the PH group (p < 0.05) (Fig. 2J and K). These findings collectively suggested that mitophagy may be overactivated in HCWP-exposed HT22 cells.

HCWP activates excessive mitophagy in HT22 cells. (A) KEGG analysis based on hippocampal tissues RNA-seq showed enrichment of mitophagy pathway. (B-E) Protein expression of autophagy-related markers LC3 and p62, with β-actin as a reference protein (n = 3). (F-I) Protein expression of Mitophagy-related markers PINK1 and Parkin, with β-actin as a reference protein (n = 3). (J, K) Confocal images of the mitophagy in HT22 cells (scale bar, 10 μm). Data are presented as mean ± standard deviation; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n.s., not significant
Mitochondrial dysfunction and morphological abnormalities occurred in HT22 cells after HCWP treatment
The observed fragmentation of mitochondria in the PH group (Fig. 2J) prompted us to further analyze the mitochondrial skeleton using Fiji software. Upon observation, we noted that following HCWP treatment, the mitochondrial morphology in the PH group transitioned from rod-shaped and filamentous to punctate and fragmented, with apparent disruption of the mitochondrial network structure compared to the CON group (Fig. 3A). These findings are consistent with our previous observations. Analysis and statistics of mitochondrial branch length using the MiNA plugin revealed that the average length of mitochondrial branches was 0.933 μm in the CON group, whereas in the PH group, it was 0.631 μm, indicating a reduction of about 1.48 times (p < 0.05) (Fig. 3B). These findings suggested that mitochondria in cells treated with HCWP exhibited a more rounded and fragmented morphology compared to those in the CON group.
A normal structure is crucial for maintaining mitochondrial function, and typically mitochondrial elongation, increased membrane potential, and improved mitochondrial function are consistent [21]. Conversely, an increase in ROS and mitochondrial disruption may indicate functional impairment [22]. Therefore, we further tested relevant indicators of mitochondrial function. We used a JC-1 probe to detect mitochondrial membrane potential (MMP). In cells with normal MMP, JC-1 forms aggregates and emits red fluorescence, while in cells with decreased MMP, JC-1 exists as monomers and emits green fluorescence. As shown in Fig. 3C and D, compared to the corresponding lipid emulsion solvent groups (CA and CH), the MMP of the propofol groups (PA and PH) decreased (the JC-1 aggregate/monomer ratio decreased by about 3.06 and 3.08 times, respectively, p < 0.05). Additionally, compared to the CON group, HCWP reduced MMP in HT22 cells (the JC-1 aggregates/monomers ratio decreased by about 6.92 times, p < 0.05). Using the DCFH-DA method to detect intracellular ROS, we found that, compared to the corresponding lipid emulsion solvent groups (CA and PA), the intracellular ROS levels in the propofol groups (CH and PH) increased by about 5.31 and 2.75 times, respectively. Furthermore, the PH group exhibited a notable increase in ROS levels, about 10.13 times higher than the CON group (p < 0.05) (Fig. 3E and F). These results suggested the occurrence of mitochondrial dysfunction in HCWP-treated HT22 cells.

HCWP activates excessive mitophagy in HT22 cells. (A) Morphological images analysis of mitochondrial skeleton using Fiji software (scale bar, 10 μm). (B) Mitochondrial branch length analysis using MiNA plugin. (C, D) Confocal images of the mitochondrial membrane potential in HT22 cells (scale bar, 20 μm) (n = 4). (E, F) Confocal images of intracellular ROS in HT22 cells (scale bar, 25 μm) (n = 6). Data are presented as mean ± standard deviation; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n.s., not significant
Mdivi-1 inhibits excessive mitophagy induced by HCWP in HT22 cells
To further explore the potential causal relationship between mitophagy and HCWP-induced ferroptosis, we conducted experiments using Mdivi-1 (a mitochondrial fission initiation protein inhibitor known to effectively inhibit mitophagy). The CCK-8 experiment showed that cell viability decreased with increasing concentration of Mdivi-1, and treatment with 20 µM Mdivi-1 resulted in significant cell damage (p < 0.05) (Fig. 4A). To eliminate the interference of high inhibitor concentrations on cells, we chose 10 µM Mdivi-1 for cell pretreatment. As shown in Fig. 4B-E, compared to the PH group, the expression of the autophagy-related protein LC3-II decreased, and the expression of p62 protein increased in the PH + Mdivi-1 group (p < 0.05). Additionally, compared to the PH group, the expression of mitophagy-related proteins PINK1 and Parkin significantly decreased in the PH + Mdivi-1 group (p < 0.05) (Fig. 4F-I). Moreover, Mdivi-1 pretreatment notably reduced lysosomal fluorescence intensity (p < 0.05) (Fig. 4J and K). These findings collectively suggested that pretreatment with Mdivi-1 reduced the autophagic flux in HCWP-exposed HT22 cells and effectively inhibited excessive mitophagy.

Mdivi-1 inhibits excessive mitophagy induced by HCWP in HT22 cells. (A) The effect of different concentrations of Mdivi-1 on the viability of HT22 cells. (B-E) The effects of Mdivi-1 pretreatment on LC3 and p62 protein expression in HCWP-induced HT22 cells (n = 3). (F-I) The effects of Mdivi-1 pretreatment on PINK1 and Parkin protein expression in HCWP-induced HT22 cells (n = 3). (J, K) Confocal images examining the effect of Mdivi-1 pretreatment on mitophagy in HT22 cells (scale bar, 10 μm). Data are presented as mean ± standard deviation; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n.s., not significant
Mdivi-1 improves mitochondrial dysfunction and morphological abnormalities in HT22 cells treated with HCWP
The balance of mitophagy plays a crucial role in maintaining mitochondrial morphology and function. Mdivi-1 pretreatment effectively inhibited excessive mitophagy in HT22 cells. Can it improve mitochondrial morphology and function? Through analysis of the mitochondrial skeleton, we observed that Mdivi-1 pretreatment reduced mitochondrial fragmentation and restored mitochondrial network structure (Fig. 5A). The average length of mitochondrial branches increased from 0.733 μm to 1.406 μm (p < 0.05) (Fig. 5B). Regarding mitochondrial function, we found that Mdivi-1 pretreatment rescued the decreased MMP level (the JC-1 ratio of aggregates/monomers in the PH + Mdivi-1 group increased by about 5.06 times compared to the PH group, p < 0.05) (Fig. 5C and D), while also reducing ROS levels (the ROS level in the PH + Mdivi-1 group decreased by about 3.02 times compared to the PH group, p < 0.05) (Fig. 5E and F). These results collectively indicated that pretreatment with Mdivi-1 improved mitochondrial morphological abnormalities and dysfunction in HT22 cells exposed to HCWP.

Mdivi-1 improves mitochondrial dysfunction and morphological abnormalities in HT22 cells treated with HCWP. (A) The effect of Mdivi-1 pretreatment on the morphology of mitochondrial skeleton (scale bar, 10 μm). (B) The effect of Mdivi-1 pretreatment on the mitochondrial branch length. (C, D) Confocal images examining the effect of Mdivi-1 pretreatment on mitochondrial membrane potential in HT22 cells (scale bar, 20 μm) (n = 4). (E, F) Confocal images examining the effect of Mdivi-1 pretreatment on intracellular ROS in HT22 cells (scale bar, 25 μm) (n = 6). Data are presented as mean ± standard deviation; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n.s., not significant
Mdivi-1 alleviates ferroptosis in HT22 cells triggered by HCWP
To explore the relationship between mitophagy and ferroptosis, we re-evaluated ferroptosis-related biomarkers after Mdivi-1 pretreatment. The results revealed that Mdivi-1 pretreatment increased HT22 cell viability (the PH + Mdivi-1 group exhibited a 21.8% increase compared to the PH group, p < 0.05) (Fig. 6A). Additionally, Mdivi-1 pretreatment decreased MDA and Fe2+ levels (p < 0.05) (Fig. 6B and C). Western blot analysis demonstrated that, compared to the PH group, Mdivi-1 pretreatment increased the expression of GPX4 and FTH1 proteins in the PH + Mdivi-1 group, while reducing the expression of ACSL4 protein (p < 0.05) (Fig. 6D-I). These findings suggested that Mdivi-1 pretreatment alleviated ferroptosis in HT22 cells triggered by HCWP.

Mdivi-1 alleviates ferroptosis in HT22 cells triggered by HCWP. (A) The effect of Mdivi-1 pretreatment on the HT22 cell activity. (B, C) The effect of Mdivi-1 pretreatment on MDA and Fe2+ levels in HT22 cells. (D-I) The effects of Mdivi-1 pretreatment on GPX4, FTH1 and ACSL4 protein expression in HCWP-exposed HT22 cells (n = 3). Data are presented as mean ± standard deviation; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, n.s., not significant
Discussion
In the current study, we first constructed the HCWP model on HT22 cells and detected biomarkers related to ferroptosis and mitophagy, confirming the presence of both phenotypes. Pretreatment with Mdivi-1 inhibited excessive mitophagy and effectively alleviated HCWP-induced ferroptosis in HT22 cells. These findings suggest that mitophagy may be a key upstream event in HCWP-induced ferroptosis. In addition, mitochondrial morphological abnormalities and dysfunction caused by HCWP were rescued by Mdivi-1, indicating that the balance of mitophagy is crucial for normal mitochondrial function and morphology, which may also be a key mechanism for Mdivi-1 to improve ferroptosis.
Oxygen is a crucial determinant of cellular function in tissues, and the hippocampus is one of the brain regions most sensitive to variations in oxygen content [23]. It also serves as the main structural basis for learning and memory abilities [24]. Zhang et al. reported that chronic hypoxia exposure could induce pathological changes in neurons [25]. Hypoxia can lead to neuronal apoptosis through oxidative stress, inflammatory response, and excitatory toxicity [26]. Propofol, a widely used anesthetic, is closely scrutinized for its safety in pediatric patients. Current research has not reached a consensus on the impact of propofol on cognitive function [9, 27]. In fact, these two factors often coexist in pediatric patients. Our observation in clinical practice indicates that children with perioperative hypoxemia exhibit significant long-term cognitive impairment after receiving propofol anesthesia. To elucidate the mechanism by which exposure to general anesthesia affects long-term cognitive function in children with hypoxemia, we conducted this study.
In 2012, Dixon et al. first introduced the concept of ferroptosis, a form of programmed cell death that is iron-dependent and characterized by the accumulation of lipid reactive oxygen species (ROS) [12]. Recent studies have shown that ferroptosis is involved in the occurrence and development of various neurological diseases. Zhang et al. reported that the overexpression and hyperphosphorylation of tau protein in Alzheimer’s disease (AD) mice can induce neuronal loss via ferroptosis, thereby leading to a decline in cognitive function [28]. Tang et al. found that Ferrostatin-1 improved diabetes-related cognitive dysfunction by inhibiting ferroptosis [29]. Our previous research has demonstrated that ferroptosis plays an important role in HCWP animal models, and that inhibiting ferroptosis can alleviate cognitive impairment in immature rats induced by HCWP [14].
As research on ferroptosis has advanced, recent studies have increasingly shown a crucial interaction between ferroptosis and autophagy: ferroptosis requires autophagy mechanisms to execute [30, 31]. Mitophagy, a form of selective autophagy, is crucial for maintaining mitochondrial quality control and homeostasis, and it significantly contributes to the regulation of ferroptosis. Lin et al. discovered that ferroptosis in renal tubular epithelial cells exacerbated cisplatin-induced acute kidney injury (AKI), whereas mitophagy alleviated cisplatin-induced ferroptosis and kidney injury through the ROS/HO-1/GPX4 axis [32]. Li et al. observed that the activation of mitophagy alleviated ferroptosis induced by the knockdown of the mitochondrial localization protein CISD3, by eliminating damaged mitochondria [33]. These studies indicated the inhibitory effect of mitophagy on ferroptosis. Conversely, Rademaker et al. reported that compound WJ460 targeting oncoprotein Myoferlin, in combination with ferroptosis activators erastin and RSL3, promoted mitophagy and ROS accumulation in pancreatic cancer cells, leading to ferroptosis [18]. Similarly, Fan et al. found that the main nutrient sensor for glucose flow (protein O-GlcNAcylation) played a critical coordinating role in ferroptosis and mitophagy, and inhibiting mitophagy can alleviate ferroptosis [34]. These studies indicated the promoting effect of mitophagy on ferroptosis. The findings from previous research collectively indicated that the regulatory relationship between mitophagy and ferroptosis was complex. Our research suggested that mitophagy promoted ferroptosis in HCWP-treated HT22 cells, potentially due to mitochondrial dysfunction caused by excessive activation of mitophagy.
Mdivi-1 is a selective inhibitor of dynamin-related protein 1 (Drp1), that can inhibit mitochondrial fission [35]. Since mitochondrial fission is a prerequisite for mitophagy, Mdivi-1 can inhibit mitophagy by regulating mitochondrial fission [36]. Recent reports have suggested that Mdivi-1 shows potential therapeutic benefits in various diseases, such as Alzheimer’s disease [37], traumatic brain injury [38], acute myocardial infarction [39], ischemia-reperfusion [40, 41], and autoimmune encephalomyelitis [42], owing to its protective effects on mitochondrial activity. Furthermore, multiple studies have reported that Mdivi-1 attenuates the dissipation of MMP, prevents ATP depletion, corrects abnormalities in electron transport chain complexes I-V, and inhibits both mitochondrial fission and mitophagy in ischemia-reperfusion (I/R)-induced brain injury [43]. Our research demonstrated that Mdivi-1 effectively mitigated mitochondrial fragmentation and attenuated mitophagy flux by inhibiting the process of mitochondrial fission. Mdivi-1 pretreatment effectively improved the mitochondrial dysfunction in HT22 cells induced by HCWP, including restoring MMP and reducing ROS levels. Our results, consistent with previous research, suggested that Mdivi-1 may have a protective effect on mitochondrial function. The improvement of mitochondrial function and morphology in HCWP-treated HT22 cells by Mdivi-1 may be its mechanism for alleviating ferroptosis.
Conclusion
In summary, our combined in vitro and in vivo research findings demonstrated that the hypoxia combined with propofol (HCWP) induced ferroptosis in hippocampal neurons, resulting in cognitive impairment in immature SD rats. The overactivation of mitophagy appeared to be a pivotal upstream mechanism triggering ferroptosis. Therefore, targeted modulation of mitophagy by Mdivi-1 might present a new therapeutic target for the prevention of cognitive impairment associated with HCWP.
Data availability
The datasets used and/or analyzed during the study are available from the corresponding author on reasonable request.
References
Granger KT, Barnett JH (2021) Postoperative cognitive dysfunction: an acute approach for the development of novel treatments for neuroinflammation. Drug Discov Today 26(5):1111–1114
Zhao Q, Wan H, Pan H, Xu Y (2024) Postoperative cognitive dysfunction-current research progress. Front Behav Neurosci 18:1328790
Lin X, Chen Y, Zhang P, Chen G, Zhou Y, Yu X (2020) The potential mechanism of postoperative cognitive dysfunction in older people. Exp Gerontol 130:110791
Le Y, Liu S, Peng M, Tan C, Liao Q, Duan K, Ouyang W, Tong J (2014) Aging differentially affects the loss of neuronal dendritic spine, neuroinflammation and memory impairment at rats after surgery. PLoS ONE 9(9):e106837
Zhu S, Sai X, Lin J, Deng G, Zhao M, Nasser MI, Zhu P (2020) Mechanisms of perioperative brain damage in children with congenital heart disease. Biomed Pharmacother 132:110957
Moller JT (1994) Anesthesia related hypoxemia. The effect of pulse oximetry monitoring on perioperative events and postoperative complications. Dan Med Bull 41(5):489–500
Snyder CM, Chandel NS (2009) Mitochondrial regulation of cell survival and death during low-oxygen conditions. Antioxid Redox Signal 11(11):2673–2683
Bosnjak ZJ, Logan S, Liu Y, Bai X (2016) Recent insights into Molecular mechanisms of Propofol-Induced Developmental Neurotoxicity: implications for the protective strategies. Anesth Analg 123(5):1286–1296
Huang J, Jing S, Chen X, Bao X, Du Z, Li H, Yang T, Fan X (2016) Propofol Administration during early postnatal life suppresses hippocampal neurogenesis. Mol Neurobiol 53(2):1031–1044
Milanovic D, Pesic V, Loncarevic-Vasiljkovic N, Pavkovic Z, Popic J, Kanazir S, Jevtovic-Todorovic V, Ruzdijic S (2016) The Fas Ligand/Fas Death receptor pathways contribute to Propofol-Induced apoptosis and neuroinflammation in the brain of neonatal rats. Neurotox Res 30(3):434–452
Wang Y, Chen Z, Zhao Y, Shi R, Wang Y, Xu J, Wu A, Johns RA, Yue Y (2013) Epigenetics as a new therapeutic target for postoperative cognitive dysfunction. Med Hypotheses 80(3):249–251
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G (2020) Ferroptosis: past, present and future. Cell Death Dis 11(2):88
Liu L, Gao W, Yang S, Yang F, Li S, Tian Y, Yang L, Deng Q, Gan Z, Tu S (2024) Ferritinophagy-mediated Hippocampus Ferroptosis is involved in cognitive impairment in immature rats Induced by Hypoxia Combined with Propofol. Neurochem Res
Shen ZF, Li L, Zhu XM, Liu XH, Klionsky DJ, Lin FC (2023) Current opinions on mitophagy in fungi. Autophagy 19(3):747–757
Kubli DA, Gustafsson AB (2012) Mitochondria and mitophagy: the Yin and Yang of cell death control. Circ Res 111(9):1208–1221
Granata S, Votrico V, Spadaccino F, Catalano V, Netti GS, Ranieri E, Stallone G, Zaza G (2022) Oxidative stress and Ischemia/Reperfusion Injury in kidney transplantation: focus on Ferroptosis, Mitophagy and New Antioxidants. Antioxid (Basel) 11(4)
Rademaker G, Boumahd Y, Peiffer R, Anania S, Wissocq T, Liegeois M, Luis G, Sounni NE, Agirman F, Maloujahmoum N et al (2022) Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol 53:102324
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE et al (2017) Ferroptosis: a regulated cell death Nexus linking metabolism, Redox Biology, and Disease. Cell 171(2):273–285
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, Jiang X (2019) Role of Mitochondria in Ferroptosis. Mol Cell 73(2):354–363 e353
Gao G, Wang Z, Lu L, Duan C, Wang X, Yang H (2017) Morphological analysis of mitochondria for evaluating the toxicity of alpha-synuclein in transgenic mice and isolated preparations by atomic force microscopy. Biomed Pharmacother 96:1380–1388
Kam WW, Banati RB (2013) Effects of ionizing radiation on mitochondria. Free Radic Biol Med 65:607–619
Zhang K, Zhu L, Fan M (2011) Oxygen, a key factor regulating cell behavior during Neurogenesis and Cerebral diseases. Front Mol Neurosci 4:5
Langston RF, Stevenson CH, Wilson CL, Saunders I, Wood ER (2010) The role of hippocampal subregions in memory for stimulus associations. Behav Brain Res 215(2):275–291
Zhang YQ, Zhang WJ, Liu JH, Ji WZ (2022) Effects of Chronic Hypoxic Environment on cognitive function and neuroimaging measures in a high-Altitude Population. Front Aging Neurosci 14:788322
Berger HR, Nyman AKG, Morken TS, Vettukattil R, Brubakk AM, Wideroe M (2017) Early metabolite changes after melatonin treatment in neonatal rats with hypoxic-ischemic brain injury studied by in-vivo1H MR spectroscopy. PLoS ONE 12(9):e0185202
Zeng K, Long J, Li Y, Hu J (2023) Preventing postoperative cognitive dysfunction using anesthetic drugs in elderly patients undergoing noncardiac surgery: a systematic review and meta-analysis. Int J Surg 109(1):21–31
Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY, Guo SQ, Wang S, Guo T, Wang ZY et al (2018) Alpha-lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S tau transgenic mice. Redox Biol 14:535–548
Tang W, Li Y, He S, Jiang T, Wang N, Du M, Cheng B, Gao W, Li Y, Wang Q (2022) Caveolin-1 alleviates Diabetes-Associated Cognitive Dysfunction through modulating neuronal ferroptosis-mediated mitochondrial homeostasis. Antioxid Redox Signal 37(13–15):867–886
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D (2020) Autophagy-dependent ferroptosis: Machinery and Regulation. Cell Chem Biol 27(4):420–435
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D (2020) Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol 66:89–100
Lin Q, Li S, Jin H, Cai H, Zhu X, Yang Y, Wu J, Qi C, Shao X, Li J et al (2023) Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int J Biol Sci 19(4):1192–1210
Li Y, Wang X, Huang Z, Zhou Y, Xia J, Hu W, Wang X, Du J, Tong X, Wang Y (2021) CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis 12(9):839
Yu F, Zhang Q, Liu H, Liu J, Yang S, Luo X, Liu W, Zheng H, Liu Q, Cui Y et al (2022) Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov 8(1):40
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR et al (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14(2):193–204
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12(1):9–14
Oliver D, Reddy PH (2019) Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 8(9)
Wu Q, Gao C, Wang H, Zhang X, Li Q, Gu Z, Shi X, Cui Y, Wang T, Chen X et al (2018) Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol 94:44–55
Ong SB, Kwek XY, Katwadi K, Hernandez-Resendiz S, Crespo-Avilan GE, Ismail NI, Lin YH, Yap EP, Lim SY, Ja K et al (2019) Targeting mitochondrial fission using Mdivi-1 in a clinically relevant large animal model of Acute myocardial infarction: a pilot study. Int J Mol Sci 20(16)
Rosdah AA, Bond ST, Sivakumaran P, Hoque A, Oakhill JS, Drew BG, Delbridge LMD, Lim SY (2017) Mdivi-1 protects human W8B2(+) cardiac stem cells from oxidative stress and simulated ischemia-reperfusion Injury. Stem Cells Dev 26(24):1771–1780
Yang M, Linn BS, Zhang Y, Ren J (2019) Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis 1865(9):2293–2302
Li YH, Xu F, Thome R, Guo MF, Sun ML, Song GB, Li RL, Chai Z, Ciric B, Rostami AM et al (2019) Mdivi-1, a mitochondrial fission inhibitor, modulates T helper cells and suppresses the development of experimental autoimmune encephalomyelitis. J Neuroinflammation 16(1):149
Nhu NT, Li Q, Liu Y, Xu J, Xiao SY, Lee SD (2021) Effects of Mdivi-1 on neural mitochondrial dysfunction and mitochondria-mediated apoptosis in Ischemia-Reperfusion Injury after Stroke: a systematic review of Preclinical studies. Front Mol Neurosci 14:778569
Acknowledgements
We would like to thank Dr. Xuemei Liao for her help in our confocal photography and image analysis.
Funding
This work was supported by the Chongqing Natural Science Foundation (CSTB2023NSCQ-MSX0434) and the National Clinical Research Center for Child Health and Disorders General Program of Clinical Medical Research (NCRCCHD-2022-GP-0X).
Author information
Authors and Affiliations
Contributions
The authors confirm contributions to the paper as follows: study conception and design: SY, ZD and ST; experimental materials procurement: YX, TA, QD, ZG; data collection: SY; data analysis: SY, YX, ZD; manuscript drafting: SY; manuscript revision: SY, ZD and ST; assistance in data collection: YY and HY. All authors reviewed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
No applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, S., Xu, Y., Ahmad, T. et al. Mdivi-1 alleviates ferroptosis induced by hypoxia combined with propofol in HT22 cells by inhibiting excessive mitophagy. Mol Biol Rep 51, 870 (2024). https://doi.org/10.1007/s11033-024-09812-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09812-y