
Abstract
Flowering time, as an important ecological trait related to photoperiod response, maturity, and final yield, is a complex trait conferred by multiple genes. To further elucidate the genetic mechanism for the flowering time, quantitative trait loci (QTLs) related to the flowering time and maturity were identified utilizing specific-locus amplified fragment sequencing (SLAF-Seq) technology. In total, we identified three QTLs on chromosomes 5, 6, and 7 from a recombinant inbred line (RIL) population of 171 individuals derived from a cross between Minsoy and Archer soybeans. Of these QTLs, one new QTL on chromosome 7, called E11, was simultaneously detected in an ~ 1.03 Mb region from the F6 and F8 generations of the RIL population, and accounted for ~ 15% of the total phenotypic variation over 2 years. The gene symbol E11e11 had been approved by the soybean genetic committee. The segregation patterns observed in residual heterozygous lines (RHLs) at the E11 locus revealed that early flowering was controlled by a single dominant gene. The gene was fine-mapped to an ~ 138 kb interval, including 11 genes based on the reference genome. Through amino acid sequence analysis, three most likely candidate genes, Glyma.07 g048500, Glyma.07 g049000, and Glyma.07 g049200, were identified. The phenotypes detected from two near-isogenic lines (NILs) revealed that NILs for E11 allele significantly promoted the flowering time and maturity than NILs for the e11 under the long-day (LD) conditions. These results suggest that E11 is a new flowering time gene that will be valuable in improving our understanding of the mechanism for the flowering time and molecular breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean [Glycine max (L.) Merrill] is a facultative short-day plant that is very sensitive to photoperiod, but cultivars can grow across the world in a wide range of latitudes from 50° N to 35° S (Watanabe et al. 2012). This is attributed to the rich genetic variability in the genes or loci conditioning the photoperiod of the flowering time. A number of major genes and QTLs have been selected during the breeding process. Thus far, 11 major genes controlling the flowering time and maturity have been reported in soybean: E1 and E2 (Bernard 1971), E3 (Buzzell 1971), E4 (Buzzel and Voldeng 1980), E5 (McBlain and Bernard 1987), E6 (Bonato and Vello 1999), E7 (Cober and Voldeng 2001), E8 (Cober et al. 2010), E9 (Kong et al. 2014), E10 (Samanfar et al. 2017), and J (Ray et al. 1995).
Of the 11 major loci, only E6 and J were reported to control the flowering time under short-day (SD) conditions (Li et al. 2017). Moreover, J was identified as the ortholog of the Arabidopsis gene EARLY FLOWERING 3 (Lu et al. 2017). The other nine genes were detected under long-day (LD) conditions. E1 has been cloned using a map-based approach and identified as a legume-specific transcription factor with a putative nuclear localization signal and a B3 DNA-binding domain (Xia et al. 2012). E2 is an ortholog of the Arabidopsis flowering gene GIGANTEA (Watanabe et al. 2011). Both E1 and E2 play core functions in the photoperiod pathway. E3 and E4, which have been confirmed as phytochrome A homologs, control the flowering time by upregulating E1 and E2 (Liu et al. 2008; Watanabe et al. 2009). E9 and E10 are homologs of FLOWERING LOCUS T and play function downstream of E1 and E2 (Kong et al. 2014; Zhao et al. 2016; Samanfar et al. 2017; Lu et al. 2017). In cultivated soybeans, there are at least three dysfunctional alleles and one hypomorph allele at the E1 locus (Xia et al. 2012; Tsubokura et al. 2014), one dysfunctional allele at the E2 locus (Watanabe et al. 2009), three dysfunctional alleles and one hypomorph allele at the E3 locus (Xu et al. 2013), and six dysfunctional alleles at the E4 locus (Tsubokura et al. 2013). The various allelic combinations at the E1, E2, E3, and E4 loci greatly contribute to the wide adaptability of soybeans (Xu et al. 2013; Tsubokura et al. 2014). However, these allelic variations generate some, not all, of the variations in the flowering time among soybean cultivars. Thus, the detection of new QTLs and genes associated with the flowering time is necessary to improve soybean adaptation and productivity.
With the great development in next-generation sequencing (NGS), it provides a powerful method for single-nucleotide polymorphism (SNP) discovery and genotyping in large population, including restriction-site associated DNA tag sequencing (RADseq, Baird et al. 2008), genotyping-by-sequencing (GBS, Elshire et al. 2011), and specific-locus amplified fragment sequencing (SLAF-seq, Sun et al. 2013). One of these, SLAF-seq performs specifically a predesign experiment to evaluate restriction enzymes and sizes of restriction fragments using the soybean reference genome sequence, which improves its overall efficiency (Sun et al. 2013). Therefore, SLAF-seq is widely used for the construction of high-density genetic mapping for QTLs.
As mentioned above, flowering and maturity processes in soybeans are not fully understood. Many QTLs controlling the flowering time are still not reported or cloned. The objectives of this study are as follows: (1) to detect more QTLs associated with the flowering time, (2) to fine-map one major locus, E11, and to identify the candidate gene(s). We hope that the results of this study will promote a better understanding of the mechanisms controlling the flowering time and identify the tightly linked molecular marker for E11 that will be helpful in molecular breeding.
Materials and methods
Field experiments
Genetic material consisting of recombinant inbred lines (RILs) was developed by a single-seed descendent method (Fehr 1987) from the cross between Minsoy (PI27890) and Archer (PI54687) (called MA population). The F6 and F8 seeds for each RIL were developed by single seed descent, and the seeds of the parental lines were sown in the field under natural day length conditions at the Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences, Harbin, China (45°43′ N, 126° 45′ E), during the month of May in 2016 and 2017. The RHL segregating families for the E11 locus (Family #13 and Family #131) were selected from RILs and planted on 9 May 2017; their progenies were planted in a line on 14 May 2018. Hybrids and lines were sown with a single seed every 20 cm in 5 m rows, spaced 60 cm apart. All trials received standard cultural practices to control insects and weeds (Lu et al. 2017).
Construction of high-density genetic maps
Total DNA was extracted from leaf tissue of MA population plants and the two parents using the CTAB extraction method. The parent plants were sequenced on an Illumina HiSeq 2000 platform (Illumina, Inc., San Diego, CA, USA), and 125-bp paired-end reads with insert sizes of around 350 bp were generated. Sequencing libraries were generated as described by Cheng et al. (2015). The MA population was genotyped using SLAF sequencing. The sequencing data of the 171 RILs were analyzed following previously reported methods (Sun et al. 2013; Li et al. 2014). Briefly, low-quality reads (quality score < 20e) were filtered out, and then raw reads were sorted for each RIL according to duplex barcode sequences (Biomarker, Beijing, China). After the barcodes and the terminal 5-bp positions were trimmed from each high-quality read, clean reads from the same sample were mapped onto soybean genome sequences (Wm82.a2.v1) using SOAP software (Li et al. 2008). Sequences mapped to the same position. The SNP of each SLAF locus was detected. The SNP loci of each SLAF locus were detected between parents, and SLAFs with more than three SNPs were filtered out first. Alleles of each SLAF locus were then defined according to parental reads with sequence depth > 30-fold, while for each offspring the reads with sequence depth > tenfold were used to define alleles. Genotype scoring was then performed using a Bayesian approach to further ensure genotyping quality. High-quality SLAF markers for genetic mapping were filtered following reported criteria (Kong et al. 2018).
As for bin mapping, markers with the same genotype were divided into bin markers using a Perl script. Based on their physical position, the markers were divided into 20 chromosomes (Chr), and HighMap software (Liu et al. 2014) was used to order the markers in every linkage group. A linkage map for MA RILs was constructed containing 5074 SNP markers and covered 3588.94 cM.
Screening of Indel markers
Indel markers were developed in this study on the basis of resequencing data from the Minsoy and Archer. Three types of software, SAMtools (v1.7) (Li 2011), Pindel (v1.0) (Ye et al. 2009), and Soapindel (v2.1) (Li et al. 2013), were used for the discovery of indel markers. To optimize the Indels, we trained the support vector machine (SVM) filter by simulative data and identified Indels with quality scores > 200. High-quality Indel markers contrasting among parents were selected for QTL analysis, fine-mapping, and NILs genotype identification. We selected eight Indels to map the E11 in the R6 (2016) and R8 (2017) populations and constructed linkage maps with Map Manager QTXb2.0 (Manly et al. 2001). Eleven indel markers were used for fine-mapping. Sixteen Indels were used to develop NILs.
QTL analysis
For the flowering time, QTLs in different environments were detected by multiple-QTL model (MQM) mapping using the MapQTL5 package (Van Ooijen 2004). The LOD threshold for declaring significant QTLs was determined using a permutation test (PT) with a significance level of p < 0.05 (n = 1000).
Fine-mapping
We genotyped 11 indels markers flanking the genes from ID7079 to ID7088 in the progeny of RIL families #13 (n = 271) and #131 (n = 109) and detected some recombinant plants. The QTL genotypes of recombinants and nonrecombinant control plants were determined by comparing their graphical genotypes constructed with Indel markers. Phenotypic segregation patterns in the progeny of the recombinants were determined by GraphPad Prism 6 Software (Berkman et al. 2018).
The candidate genes for E11 locus
The candidate genes for E11 loci were categorized; the function of which was determined using the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html). The Blast2GO 4.0 (BioBam Bioinformatics S.L., Valencia, Spain) or Phytozome database was used to determine the GO ID of candidate genes. Default parameter settings were employed.
Phenotype statistics
Days to flowering and maturity were recorded at the R1 stage (days from emergence to first open flower appeared on 50% of the plants) and the R8 stage (days from emergence to 95% of pods exhibiting mature colors) (Fehr et al. 1971). Chi-square (χ2) tests were conducted to detect segregation distortion. A one-way analysis of variance (ANOVA) was used to detect significant marker-phenotype associations between polymorphic DNA markers and days to the flowering time and maturity.
Results
Phenotypic analysis of flowering time in soybean RIL populations
In order to find new QTLs associated with the flowering time, we grew the RIL population of 171 individuals derived from the cross between Minsoy and Archer in Harbin during 2016 and 2017. Minsoy flowered averagely ~ 30.6 days after emergence (DAE) and matured 91 DAE, while Archer flowered ~ 33.6 DAE and matured 119 DAE on average (Fig. 1a, b). Frequency distributions of the flowering time for different years in the segregating populations of the cross are presented in Fig. 1c, d. The F6 and F8 RIL populations showed a continuous and normal distribution, with days to flowering ranging from 28 to 54 DAE in F6 RILs during 2016 and from 32 to 58 DAE in F8 RILs during 2017. These results indicated that the flowering time was mainly controlled by genetic factors and inherited in a quantitative manner. Moreover, transgression segregations were observed in the RIL population, suggesting that of the two parents might contain recessive genes that affect the late-flowering trait.

Phenotype of Minsoy, Archer, and RILs in the yield. a Flowering time of the parents in 2016 and 2017. b Maturity of the parents over 2 years. c Segregation of flowering time in the MA F6 (2016) population. d Segregation of flowering time in the MA F8 (2017) population. Arrows indicate the days to flowering of the parent soybeans, Minsoy (M) and Archer (A). X-axis represents number of plants flowering at the same time. Y-axis represents days to flowering (DAE)
QTL mapping for the flowering time using high-density genetic map
Orf et al. (1999) analyzed RIL populations derived from the cross between Minsoy and Archer and found that four QTLs for the flowering time were located on linkage group (LG) C2 (Chr6), LG M (Chr7), and LG L (Chr19). The QTL on Chr6 most likely corresponds to E1, which account for 31% of the observed variation in the flowering time; this is in accordance with previous findings that E1 has the largest effect on soybean flowering (Cober et al. 1996; Liu et al. 2007; Xia et al. 2012). The second QTL on Chr7 can explain 26% of the observed variation, which is a new QTL for the flowering time.
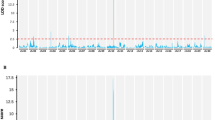
A high-density genetic map comprising of 5074 SLAF markers was used for QTL mapping (Fig. 2a) in the study. Based on 1000 permutations, a LOD score of 2.0 was used as the threshold for declaring the presence of an additive QTL. Using this genetic map and R1 data in 171 lines, three flowering time QTLs (qTOF5, qTOF6.1, and qTOF7) were consistently identified over 2 years (2016 and 2017). Three separate QTLs (qTOF1, qTOF6.2, and qTOF13) were located on Chr1, 6, and 13, respectively, and only appeared for 1 year (Table 1; Fig. 2b; Fig. 2c).

Genetic map of RIL populations and QTL mapping for flowering time. a Genetic map of the MA RIL population constructed with HighMap software. Chr, chromosome. b QTLs of the flowering time trait (R1) using 2017 data. c QTLs of the flowering time trait (R1) using the 2016 data
Of these QTLs, one major QTL on Chr6 (qTOF6.1) was detected over the 2 years and accounted for 9.8% of the observed variation and had the highest LOD score (3.81) in F8 RILs (Table 1). The genotypes for Minsoy and Archer soybeans were determined using allele-specific DNA markers for the E1, E2, E3, and E4 genes (Supplement Table S1). The genotypes for Minsoy and Archer are E1e2e3E4 and e1-ase2E3E4, respectively. The difference of the E1 genotype between parents further indicates that the QTL on Chr 6 is the E1 gene, which is consistent with the findings of Orf et al. (1999).
For the flowering time, another major QTL on Chr7 (qTOF7, named E11) was repeatedly identified on soybean Chr7 (Fig. 2b; Fig. 2C). This QTL accounted for 8% and 7.7% of the observed variation in F6 and F8 RILs, respectively, with the highest LOD scores (3.1 and 2.99, respectively; Table 1). Thus far, there are no known flowering time genes located on Chr7 in soybean. Therefore, E11 is a new locus for the flowering time and should be subject to further characterization.
A single gene on E11 locus
To map E11 more precisely, eight Indel markers on Chr7 were used, contrasting between parents, were used on the basis of resequencing data (Supplement Table S2). The highest LOD scores (4.91 and 5.21) were detected near ID7088 in R6 (2016) and ID7079 in R8 (2017), implying that E11 might locate between ID7079 and ID7088 that are 1.03 Mb apart. In F6 and F8, this QTL accounted for 15.2% and 15.0% of the total phenotypic variance in the flowering time, respectively. The additive effect of the MA allele was 2.81 d in the F6 population and 2.05 d in the F8 population. Using these two markers, two RHL families, #13 and #131, were found to be heterozygous at markers ID7088 and ID7079, which suggests that these two families might segregate at the E11 locus. Family #13 possessed homozygous E1 allele with 271 progenies and family #131 possessed homozygous e1-as allele with 109 progenies (Supplement Table S1). In 2017, we grew the 271 plants of family #13 and 109 plants of family #131 in Harbin to test the segregation patterns of the flowering time and maturity, and both exhibited a bimodal distribution for the flowering time and maturing time (Supplement Figs. S1, S2).
The results revealed that the individuals of family #13 were clearly grouped into two groups: an early or intermediate flowering (n = 199) group and a late-flowering (n = 72) group. When they were separated at 43 DAE, the observed frequencies fit a monogenic 3:1 ratio (χ2 = 0.36, p = 0.55). A similar result was obtained for early maturity (n = 176) and late maturity (n = 60); the observed frequencies fit a 3:1 ratio (χ2 = 0.02, p = 0.88; Table 2). In the population of family #131, with separation at 41 DAE, plants were grouped into two groups: an early-flowering (n = 79) group and a late-flowering (n = 30) group. The observed frequencies fit a monogenic 3:1 ratio (χ2 = 0.37, p = 0.54). While the maturity was grouped into either early maturity (n = 79) or late maturity (n = 26) by separation at 127 DAE; the observed frequencies also fit a 3:1 ratio (χ2 = 0.003, p = 0.96; Table 2). These F2 segregation tests demonstrate that the single E11 locus was segregated for the flowering time and maturity in both families #13 and #131, implying that the dominant E11 allele might condition the early flowering time and early maturity.
To confirm whether the single E11 locus conditions early flowering and maturity, we further planted the F3 progenies of #131 and #13 F2 plants in the field in 2018 and evaluated their segregation patterns in the flowering time and maturity. The results revealed that in family #13, 59 of 236 rows showed homozygous early flowering, while 121 rows segregated for the flowering time, and another 56 rows were homozygous for late flowering. In family #131, 27 of 105 rows produced only early-flowering plants and were considered to be homozygous for the early-flowering allele, while 53 segregated for the flowering time, and another 25 were late-flowering plants and were considered to be homozygous for the late-flowering allele. These segregation ratios for families #13 and #131 fit a 1:2:1 ratio (F3-#13: n = 236, χ2 = 0.23, p = 0.89; F3-#131: n = 105, χ2 = 0.09, p = 0.96; Table 3). A similar result was observed in the maturing time as well and fits a 1:2:1 ratio (F3-#13: n = 236, χ2 = 0.09, p = 0.96; F3-#131: n = 105, χ2 = 0.31, p = 0.85; Table 4). These results strongly suggest that a single dominant gene, E11, controls early flowering time, and maturity under natural LD environments.
Fine-mapping of E11 locus
According to the mapping results, E11 was mapped between ID7079 and ID7088 on Chr7 (Fig. 3). To define the E11 gene, 11 polymorphic Indel markers (Supplement Table S4) were further genotyped to screen the recombinants from two families, #13 and #131. Based on the segregation patterns observed in the progenies of family #13, five recombinants were detected (#13–131-5, #13–249, #13–188, #13–121, and #13–67). Combined with the flowering time detected during 2017, which was confirmed based on the segregation patterns in the progeny during 2018, an interval between markers M3 and M8 was delimited (Fig. 4a). In family #131, four recombinants were found (#131–105, #131–20, #131–62, and #131–102). The flowering time and segregation patterns helped identify the interval between markers M1 and M7 (Fig. 4b). Integrated with the above intervals, E11 was delimited to an interval of ~ 138 kb between markers M3 and M7 (Fig. 4C). Additionally, markers M4, M5, and M6 co-segregated with days to flowering. Overall, these results confirm that the candidate gene, E11, is located in a ~ 138 kb region.

Genetic mapping of the E11 locus. The E11 gene was initially delimited near ID7088 or ID7079 using the F6 and F8 RIL populations in a 2016 and b 2017

Fine mapping of the E11 gene. a Graphical genotypes of soybean recombinants carrying crossovers. White bars represent the homozygote for the e11 allele from Archer, black bars represent the homozygote for the E11 allele from Minsoy, and cross-hatched bars represent the heterozygote. Fine mapping of E11 gene on Chr7 using the recombinants from #13. Five recombinants (#13–131-5, #13–249, #13–188, #13–121 and #13–67), and four non-recombinant control plants were genotyped at 11 Indels. Chromosome positions of markers were from the soybean reference genome, indicated at the top of the markers. Days to flowering of the recombinants were evaluated in the LD conditions. The phenotypic segregation of family #13 is shown in right boxplot format. The interquartile region, median, and range are indicated by the box, the bold vertical line, and the horizontal line, respectively. The E11 gene was delimited between M3 and M8 on Chr7 (red frame). b Fine mapping of the E11 gene on Chr7 using the recombinants from family #131. The information of chart is as above Fig. 4a. With F2 recombinants from family #131, we were able to delimit the e11 gene between M1 and M7 in Chr7. c The E11 gene was located between M3 and M7 in Chr7 according to Fig. 4a and Fig. 4b. Eleven open reading frames (arrows) were predicted in a genomic region of ~ 138 kb delimited between M3 and M7
Glyma.07 g4850, Glyma.07 g049000, and Glyma.07 g049100 as candidates for E11
According to the Williams, 82 genome sequence and 11 genes are located in ~ 138 kb region (Fig. 4c; Table 5). Specifically, there are three genes for O-methyltransferase I (Glyma.07 g048700, Glyma.07 g048800, and Glyma.07 g048900), one gene for pyrophosphorylase I (Glyma.07 g048300), one gene for protein kinase superfamily protein (Glyma.07 g048400), one gene for 2-oxoglutarate (2OG), and Fe(II)-dependent oxygenase superfamily protein (Glyma.07 g048600), one gene for photosystem I P subunit (Glyma.07 g049000), one gene for metal tolerance protein A2 (Glyma.07 g049200), one gene encoding an AP2/B3-like transcriptional factor (Glyma.07 g048200), one gene for Homeodomain-like superfamily protein (Glyma.07 g048500), and one encoding basic helix-loop-helix (bHLH) DNA-binding superfamily protein (Glyma.07 g049100). According to the theory of transcriptional networks that mainly control the flowering time (Fornara et al. 2010; Rychel et al. 2019), Glyma.07 g048300, Glyma.07 g048400, Glyma.07 g048600, Glyma.07 g048700, Glyma.07 g048800, and Glyma.07 g048900 do not participate in flowering regulation. Sequence analysis of the remaining five genes based on resequencing data of Minsoy and Archer, we found that Glyma.07 g048500, Glyma.07 g049000, and Glyma.07 g049200 differed in CDS between the two parents. Therefore, Glyma.07 g48500, Glyma.07 g049000, and Glyma.07 g049200 were considered as candidate genes of the E11 locus. Functional characterization of these genes should be performed to further validate their functions on the soybean flowering time and maturity in future studies.
E11 results in early flowering and maturity
We confirmed the association between the genotype at E11 and the flowering time using NILs for the Minsoy allele (NIL-E11) and the Archer allele (NIL-e11). In all of the segregating families (namely, F7 and F8), the average flowering time trait was homozygous for the E11 allele (such as #13–27 and #13–28) was earlier than those of plants homozygous for the e11 allele (such as #13–142 and #13–183) in the yield, suggesting that the E11 allele behaved as a dominant allele (Fig. 4). Two NILs derived from the heterozygous plant (#13–249 and #131–20) at E11 (Supplementary Fig. S3). The flowering and maturity time under LD conditions were significantly different (p < 0.001) between the NIL-E11 and NIL-e11 alleles in all sets of NILs tested; plants that were homozygous for the NIL-E11 allele from Minsoy flowered on average 9–10 days earlier than those that were homozygous for the NIL-e11 allele from archer in the field condition. The maturity date of NILs for E11 allele was on average 12–15 day earlier than NILs for e11 allele (Fig. 5). In conclusion, the results demonstrate that the NIL-E11 allele significantly promoted earlier flowering time and maturity, confirming that E11 plays a central role in flowering regulation.

Phenotypes of the NIL-E11 and NIL-e11 under LD conditions: a flowering time and b maturity
Discussion
In this study, we described the molecular dissection of an important QTL (named E11), for the flowering time under LD conditions by the cross between early-maturing soybean cultivars. Genetic analysis of the flowering time in residual heterozygous lines (#13 and #131) segregated only for E11 and possessed the homozygous E1 genotype, revealing that E11 controls the flowering time and maturity regardless of E1. As is known, E1 has the largest effect on soybean flowering under LD conditions (Cober et al. 1996; Liu et al. 2007; Xia et al. 2012; Tsubokura et al. 2014).
Minsoy and Archer soybeans were used as parents in this study, and the E1 genotype of the two parents is different (Supplement Table S1), but their flowering time has no obvious difference (Fig. 1a), which suggests that there may be undiscovered genes that regulate the flowering time. The RIL population of 171 individuals was derived from crossing Minsoy and Archer soybeans in order to clone new genes for the flowering time. Although the two parents were early-maturing soybean cultivars, significant transgression segregations were observed in RILs outside of the parents over the course of 2 years (Fig. 1c, d). The F6 and F8 populations showed a continuous and normal distribution, which indicates that more than two genes control the flowering time in this RIL population. Using SLAF-genetic mapping and the corresponding flowering dates, we mapped three stable QTLs (qTOF5, qTOF6.1, and qTOF7) on Chr5, 6, and 7, one QTL (qTOF13) on Chr13 during 2017, and two QTLs (qTOF1 and qTOF6.2) on Chr1 and Chr6 during 2016 (Table 1; Fig. 2). These flowering time loci will contribute to a better understanding of flowering time regulation in soybeans. Additionally, these results suggest that the crosses did not limit parents with large differences in phenotypes when a population was developed.
Of these QTLs, qTOF6.1 and qTOF7 were also reported on by Orf et al. (1999), while other QTLs (qTOF1, qTOF5, qTOF6.1, and qTOF13) were not detected. Flowering time QTLs in the same region as the major QTL, qTOF6.1, was previously detected in many studies, which accounted for the higher ratio of the observed variation in different populations (Mansur et al. 1993; Orf et al. 1999; Githiri et al. 2007). The region near qTOF6.1 also harbors the E1 gene. Combined with the difference of E1 genotype between parents, we further revealed that qTOF6.1 was E1 gene. This finding of E1 implies that our RIL population could be further researched in order to study other new genes that may also control flowering time.
The qTOF7 locus was called E11, which is the second major site other than E1 that largely influenced flowering time in the MA population. The gene symbol of E11e11 had been approved by the soybean genetic committee. This QTL was consistently detected in different populations (Orf et al. 1999; Liu et al. 2018), indicating its importance in regulating soybean flowering time. Hence, this QTL was selected for detailed analysis in the present study. Based on the vital role of E1 under LD conditions, two RHLs were selected for cloning the E11 gene; family #13 had the E1 allele and family #131 had the e1as allele, which aimed to remove the effect of E1 on flowering time.
The segregation patterns observed in RHLs for E11 revealed that early flowering was controlled by a single dominant gene. The gene was fine-mapped to an ~ 138 kb interval. In this region, there are 11 annotated genes, including six genes for function enzymes (Glyma.07 g048300, Glyma.07 g048400, Glyma.07 g048600, Glyma.07 g048700, Glyma.07 g048800, and Glyma.07 g048900), three genes encoding transcription factors (Glyma.07 g048200, Glyma.07 g048500, and Glyma.07 g049100), one gene encoding photosystem I P subunit (Glyma.07 g049000), and one gene encoding metal tolerance protein A2 (Glyma.07 g049200) (Table 5). Through amino acid sequence analysis, three candidate genes were identified: Glyma.07 g48500, the best BLAST hit to LATE ELONGATED HYPOCOTYL (LHY); Glyma.07 g049000, a homologous gene of CURVATURE THYLAKOID 1B (CURT1B); and Glyma.07 g49200, an ortholog of metal tolerance protein 3 (MTP3) in Arabidopsis.
Previous studies on homologous genes suggest that Glyma.07 g48500 is most likely a candidate gene for E11. In Arabidopsis, loss-of-function mutant, lhy-1, flowered earlier than wild-type plants under SD and LD conditions, exhibiting an effect independently of photoperiod (Schaffer et al. 1998; Spensley et al. 2009). Meanwhile, LHY, CIRCADIAN CLOCK ASSOCIATED1 (CCA1), and TIMING OF CAB EXPRESSION1 (TOC1) formed core oscillator of the circadian clock (Alabadí et al. 2001; Cao et al. 2017; Xu et al. 2018). As one of the core members in regulating circadian clock, mutation of LHY disrupted circadian clock regulation of gene expression and leaf movements, causing dwarfing of plants and reduction in yields (Mizoguchi et al. 2002). These investigations suggest that LHY in soybeans may be important in the regulation of flowering time and maturity. However, the CURT1B protein was previously located in thylakoids and designated as a “thylakoid membrane phosphoprotein of 14 kD” (TMP14), which is a novel submit of plant photosystem I (Hansson and Vener 2003; Armbruster et al. 2013). Additionally, the MTP3 protein contributes to basic cellular zinc tolerance and controls zinc partitioning, particularly under conditions with high rates of zinc influx into the root symplasm. Moreover, MTP3–GFP fusion proteins are localized to the vacuolar membrane when expressed in Arabidopsis (Arrivault et al. 2006).
As an additional effort to examine the specific effects of E11, we compared phenotypes of the two NILs with a separation of genotypes between M3 and M7 (Supplement Fig. S3). The results showed that NIL-E11 plants significantly promoted flowering time and maturity as opposed to NIL-e11, confirming that the E11 gene could greatly control flowering time and maturity in soybeans under LD conditions (Fig. 5). In previous studies, E11 was reported to be related to flowering time and maturity date, as well as yield-related traits, such as seed weight and seed number (Orf et al. 1999; Wang et al. 2004; Panthee et al. 2007; Han et al. 2012). Meanwhile, we found that the yield and grains of NIL-E11 plants were lower than NIL-e11 plants (data not presented). As is known, many major genes and QTLs for flowering time often influence agronomic traits other than flowering time and maturity, such as plant height and yield (Cober and Morrison 2010). Taken together, E11 was characterized as having a major role in the regulation of flowering time and maturity, which can be important for soybean breeding.
For the flowering time, dominant alleles at E6, E9, and J promote early flowering (Kong et al. 2014; Li et al. 2017). This study demonstrated that E11 was similar to E9, such that the dominant allele promoted early flowering under LD conditions. Additionally, J and E6 were characterized as having a role in the promotion of flowering under induced SD conditions, differing from E11, which promotes flowering under LD conditions. Further research is needed to confirm the effects of E11 on the regulation of flowering time. The new early flowering gene, E11, will prove useful for the molecular breeding of early maturity and the stable productivity of soybean under high latitude environments.
References
Alabadí D, Oyama T, Yanovsky MJ, Harmon FG, Más P, Kay SA (2001) Reciprocal regulation between TOC1 and LHY/CCA1 within the Arabidopsis circadian clock. Science 293:880–883
Armbruster U, Labs M, Pribil M, Viola S, Xu W, Scharfenberg M, Hertle AP, Rojahn U, Jensen PE, Rappaport F, Joliot P, Dörmann P, Wanner G, Leister D (2013) Arabidopsis CURVATURE THYLAKOID1 proteins modify thylakoid architecture by inducing membrane curvature. Plant Cell 25:2661–2678
Arrivault S, Senger T, Krämer U (2006) The Arabidopsis metal tolerance protein AtMTP3 maintains metal homeostasis by mediating Zn exclusion from the shoot under Fe deficiency and Zn oversupply. Plant J 46:861–879
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376
Berkman SJ, Roscoe EM, Bourret JC (2018) Comparing self-directed methods for training staff to create graphs using Graphpad prism. J Appl Behav Anal 52:188–204
Bernard RL (1971) Two major genes for time of flowering and maturity in soybeans. Crop Sci 11:242–247
Bonato ER, Vello NA (1999) E6 a dominant gene conditioning early flowering and maturity in soybeans. Genet Mol Biol 22:229–232
Buzzel RI, Voldeng HD (1980) Inheritance of insensitivity to long day length. Soybean Genet News 17:26–29
Buzzell RI (1971) Inheritance of a soybean flowering response to fluorescent day length conditions. Can J Genet Cytol 13:703–707
Cao D, Takeshima R, Zhao C, Liu B, Jun A, Kong F (2017) Molecular mechanisms of flowering under long days and stem growth habit in soybean. J Exp Bot 68:1873–1884
Cheng CK, Cheung MK, Nong W, Law PT, Qin J, Ling JM, Kam KM, Cheung WM, Kwan HS (2015) Next generation genome sequencing reveals phylogenetic clades with different level of virulence among Salmonella Typhimurium clinical human isolates in Hong Kong. BMC Genomics 16:688
Cober ER, Morrison MJ (2010) Regulation of seed yield and agronomic characters by photoperiod sensitivity and growth habit genes in soybean. Theor Appl Genet 120:1005–1012
Cober ER, Voldeng HD (2001) A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Sci 41:698–701
Cober ER, Tanner JW, Voldeng HD (1996) Genetic control of photoperiod response in early-maturing, near-isogenic soybean lines. Crop Sci 36:601–605
Cober ER, Molnar SJ, Charette M, Voldeng HD (2010) A new locus for early maturity in soybean. Crop Sci 50:524–527
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Fehr WR (1987) Principles of cultivar development. Macmillan, London
Fehr WR, Caviness CE, Bumood DT, Pennington JS (1971) Stage of development descriptions for soybeans, Glycine max L. Merrill. Crop Sci 11:929–931
Fornara F, de Montaigu A, Coupland G (2010) SnapShot: control of flowering in Arabidopsis. Cell 141:550
Githiri SM, Yang D, Khan NA, Xu D, Komatsuda T, Takahashi R (2007) QTL analysis of low temperature induced browning in soybean seed coats. J Hered 98:360–366
Han YP, Li DM, Zhu D, Li HY, Li XP, Teng WL, Li WB (2012) QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theor Appl Genet 125:671–683
Hansson M, Vener AV (2003) Identification of three previously unknown in vivo protein phosphorylation sites in thylakoid membranes of Arabidopsis thaliana. Mol Cell Proteomics 2:550–559
Kong F, Nan H, Cao D, Li Y, Wu F, Wang J, Lu S, Yuan X, Cober ER, Abe J, Liu B (2014) A new dominant gene E9 conditions early flowering and maturity in soybean. Crop Sci 54:2529–2535
Kong L, Lu S, Wang Y, Fang C, Wang F, Nan H, Su T, Li S, Zhang F, Li X, Zhao X, Yuan X, Liu B, Kong F (2018) Quantitative trait locus mapping of flowering time and maturity in soybean using next-generation sequencing-based analysis. Front Plant Sci 9:995
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 4:713–714
Li S, Li R, Li H, Lu J, Li Y, Bolund L, Schierup MH, Wang J (2013) SOAPindel: efficient identification of indels from short paired reads. Genome Res 23:195–200
Li B, Tian L, Zhang J, Huang L, Han F, Yan S, Wang L, Zheng H, Sun J (2014) Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genomics 15:1086
Li X, Fang C, Xu M, Zhang F, Lu S, Nan H et al (2017) Quantitative trait locus mapping of soybean maturity gene E6. Crop Sci 57:1–8
Liu B, Fujita T, Yan ZH, Sakamoto S, Xu D, Abe J (2007) QTL mapping of domestication-related traits in soybean (Glycine max). Ann Bot 100:1027–1038
Liu B, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J (2008) Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome a gene. Genetics 180:995–1007
Liu D, Ma C, Hong W, Huang L, Liu M, Liu H, Zeng H, Deng D, Xin H, Song J, Xu C, Sun X, Hou X, Wang X, Zheng H (2014) Construction and analysis of high-density linkage map using high throughput sequencing data. PLoS One 9:e98855
Liu D, Yan Y, Fujita Y, Xu D (2018) A major QTL (qFT12.1) allele from wild soybean delays flowering time. Mol Breed 38:45
Lu SJ, Zhao XH, Hu YL, Liu SL, Nan HY, Li XM, Fang C, Cao D, Shi XY, Kong LP, Su T, Zhang FG, Li SC, Wang Z, Yuan XH, Cober ER, Weller JL, Liu BH, Hou XL, Tian ZX, Kong FJ (2017) Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat Genet 49:773–779
Manly KF, Cudmore RH Jr, Meer JM (2001) Map manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
Mansur LM, Lark KG, Kross H, Oliveira A (1993) Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean (Glycine max L.). Theor Appl Genet 86:907–13
McBlain BA, Bernard RL (1987) A new gene affecting the time of flowering and maturity in soybeans. J Hered 78:160–162
Mizoguchi T, Wheatley K, Hanzawa Y, Wright L, Mizoguchi M, Song HR, Carré IA, Coupland G (2002) LHY and CCA1 are partially redundant genes required to maintain circadian rhythms in Arabidopsis. Dev Cell 2:629–641
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG (1999) Genetics of soybean agronomic traits: I comparison of three related recombinant inbred populations. Crop Sci 39:1642–1651
Panthee DR, Pantalone VR, Saxton AM, West DR, Sams CE (2007) Quantitative trait loci for agronomic traits in soybean. Plant Breed 126:51–57
Ray JD, Hinson K, Mankono JEB, Malo MF (1995) Genetic control of a long-juvenile trait in soybean. Crop Sci 35:1001–1006
Rychel S, Książkiewicz M, Tomaszewska M, Bielski W, Wolko B (2019) FLOWERING LOCUS T, GIGANTEA, SEPALLATA, and FRIGIDA homologs are candidate genes involved in white lupin (Lupinus albus L.) early flowering. Mol Breed 39:43
Samanfar B, Molnar SJ, Charette M, Schoenrock A, Dehne F, Golshani A, Belzile F, Cober ER (2017) Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean. Theor Appl Genet 130:377–390
Schaffer R, Ramsay N, Samach A, Corden S, Putterill J, Carré IA, Coupland G (1998) The late elongated hypocotyl mutation of Arabidopsis disrupts circadian rhythms and the photoperiodic control of flowering. Cell 93:1219–1229
Spensley M, Kim JY, Picot E, Reid J, Ott S, Helliwell C, Carré IA (2009) Evolutionarily conserved regulatory motifs in the promoter of the Arabidopsis clock gene LATE ELONGATED HYPOCOTYL. Plant Cell 21:2606–2623
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H, Xu C, Song J, Huang L, Wang C, Shi J, Wang R, Zheng X, Lu C, Wang X, Zheng H (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One 8:e58700
Tsubokura Y, Matsumura H, Xu M, Liu B, Nakashima H, Anai T, Kong F, Yuan X, Kanamori H, Katayose Y, Takahashi R, Harada K, Abe J (2013) Genetic variation in soybean at the maturity locus E4 is involved in adaptation to long days at high latitudes. Agronomy 3:117–134
Tsubokura Y, Watanabe S, Xia Z, Kanamori H, Yamagata H, Kaga A, Katayose Y, Abe J, Ishimoto M, Harada K (2014) Natural variation in the genes responsible for maturity loci E1, E2, E3 and E4 in soybean. Ann Bot 113:429–441
Van Ooijen JW (2004) MapQTL® 5, Software for the mapping of quantitative trait loci in experimental populations. Kyazma BV, Wageningen
Wang D, Graef GL, Procopiuk AM, Diers BW (2004) Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor Appl Genet 108:458–467
Watanabe S, Hideshima R, Xia Z, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T, Tabata S, Harada K (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182:1251–1262
Watanabe S, Xia Z, Hideshima R, Tsubokura Y, Sato S, Yamanaka N, Takahashi R, Anai T, Tabata S, Kitamura K, Harada K (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188:395–407
Watanabe S, Harada K, Abe J (2012) Genetic and molecular bases of photoperiod responses of flowering in soybean. Breed Sci 61:531–543
Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, Anai T, Sato S, Yamazaki T, Lü S, Wu H, Tabata S, Harada K (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci U S A 109:E2155–E2164
Xu M, Xu Z, Liu B, Kong F, Tsubokura Y, Watanabe S, Xia Z, Harada K, Kanazawa A, Yamada T, Abe J (2013) Genetic variation in four maturity genes affects photoperiod insensitivity and PHYA-regulated post-flowering responses of soybean. BMC Plant Biol 13(9):91
Xu G, Jiang Z, Wang H, Lin R (2018) The central circadian clock proteins CCA1 and LHY regulate iron homeostasis in Arabidopsis. J Integr Plant Biol 61:168–181
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25:2865–2871
Zhao C, Takeshima R, Zhu J, Xu M, Sato M, Watanabe S, Kanazawa A, Liu B, Kong F, Yamada T, Abe J (2016) A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a flowering locus T orthologue. BMC Plant Biol 16:20
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was funded by the Natural Key R&D Program of China (2017YFE0111000 and 2016YFD0100400), the National Natural Science Foundation of China (No.31725021, 31701445, 31801384, 31771815), and the National Natural Science Foundation of Heilongjiang Province (No. QC2014C036).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors declare that this study complies with the current laws of the countries in which the experiments were performed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 182 kb)
ESM 2
Fig. S1 Frequency distribution of days to flowering in two RHL families, (A) #13 and (B) #131, grown during 2017 in Harbin, China. Fig. S2 Frequency distribution of days to maturity in two RHL families, (A) #13 and (B) #131, grown during 2017 in Harbin, China. Fig. S3 The genotypes of the NILs for E11 derived from the cross between Minsoy and Archer. Molecular markers and their genomic positions were shown in Chr7. (PDF 98 kb)
Rights and permissions
About this article

Cite this article
Wang, F., Nan, H., Chen, L. et al. A new dominant locus, E11, controls early flowering time and maturity in soybean. Mol Breeding 39, 70 (2019). https://doi.org/10.1007/s11032-019-0978-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-019-0978-3