
Abstract
Circular RNA (circRNA), a novel type of non-coding RNA that consists of a circular loop, has been demonstrated to act as a “sponge” for microRNAs (miRNAs). However, the role of circRNAs in keloid remains unknown. In this study, we investigated circRNA expression profiles in keloid to identify potential diagnostic and therapeutic circRNAs. We performed a circRNA microarray assay to determine circRNA expression in keloid and paired normal skin tissues. Quantitative reverse transcription polymerase chain reaction was used to evaluate the expression levels of candidate circRNAs. The most significantly over-expressed circRNA was used to predict putative miRNA targets and the binding sites of miRNAs with this circRNA. Finally, we constructed a circRNA–miRNA interaction network and carried out gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. We found 52 significantly upregulated and 24 downregulated circRNAs in keloid compared with normal skin tissue. We confirmed that hsa_circ_0057452, hsa_circ_0007482, hsa_circ_0020792, hsa_circ_0057342, and hsa_circ_0043688 were significantly upregulated in keloid tissues. Analysis of the circRNA–miRNA interaction network revealed that circRNAs could interact with miRNAs, including miRNA-29a, miRNA-23a-5p and miRNA-1976. GO and KEGG analyses indicated that these target genes were involved in biological functions and signaling pathways that may play vital roles in the pathogenesis of keloid. This study revealed that circRNAs are potentially implicated in the development of keloid and could serve as novel diagnostic and therapeutic targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Keloid is a fibroproliferative dermal tumor that results from aberrant wound healing in predisposed individuals. Keloid is characterized by the hyper-proliferation of fibroblasts and excessive deposition of extracellular matrix, especially collagen. Keloid often extends beyond the boundaries of the original injury [1, 2]. Keloid lesions can cause significant pain, itching, skin deformity, and even movement dysfunction if located over a joint, which can lead to serious physical and psychological impacts on patients. Although several treatment methods are available for keloid, such as radiation, laser ablation, and intralesional injection, none provide satisfactory results in all patients [1, 3, 4]. Thus, there is a need for improved therapeutic approaches. However, the development of new therapies is limited by our lack of understanding of the molecular mechanism underlying the formation and progression of keloid.
Circular RNAs (circRNAs) were identified recently as a new class of non-coding RNA that form covalently closed loops by back-splicing (Fig. 1) [5,6,7,8]. Compared with linear RNAs, circRNAs are more stable and conserved as they are protected from RNase digestion because their 3′ and 5′ ends are covalently joined [9]. Studies have demonstrated that circRNAs can interfere with gene transcription, especially in mammals. circRNAs serve as “sponges” for microRNAs (miRNAs), sequester particular miRNA families, and suppress the ability of miRNAs to bind to their mRNA targets [8, 10, 11]. circRNAs have also been reported to play significant roles in the development and progression of various diseases, including cardiovascular disease, Alzheimer’s disease, and cancer [12, 13]. To date, it has been reported that the abnormal expression of circRNAs in fibrotic diseases, including hypertrophic scars and liver cirrhosis, which may be further screened biomarkers related to fibrotic diseases, and elucidated the role of epigenetics in the complex pathogenesis of fibrosis. However, there is little information about the relationship between circRNAs and keloid, and further study is necessary to clarify the potential functions of circRNAs in keloid.

Biogenesis of circRNA and its regulation on microRNA. Introns and exons were spliced and spliced to generate linear RNA and circular RNA (circRNA), in which circRNA biogenesis included three patterns, including exon circRNA (ecircRNA), exon–intron circRNA (elciRNA) and intron circRNA (ciRNA). Mature microRNA (miRNA) can inhibit the synthesis of proteins by targeting and degrading linear RNA. circRNA is considered to be a sponge of miRNA, which can down-regulate its expression by adsorption of miRNA, thus interfering with the above effects of miRNA
In this study, we used a microarray assay to evaluate the circRNA expression profile in three keloid and paired normal skin tissues. We found that 76 circRNAs were significantly altered in keloid tissue compared with normal tissue. Five of these differentially expressed circRNAs were further validated by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Then, circRNA–miRNA network and bioinformatics analyses were used to determine the biological function and signaling pathways of the target genes of the most significant upregulated circRNA. Our results indicated that circRNAs might play a crucial role in the formation and progression of keloid.
Materials and methods
Sample preparation
Patients with keloid were recruited from the Second Affiliated Hospital of Kunming Medical University from October 2014 to May 2015. Medical records were examined to identify patients’ medical history and clinical features. Before inclusion, all keloid tissue was confirmed by clinical or pathological examination, and the course of disease was more than 1.5 years. The site of the lesion is limited to the anterior chest, ear lobe and lower extremities. No patient had received any topical or systemic therapy for at least 2 months before undergoing a skin biopsy. The skin tissue of the healthy control group was taken from patients with trauma or cosmetic surgery, and the risk of keloid in these controls was excluded. We listed the relevant information for the sample in Table 1. Skin biopsy tissues were stored temporarily in RNAlater (QIAGEN, Valencia, CA) until RNA extraction. All participants provided written informed consent, and the study was approved by the Ethics Committee of Kunming Medical University.
RNA isolation
Total RNA was extracted from keloid and normal skin tissues using an RNA isolation kit (Ambion, Inc., Carlsbad, CA) following the manufacturer’s instructions. The samples were checked for quantity and quality on a NanoDrop-1000 spectrophotometer (Thermo Scientific, Wilmington, DE) and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA), and then stored at − 80 °C.
circRNA microarray
Total RNA was treated with ribonuclease R (Epicenter, Madison, WI) to remove linear RNA species. Subsequently, the enriched circRNAs were amplified and transcribed into fluorescently-labeled circRNAs using random priming. The labeled circRNAs were purified with an RNeasy Mini Kit (QIAGEN, Valencia, CA), and the concentration of the labeled circRNAs was assessed using a NanoDrop ND-1000. A mixture of 1 μg labeled circRNAs, 5 μL blocking agent (10 ×), and 1 μL fragmentation buffer (25 ×) was heated at 60 °C for 30 min. Then, 25 μL hybridization buffer (2 ×) was added to dilute the labeled circRNAs. Hybridization solution was added to the gasket slide and incubated at 65 °C for 17 h. Finally, the hybridized array was washed, fixed, and scanned using an Agilent scanner.
qRT-PCR
Total RNA was extracted from skin tissues and‘RT reactions were performed with an RNA First-Strand cDNA Synthesis Kit (Tiangen Biotech Co., Ltd., Beijing, China), followed by PCR with a qPCR Detection Kit (Tiangen Biotech Co., Ltd.). All of the primers used in this study are listed in Table 2. The cycling conditions were as follows: 95 °C for 15 min, and then 40 cycles at 94 °C for 20 s and 60 °C for 34 s. Fold changes were calculated with the comparative threshold cycle (2−ΔΔCt) method. Human β-actin was used as a normalization control. Data analyses were performed via GraphPad Prism version 6.00.
Predicting miRNA binding sites of circRNAs
The targeted miRNAs and corresponding miRNA response elements of circRNAs were obtained using circBank (http://www.circbank.cn/) and circBase (http://www.circbase.org/). We firstly identified each circRNA by querying the circBase database [14]. Next, the targeted miRNAs of the corresponding circRNA were screened using the circBank database [15].
In our previous studies, miRNAs from the same sample have been probed by microarray [16]. In this study, we further integrated the predicted circRNAs-targeted miRNAs with the microarray results to obtain the intersection miRNAs [17].
Obtaining target genes of integrated miRNAs
Target genes of integrated miRNAs were detected by targetScan (http://www.targetscan.org/vert_71/) and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) [18, 19]. The interaction network of circRNAs and miRNAs was delineated using Cytoscape 3.01 [20].
Bioinformatics analysis
Gene ontology (GO) analysis was performed to predict the potential functions, including biological process, molecular function, and cellular component, of the target genes using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) version 6.8. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations were used for pathway analysis of the target genes by the KEGG Ontology-Based Annotation System (KOBAS) version 2.0 [21, 22].
Statistical analysis
Data analysis was performed Using Statistical Program for Social Sciences (SPSS) version 18.0 software (SPSS, Chicago, IL, USA). All data are presented as the mean ± standard error of the mean and analyzed with Student’s t test. Figures were created using GraphPad Prism version 6.0. P < 0.05 was considered statistically significant.
Results
circRNA differential expression profile in keloid
We detected a total of 91,334 circRNAs in three pairs of keloid and normal healthy skin tissues by microarray analysis. The differentially expressed circRNAs were selected based on P-value and fold change filtering. Our results revealed that 76 circRNAs were identified with a fold change ≥ 2.0 and P-value < 0.05, among which 52 circRNAs were upregulated and 24 were downregulated. Figure 2 and Supplemental Tables 1 and 2 list the differentially expressed circRNAs and the most upregulated and downregulated circRNAs.

Differential expression of circRNAs between keloid and normal skin tissues. a Differentially expressed circRNAs were showed by volcano plots. The red (up-regulated) or green (down-regulated) points indicated > 2 fold-change (P < 0.05). b Hierarchical cluster analysis of all the differential circRNAs. Red bands indicated high expression and green indicated low expression. c Distribution of circRNAs on human chromosomes, and red histogram showed the number of parental genes incoding collagen. d Characteristics of differentially expressed circRNAs. (Color figure online)
Interestingly, of the 76 differentially expressed circRNAs, the parental genes of 31 circRNAs were derived from sequences encoding collagen. Among them, the parental genes of 9 circRNAs are derived from chromosome 2 (Fig. 2c). We also found that the majority of these dysregulated circRNAs were exonic, followed by antisense circRNAs (Fig. 2d).
Verification of selected circRNAs
To validate the microarray data, the five most significantly differentially expressed circRNAs (hsa_circ_0057452, hsa_circ_0007482, hsa_circ_0020792, hsa_circ_0057342, and hsa_circ_0043688) were selected for qRT-PCR verification in keloid and perilesional tissues of 14 patients and 14 normal healthy samples. The expression patterns of these five circRNAs were consistent with the results of the microarray analysis (Fig. 3).

Validation of selected circRNAs by quantitative real-time PCR
circRNA–miRNA network
According to the qRT-PCR results, we selected these five upregulated circRNAs to construct a circRNA–miRNA interaction network. Through the circBank and targetScan databases, we found that 5 kinds of circRNAs could target 726 miRNAs. Among them, miRNA-4768-3p can be adsorbed by four circRNAs, including hsa_circ_0057452, hsa_circ_0007482, hsa_circ_0020792 and hsa_circ_0057342.
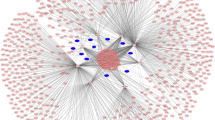
In previous studies, we analyzed the miRNA expression profiles of the above samples using microarray. We have detected 264 differentially expressed miRNAs, and found that 125 of them were down-regulated. To further screen the target miRNAs of circRNAs in keloid tissue, we integrated the predicted miRNAs and the down-regulated miRNAs in the microarray results. We first standardized the name of each miRNAs and screened out the same miRNAs in both groups. Finally, the intersection of the predicted miRNAs and the microarray is detected. Of the predicted 726 miRNAs of 5 circRNAs, a total of 24 miRNAs were also explored by the microarray and two miRNAs, including miRNA-5010-5p and miRNA-3177-3p, can simultaneously be adsorbed by two circRNAs (Fig. 4a).

Prediction for circRNA–miRNA interactions and binding sites. a Interaction analysis of 5 circRNAs and miRNAs. b Binding sites between circRNA and miRNAs
Based on the sequence alignment of the targetScan software, we obtained the seed sequences for each circRNA to adsorb the corresponding miRNAs (Fig. 4b). Among them, has_circRNA_0057342 has three sites that can adsorb miRNA-29a by sponge. The other two miRNAs (miRNA-23a-5p and miRNA-1976) have three sites on the has_circ_0020792 sequence, respectively.
GO and pathway analyses of the target genes
To predict the potential target genes of these 24 miRNAs, we used three databases (targetScan, miRanda, and miRBase) and identified a total of 2138 target genes. We next determined the potential functions of these target genes, GO and pathway analyses were performed using DAVID and KOBAS (Fig. 5). Biological process analysis showed that these target genes could regulate transcription, DNA-templated regulation of transcription, DNA-templated negative regulation of transcription from RNA polymerase II promoters, and positive regulation of cell proliferation. Molecular function analysis showed that these targeted genes were primarily involved in protein binding, metal ion binding, DNA binding, ATP binding, and nucleic acid binding. The cellular component may be linked with nucleus, cytoplasm, cytosol, membrane, and intracellular function. Pathway analysis revealed that these target genes were involved in cancer, viral carcinogenesis, cAMP signaling, and cell cycle pathways, which may play vital roles in the pathogenesis of keloid.

Gene Ontology and KEGG pathway a Top 10 classes of GO enrichment terms. b Top 10 classes of KEGG pathway enrichment terms
Discussion
Increasing evidences suggested that circRNAs perform crucial roles by regulating physiological and pathological processes [23]. Recent studies have demonstrated that circRNAs are involved in several fibrotic diseases, including hepatic fibrosis and cardiac fibrosis. Chen et al. revealed the expression pattern and regulatory capacity of differentially expressed circRNAs in radiation-induced liver fibrosis and found that 179 circRNAs were upregulated and 630 were downregulated [24]. In myocardial fibrosis, a total of 43 differentially expressed circRNAs were identified, consisting of 24 upregulated and 19 downregulated circRNAs [25]. Twenty-nine significantly differentially expressed circRNAs were identified in photoaged human dermal fibroblasts, of which 12 were upregulated and 17 were downregulated [26]. However, the functions of dysregulated circRNAs in keloid remain unclear. In our study, we screened the expression pattern of circRNAs in keloid using microarray analysis and identified 52 upregulated and 24 downregulated circRNAs. Furthermore, five circRNAs were selected for qRT-PCR analysis to evaluate their expression levels in 14 pairs of keloid and normal tissue. We found that the expression patterns of hsa_circ_0057452, hsa_circ_0007482, hsa_circ_0020792, hsa_circ_0057342, and hsa_circ_0043688 were consistent with the microarray results. Therefore, our findings indicated that these circRNAs may be biomarkers of keloid and may also be involved in the occurrence and development of keloid.
Several studies have explored the mechanisms of circRNAs in the pathogenesis of fibrosis. circRNA_000203 was found to be upregulated in diabetic mouse myocardium as well as angiotensin II (Ang-II)-induced mouse cardiac fibroblasts and it can enhance the expression of fibrosis-associated genes by suppressing the function of miRNA-26b-5p [27]. Another study also suggested that circRNA_010567 promoted mycocardial fibrosis of mice by downregulating miRNA-141 [25]. In our previous studies, miRNA microarray studies have been carried out in the same tissues (3 keloids and 3 normal tissues), and the differential expression of miRNAs, has been screened out, of which 125 have been down-regulated. Due to the sponge adsorption of miRNA by circRNA, the expression of circRNA and miRNA was negatively correlated. The results of this study showed that of target miRNAs that the five upregulated circRNAs, 24 down-regulated miRNAs were simultaneously detected in miRNA microarray of same samples [16]. This suggests that these circRNA–miRNA interaction pathways may play a key role in the development of keloids.
Previous studies have suggested that miRNA-23a and miRNA-29a play crucial roles in fibrotic diseases, including keloid, liver fibrosis and renal fibrosis [28,29,30]. In our study, the selected has_circ_0020792 has three sites for miRNA-23a adsorption, and has_circRNA_0057342 also has also three sequences that match miRNA-29a. Therefore, the two pathways, has_circ_0020792-miRNA-23a and has_circRNA_0057342-miRNA-29a, may be the pivotal signaling pathways regulating keloid development.
GO enrichment analysis indicated that the genes targeted by the most differentially expressed circRNA functioned in several biological processes, cellular components, and molecular functions that are associated with extracellular matrix and collagen metabolism. KEGG pathway analysis demonstrated that these target genes were involved in cancer, cAMP signaling, and cell cycle pathways. Previous research suggested that abnormal extracellular matrix-receptor interactions, the PI3 K/Akt signaling pathway, and focal adhesion can damage cell adhesion, leading to a disorder in the arrangement and proliferation of fibroblasts [31,32,33]. Multiple signaling pathways, including cell cycle and cAMP signaling, are involved in the occurrence and development of keloid [34, 35]. Taken together, these findings indicated that circRNAs play a critical role in the pathogenesis and development of keloid, and may serve as new diagnostic and therapeutic targets.
However, there are some limitations that need to be considered when evaluating the results of this study. The key limitation of the circRNA differential expression profile analysis used here is the small sample size, which could lead to a bias in the results. In addition, we used skin tissue rather than specific cell types isolated from skin tissue, such as fibroblasts and keratinocytes. Finally, further cellular and animal studies are needed to confirm the functions of the differentially expressed circRNAs.
Conclusion
Our study showed the aberrant circRNA profile of keloid. The circRNA–miRNA interaction network and GO and pathway analyses support the hypothesis that dysregulated circRNAs may play a role in the pathogenesis and development of keloid. However, the functions of circRNAs in keloid still need to be studied further.
References
Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J (2016) Keloids: the paradigm of skin fibrosis—Pathomechanisms and treatment. Matrix Biol 51:37–46
Bran GM, Goessler UR, Hormann K, Riedel F, Sadick H (2009) Keloids: current concepts of pathogenesis (review). Int J Mol Med 24(3):283–293
Seifert O, Mrowietz U (2009) Keloid scarring: bench and bedside. Arch Dermatol Res 301(4):259–272
Al-Attar A, Mess S, Thomassen JM, Kauffman CL, Davison SP (2006) Keloid pathogenesis and treatment. Plast Reconstr Surg 117(1):286–300
AbouHaidar MG, Venkataraman S, Golshani A, Liu B, Ahmad T (2014) Novel coding, translation, and gene expression of a replicating covalently closed circular RNA of 220 nt. Proc Natl Acad Sci USA 111(40):14542–14547
Jeck WR, Sharpless NE (2014) Detecting and characterizing circular RNAs. Nat Biotechnol 32(5):453–461
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO (2013) Cell-type specific features of circular RNA expression. PLoS Genet 9(9):e1003777
Ebbesen KK, Kjems J, Hansen TB (2016) Circular RNAs: identification, biogenesis and function. Biochem Biophys Acta 1859(1):163–168
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495(7441):384–388
AbouHaidar MG et al (2016) Correction for Novel coding, translation, and gene expression of a replicating covalently closed circular RNA of 220 nt. Proc Natl Acad Sci USA 113(35):E5252–E5253
Boeckel JN, Jae N, Heumuller AW, Chen W, Boon RA, Stellos K et al (2015) Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res 117(10):884–890
Hsiao KY, Sun HS, Tsai SJ (2017) Circular RNA-new member of noncoding RNA with novel functions. Exp Biol Med 242(11):1136–1141
Glazar P, Papavasileiou P, Rajewsky N (2014) circBase: a database for circular RNAs. RNA 20(11):1666–1670
Liu M, Wang Q, Shen J, Yang BB, Ding X (2019) Circbank: a comprehensive database for circRNA with standard nomenclature. RNA Biol 16(7):899–905
Zhong L, Bian L, Lyu J, Jin H, Liu Z, Lyu L et al (2018) Identification and integrated analysis of microRNA expression profiles in keloid. J Cosmet Dermatol 17(5):917–924
Su LC, Xu WD, Liu XY, Fu L, Huang AF (2019) Altered expression of circular RNA in primary Sjogren’s syndrome. Clin Rheumatol. https://doi.org/10.1007/s10067-019-04728-6
Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4:e05005
Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW et al (2018) miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 46(D1):D296–D302
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57
da Huang W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13
Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T et al (2015) Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 27(5):8057
Chen Y, Yuan B, Wu Z, Dong Y, Zhang L, Zeng Z (2017) Microarray profiling of circular RNAs and the potential regulatory role of hsa_circ_0071410 in the activated human hepatic stellate cell induced by irradiation. Gene 20(629):35–42
Zhou B, Yu JW (2017) A novel identified circular RNA, circRNA_010567, promotes myocardial fibrosis via suppressing miR-141 by targeting TGF-beta1. Biochem Biophys Res Commun 487(4):769–775
Peng Y, Song X, Zheng Y, Wang X, Lai W (2017) Circular RNA profiling reveals that circCOL3A1-859267 regulate type I collagen expression in photoaged human dermal fibroblasts. Biochem Biophys Res Commun 486(2):277–284
Tang CM, Zhang M, Huang L, Hu ZQ, Zhu JN, Xiao Z et al (2017) CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci Rep 12(7):40342
Zhang A, Li M, Wang B, Klein JD, Price SR, Wang XH (2018) miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J Cachexia Sarcopenia Muscle 9(4):755–770
Hyun J, Choi SS, Diehl AM, Jung Y (2014) Potential role of Hedgehog signaling and microRNA-29 in liver fibrosis of IKKbeta-deficient mouse. J Mol Histol 45(1):103–112
Deng Z, He Y, Yang X, Shi H, Shi A, Lu L et al (2017) MicroRNA-29: a crucial player in fibrotic disease. Mol Diagn Ther 21(3):285–294
Kolodziej CM, Kim SH, Broyer RM, Saxer SS, Decker CG, Maynard HD (2012) Combination of integrin-binding peptide and growth factor promotes cell adhesion on electron-beam-fabricated patterns. J Am Chem Soc 134(1):247–255
Cirillo N, Hassona Y, Celentano A, Lim KP, Manchella S, Parkinson EK et al (2017) Cancer-associated fibroblasts regulate keratinocyte cell-cell adhesion via TGF-beta-dependent pathways in genotype-specific oral cancer. Carcinogenesis 38(1):76–85
Colombo F, Meldolesi J (2015) L1-CAM and N-CAM: from adhesion proteins to pharmacological targets. Trends Pharmacol Sci 36(11):769–781
Sandulache VC, Parekh A, Li-Korotky H, Dohar JE, Hebda PA (2007) Prostaglandin E2 inhibition of keloid fibroblast migration, contraction, and transforming growth factor (TGF)-beta1-induced collagen synthesis. Wound Repair Regen 15(1):122–133
Zhang J, Xu D, Li N, Li Y, He Y, Hu X et al (2017) Downregulation of microRNA-31 inhibits proliferation and induces apoptosis by targeting HIF1AN in human keloid. Oncotarget 8(43):74623–74634
Acknowledgements
Supported by grants from the National Natural Science Foundation of China (NSFC; Grant No. 81960354 and 81560502), the National Natural Science Foundation of Yunnan Province (Grant No.2017FB116 and 2018FE001(-235)), the Talent Project of Yunnan Province (2019HB024) and 100 Talents Program of Kunming Medical University (Lechun Lyu). The authors acknowledge the editors and reviewers for their positive and constructive comments and suggestions on our study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shi, J., Yao, S., Chen, P. et al. The integrative regulatory network of circRNA and microRNA in keloid scarring. Mol Biol Rep 47, 201–209 (2020). https://doi.org/10.1007/s11033-019-05120-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-05120-y