
Abstract
Understanding the transition to the reproductive period is important for crop breeding. This information can facilitate the production of novel varieties that are better adapted to local environments or changing climatic conditions. Here, we report the development of a high-density linkage map based on genotyping-by-sequencing (GBS) for the genus perilla. Through GBS library construction and Illumina sequencing of an F2 population, a total of 9607 single-nucleotide polymorphism (SNP) markers were developed. The ten-group linkage map of 1309.39 cM contained 2518 markers, with an average marker density of 0.56 cM per linkage group (LG). Using this map, a total of six QTLs were identified. These quantitative trait loci (QTLs) are associated with three traits related to flowering time: days to visible flower bud, days to flowering, and days to maturity. Ortholog analysis conducted with known genes involved in the regulation of flowering time among different crop species identified GI, CO and ELF4 as putative perilla orthologs that are closely linked to the QTL regions associated with flowering time. These results provide a foundation that will be useful for future studies of flowering time in perilla using fine mapping, and marker-assisted selection for the development of new varieties of perilla.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Perilla is one genus in the mint family, Lamiaceae, and is a traditional oil crop in Korea and a medicinal herb in Asia [1]. Perilla seeds are a good source of polyunsaturated fatty acids, including alpha-linolenic acid (ALA), linoleic acid and oleic acid. Perilla seed oil consists of 54–64% omega-3 (ALA) fatty acids, which is a higher proportion than found in other plant oils. Perilla oil also contains omega-6 (linoleic acid; ~ 14%) and omega-9 (oleic acid) fatty acids. These polyunsaturated fatty acids are beneficial to human health and contribute to the prevention of several diseases, including cardiovascular disorders, cancer, inflammation, and rheumatoid arthritis [2, 3].
Oil accumulation occurs at different stages of seed development across different species. For example, according to Msaada et al. [4], coriander seeds start to accumulate lipids from the 5th day after flowering, and the maximal oil content is observed when the seeds reach their maximum maturity stage. In contrast, Perilla frutescens seeds accumulate lipids very slowly during the first stages of maturity [5], and the maximal rate of lipid accumulation is observed between 15 and 20 days after flowering. Stability in lipid accumulation is reported as a characteristic of the last phase of P. frutescens seed maturation and occurs between 20 and 31 days after flowering. A comparable trend in lipid accumulation has been observed in oil seed rape (Brassica napus): rapid lipid accumulation is observed between 25 and 35 days after flowering with slower lipid accumulation observed during the first and the last stages of maturity [6]. Understanding of flowering and maturity can be one of important factors to improve economical value of oilseed crop, perilla.
Perilla is one of the typical short-day (SD) plants [7]. Flowering time (FTi) and maturity are greatly influenced by day length in many plants [8, 9]. The molecular mechanisms of flowering and the genes involved in the pathway have been reported in Arabidopsis thaliana as a model plant [10]. Arabidopsis FLOWERING LOCUS T (FT) and one of its rice orthologues, HEADING DATE 3a (Hd3a), are key flowering integrators, which encode a florigen that is transported from leaves to shoots or lateral apical meristems through the phloem in a regulated manner and induce the development of floral meristems [11, 12]. The amount of FT transcript, which is directly induced by the transcriptional activator CONSTANTS (CO) protein, strongly influences the timing of flowering. The circadian clock and light signaling tightly control CO protein activity throughout the day in the companion cells of the leaf phloem [13]. Several studies on QTLs related to flowering and maturity also have been detected in soybean [14], rice [15], wheat [16], and other plants [17]. However, there are no reports on the flowering-related QTLs or related genes in perilla, a short-day plant.
The genetic map is required to perform QTL analysis, but there is no published data in perilla so far. Although, several RAPD [18] and SCAR [19] markers and around a hundred of SSR markers [1, 20,21,22,23,24] have been reported to classify different perilla species, but it is insufficient number to construct high-density genetic map of perilla. Additional molecular markers need to be developed to construct a high-density genetic map of perilla. SNPs are the most abundant class of polymorphisms in most genomes of living organisms, and are one of the most efficient markers to find candidate genes associated with QTLs [25]. Additionally, GBS is a useful and cost-effective tool for the resequencing of bulk samples, and can be applied to various areas of plant genetics and breeding, including SNP discovery, high-density genetic mapping, and QTL analysis especially for little studied plant [26,27,28,29,30,31,32,33].
In this study, therefore, we first constructed a high-density genetic map via GBS with 96 F2 plants from an interspecific crossed between P. citriodora and P. hirtella. We developed the F2 population using two different species for two purposes. First, we wanted to construct a high-density genetic map for anchoring the scaffolds of perilla genome. Therefore, selecting two species which can be able to cross interspecies and has a lot of polymorphic SNP between them. The second purpose was to analyze QTLs of agronomic traits. Using this linkage map, we then tried to identify QTLs related to flowering and seed maturity.
Materials and methods
Population mapping and phenotyping
A segregating population of 96 F2 plants derived from an interspecific cross between P. citriodora (P1) and P. hirtella (P2) was cultivated, grown, and evaluated for mapping at the National Institute of Crop Science, RDA (Miryang, Korea) [34]. Phenotypic data was evaluated for the following three traits: day to visible flower bud (DtoFB), day to flowering (DtoF), and day to maturity (DtoM). Days to visible flower bud (DtoFB) is days until bud showed up on shoot tip since the day of sowing, days to flowering (DtoF) is days from the day of sowing to when the first flower opens on shoot tip, and days to maturity (DtoM) is days from the day of sowing until the first seeds color turned into brown. The difference between parents was relatively small, but there was a greater variation in the F2 population. Pearson’s correlation coefficients and statistical analyses of measured traits were performed using Minitab 18 (State College, PA, USA) and SAS 9.3 (SAS Institute, Cary, NC, USA).
DNA extraction and GBS library preparation
Fresh young leaves were collected, and DNA was extracted through a modified cetyltrimethylammonium bromide (CTAB) method [35]. The DNA was quantified using a Thermo Scientific Nanodrop 8000 spectrophotometer (Fisher Scientific; Hampton, NH, USA). Genomic DNA from 96 F2 lines and parents was used to prepare the libraries for GBS. The libraries were prepared by restriction digestion of DNA for each of the F2 lines, followed by ligation with barcoded adapters. Ninety-six different barcode sequences were used to tag the samples [36]. The GBS libraries were constructed using the restriction enzymes PstI (CTGCAG) and MspI (CCGG) using a protocol modified from Elshire et al. [36]. The libraries were pooled and sequenced using Illumina TrueSeq Ver3.0 paired-end sequencing with 101-bp read lengths on the Illumina platform HiSeq 2000. Ninety-six samples were sequenced in one lane.
Sequencing and SNP genotyping
After sequencing, the raw reads were de-multiplexed according to the barcode sequences and trimmed using a Python script. This script split the raw Illumina FASTQ file into 96 separate FASTQ files based on the barcode sequences associated with each sample, and filtered out reads that contain any ambiguous bases in the barcodes [26]. The reads that contained only the common adapter also were trimmed using Cutadapt software [37]. The de-multiplexed reads were trimmed using the Solexa QA package v.1.13 [38]. It is common for the quality of the bases to decrease at the ends of Illumina reads; therefore, the ends were trimmed when the Phred quality score dropped below Q = 20 (or 0.05 probability of error). Additionally, all 5′ and 3′ stretches of ambiguous N nucleotides were trimmed. Poor quality sequence reads and reads shorter than 25 bases were discarded.
The Burrows–Wheeler Aligner program (BWA; 0.6.1–r104) [39] was applied to clean reads of the draft genome sequence of P. citriodora (unpublished data) to align the SNP calling. The mapped reads were extracted for further analyses from the resulting BAM file using SAMtools v.0.1.16 [40]. The high mapping quality ensured reliable (i.e., unique) mapping of the reads, which is important for variant calling. Using the SAM tools varFilter command, SNPs were called only for variable positions with a minimal mapping quality (i.e., -Q) of 30. The minimum and maximum of the read depths were set to 3 and 100, respectively. An in-house script that considered bi-allelic loci was used to select the significant sites in the called SNP positions [41].
Linkage map construction
A linkage map of perilla was constructed using IciMapping ver.4.1 [42] using 2518 filtered SNPs detected in F2 progenies derived from a cross between P. citriodora and P. hirtella. SNPs were grouped into ten linkage groups (LGs) with logarithm of the odds (LOD) threshold values ≥ 3. The ordering of the 2518 SNPs distributed over these 10 LGs was performed using the nnTwoOpt algorithm [43]. Rippling to fine tune the marker order was performed by the Sum of Adjacent Recombination Fractions (SARF) with a window size of 5 as the rippling criteria. The genetic distances of the SNP markers, which were based on the recombination rates, were converted using Kosambi’s mapping function [44]. The final linkage map was drawn using MapChart ver2.3 [45].
QTL analysis
QTL mapping of the three traits was performed based on the composite interval mapping (CIM) method with Windows QTL Cartographer ver2.5 [46, 47] using forward–backward stepwise regression. The LOD threshold for significance (P = 0.05) was determined via a 1000-permutation test. The software also estimated the percentage of phenotypic variance and additive effects as well as the dominance effect explained by each QTL for each trait. The gene action was determined from the calculated value of the dominance to additive effect ratio and analyzed as described by Stuber et al. [48].
Orthologous genes
To predict orthologous genes related to flowering time (FTi) in perilla, TBLASTN was performed using BLAST 2.2.82. Protein sequences of 20 FTi genes from A. thaliana and perilla draft genome sequences were used. The FTi genes were selected from Blumel et al. [49]. Among these 20 FTi genes, 15 genes were functional genes orthologous with genes from other crops or species and 10 genes were homologous with A. thaliana genes. The BLAST results were filtered using an e-value of 1e−10 and a query coverage ≥ 50% to predict ortholog genes. The position of ortholog genes in genetic map was determined using markers close to the physical location found.
Results
Evaluation of phenotypic data
There were significant differences in the phenotypes of DtoFB, DtoF, and DtoM within the F2 generation mapping population and parents. Descriptive statistics are summarized in Table 1. Normal frequency distributions were observed for all three traits (Fig. S1). DtoF was significantly correlated with the other two traits (Table 2). All three traits were positively correlated with each other. The highest degree of correlation was observed between DtoFB and DtoF (r = 0.81). P1 takes 5 more days to appear flower bud than P2.
Genome-wide identification of SNPs using GBS
The genomic DNA of 96 F2 lines was digested with the restriction enzymes PstI and MspI. A 96-plex GBS library was constructed with the F2 lines. Sequencing was carried out in one lane on 51.9 Gbp (514,560,106 reads) with a HiSeq 2000 platform (Table S1). Following the application of filtering criteria, such as removal of the barcode and PstI and MspI overhang sequences, 503,215,708 (97.8%) of the sequencing reads were de-multiplexed. As a result, 459,068,228 (91.15%) high-quality trimmed reads had a Phred quality score ≥ 20 after removing ambiguous nucleotides (Table S2). The trimmed data was aligned to the draft genome sequence of P. citriodora. Our in-house GBS analysis pipeline [26, 50] was used for SNP mining from the sequence data [41]. A total of 91,132 raw SNPs was identified in 96 F2 lines (Table S2). These SNPs were filtered to identify putative markers using the criteria of 30% missing values across the genotyped individual and MAF ≥ 0.05, which yielded a total of 14,223 SNPs. Next, 9607 polymorphic SNPs were detected between P. citriodora and P. hirtella as the parents of the mapping population. After generating a bin map of the genotyping and SNP selections using 50 kb as the window size, 2518 SNP markers were finally identified.
Construction of a high-density linkage map
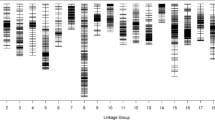
A linkage map comprised of 2518 markers on 10 LGs (Fig. 1, Table 3) was constructed from the GBS data. The total length of the linkage map was 1309.39 cM. LG01 (271.86 cM) was the largest LG and LG10 (45.8 cM) was the smallest LG. The number of markers per LG varied from 60 (LG10) to 661 (LG01), with an average of 251.8 markers per LG. The average marker density was 0.56 cM. LG06 was the densest LG (0.3 cM) and LG02 was the least dense LG (0.85 cM). A summary of the constructed linkage map is presented in Table 3 and illustrated in Fig. 1.

SNP distribution. The distribution of single-nucleotide polymorphism (SNP) markers using a linkage map of perilla constructed using the F2 population derived from the parental lines P. citriodora and P. hirtella. QTLs associated with the three traits (DtoFB, DtoF, DtoM) were shown on the left side of linkage map. Black box indicates QTLs for DtoFB, white box for DtoF, box with check pattern for DtoM. The location of the orthologue of the perilla that regulates the FTi is indicated by the underlined TAIR symbol
QTL mapping
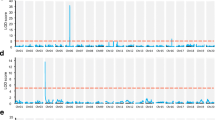
QTL mapping was performed for the three flowering-related traits on the integrated genotype and phenotype data using the CIM method. A total of six QTLs were identified for the three traits, which were distributed on LG01, LG02, and LG08 (Fig. 2). The LOD thresholds ranged from 6.49 (qDFB2) to 13.57 (qDFB1) in DtoFB, 3.98 (qDF8.2) to 7.37 (qDF2) in DtoF and 4.50 (qDM2) in DtoM. The identified QTLs have been summarized in Table 4.

Quantitative trait loci. QTL associated with three flowering-related traits [days to visible flower bud (DtoFB), days to flowering (DtoF), and days to maturity (DtoM)] in the F2 population derived from the parental lines P. citriodora and P. hirtella in the LGs identified via CIM. The LOD threshold is represented by the horizontal line. a Days to visible flower bud, b days to flowering, c days to maturity
DtoFB
Two QTLs were detected for DtoFB on LG01 and LG02 with LOD thresholds of 13.57 (qDFB1) and 6.49 (qDFB2), respectively (Fig. 2a). At all the loci, the alleles from parent P. citriodora (P1) favored DtoFB. The QTLs explained 44.07% (qDFB1) and 26.69% (qDFB2) of the phenotypic variance, respectively. This finding suggests that the P. citriodora allele may delay DtoFB.
DtoF
Three QTLs were detected for DtoF on LG02 and LG08 with LOD thresholds of 7.37 (qDF2), 4.20 (qDF8.1), and 3.98 (qDF8.2), respectively (Fig. 2b). The QTLs explained 36.13% (qDF2), 12.90% (qDF8.1), and 14.03% (qDF8.2) of the phenotypic variance, respectively. Positive additive effects derived from P. citriodora (P1) were observed for qDF2, whereas negative additive effects derived from P. hirtella (P2) were observed for qDF8.1 and qDF8.2.
DtoM
One QTL was detected for DtoM on LG02 with an LOD threshold of 4.50 (qDM2) (Fig. 2c). This QTL explained 19.34% of the phenotypic variance. A positive additive effect derived from P. citriodora (P1) was observed for qDM2, and was associated with a delay in DtoM.
One-way analysis of variance (ANOVA) for QTL markers
The effects of markers in these six QTL regions were confirmed via one-way ANOVAs. Fifty-eight markers had LOD value that exceeded the LOD threshold for each QTL: 18 markers each for qDFB1, qDFB2, and qDF2; one marker for qDF8.1; one marker for qDF8.2; and one marker for qDM2 (Table S3). Markers in qDFB1 and qDFB2 were significantly different between P. citriodora and P. hirtella with R2 values ranging from 29.68 to 44.07% and 12.37 to 31.38%, respectively. DtoFB was significantly later in P. citriodora by about 6–7 days compared to P. hirtella (P =0.000). qDF2, qDF8.1, and qDF8.2 markers also were significantly different between P. citriodora and P. hirtella. qDF2 markers had R2 values ranging from 18.26 to 38.41%, whereas the R2 values for qDF8.1 and qDF8.2 markers were 12.90% and 14.03%, respectively. qDF2 in P. citriodora was associated with a significant 4- to 5-day delay in DtoF. qDF8.1 and qDF8.2 in P. hirtella also were associated with a significant delay of DtoF of about 5 days. Lastly, qDM2 was significantly different between P. citriodora and P. hirtella (R2 = 19.34%) such that P. citriodora matures 4 days later than P. hirtella.
Orthologous genes
Orthologous genes that regulate FTi were confirmed using BLAST to estimate the candidate genes from QTLs that are related to DtoFB, DtoF, and DtoM. Protein sequences from 20 A. thaliana FTi genes were collected from the NCBI database. To estimate the FTi gene and copy number in perilla, TBLASTN was performed between the gene protein sequence and the perilla draft genome sequence. After filtering the results (e-value of 1e-10 and 50% query coverage), all of the FTi genes except ELF3 and FLC (AGL15, FLF) were selected. Each gene had at least one copy and TFL1 had six copies in perilla (Table S4). Among whole-copy genes, 2 genes were on QTL regions: ELF4, which is in the qDFB1 region of LG01, and GI, which is in the qDF2 and qDFB2 regions of LG02. AP1 was not in a QTL region but was close to qDF8.1. CO and COL5 also were close to qDF8.2 (Fig. 1). The results from resequencing analysis of parents showed that six SNPs were found in ELF4 and fourteen SNPs in GI (Table 5). Looking at the effect of SNP on the translation of amino acids, one SNP showed a non-synonymous change in the ELF4 gene and two SNPs in the GI gene.
Discussion
This study identified a large number of molecular markers and QTLs involved in flowering and maturation in the relatively little studied genus Perilla using GBS. The genome for perilla has not yet been decoded, and only a few markers (e.g., RAPD, SCAR and SSR) are available [18,19,20,21,22,23]. Significant effort is required to develop new molecular markers to construct high-quality linkage maps for perilla. The GBS method used in this study on the F2 group of perilla enabled the identification of sufficient polymorphic SNPs and genotyping to be performed simultaneously. This allowed us to create a linkage map with a fairly high density and without needing to check or apply existing markers. GBS, which has been applied to other crops, therefore is considered an effective and widely applicable method for genotyping various crops [26,27,28,29,30,31,32,33].
The oil content and composition of perilla seeds are known to change depending on the seed development stage [5], whereas the lipid content of seeds, particularly the fatty acid composition of 18:3 α-linolenic acid and 18:2 linoleic acid, changes little in the early period after flowering but sharply as maturity begins. More specifically, at the beginning of seed formation, the contents of the two components are similar. As maturity begins, 18:3 α-linolenic acid increases and 18:2 linoleic acid decreases. The maturation period therefore can be a very important trait in the breeding program. In the present study, it was confirmed that the flowering period is highly correlated with seed maturity (Table 2). The QTL analysis provides a more detailed explanation of this correlation. For example, the QTLs for DtoFB and DtoF are located in the same LGs, LG01 and LG02. The loci with the highest LODs, however, are located at different positions. In other words, the major QTL associated with the DtoF are located in LG02, but the major QTL associated with the DtoFB is located in LG01. Similarly, the QTL for DtoM also was located in LG01 and LG02, but the LOD was relatively low (LOD = 4.00). This finding suggests that there is less genetic influence on DtoM than on the peak time to DtoFB or DtoF.
In Arabidopsis, CO is a key integrator in the induction of FT expression [51]. Daily CO expression profiles are regulated mainly by the circadian clock-regulated proteins including FLAVIN-BINDING, F-BOX1 (FKF1), KELCH REPEAT, GI, and CYCLING DOF FACTOR (CDF) [52,53,54]. FKF1 and GI proteins form a complex that degrades CDF proteins which repress transcription of CO during the morning, and that up-regulates the expression of CO at the end of the day under LD conditions [53]. GI also directly interacts with EARLY FLOWERING 4 (ELF4), which sequesters GI from the nucleoplasm, where GI binds the promoter of CO, to discrete nuclear bodies [55]. This subnuclear localization of GI by ELF4 affects the level of CO expression. Light signaling pathways and the circadian clock co-ordinate the control of CO activity to induce FT [10]. The functions and roles of SD plants, legume orthologues of CO-like (COL) genes in the control of flowering may differ from those of LD plant Arabidopsis CO. GmCOL1a and GmCOL1b suppressed flowering under LD conditions in soybean [56]. It is suggested that the role of GmCOL1a and GmCOL1b, in the regulation of GmFT2a and GmFT5a may be similar to the role of Hd1 in the regulation of H3a in rice. Hd1 activates the expression of Hd3a under SD conditions, but suppresses it under LD conditions [8, 57, 58]. As a SD plant, three important modules that inhibit flowering in LD conditions in soybean were reported: the PHYA-E1, GI-CO, and miRNA-dependent modules [59]. In this study, the combination of QTL analysis and ortholog gene comparison enabled candidate genes to be identified more easily, such as PcELF4 and PcGI. This result indicated that GI-CO module suggested in soybean may be involved in the regulation of flowering time in perilla.
Gene-based molecular markers are helpful for molecular breeding. In the future, if these perilla genes are verified by molecular biological and genetic studies through mutants, they are expected to be used as molecular markers related to the flowering period of perilla.
References
Park Y-J, Dixit A, Ma K-H, Lee J-K, Lee M-H, Chung C-S, Nitta M, Okuno K, Kim T-S, Cho E-G, Rao VR (2007) Evaluation of genetic diversity and relationships within an on-farm collection of Perilla frutescens (L.) Britt. using microsatellite markers. Genet Resour Crop Evol 55(4):523–535. https://doi.org/10.1007/s10722-007-9258-x
Asif M (2011) Health effects of omega-3,6,9 fatty acids: Perilla frutescens is a good example of plant oils. Orient Pharm Exp Med 11(1):51–59. https://doi.org/10.1007/s13596-011-0002-x
Kim HU, Lee KR, Shim D, Lee JH, Chen GQ, Hwang S (2016) Transcriptome analysis and identification of genes associated with omega-3 fatty acid biosynthesis in Perilla frutescens (L.) var. frutescens. BMC Genom 17:474. https://doi.org/10.1186/s12864-016-2805-0
Msaada K, Hosni K, Taarit MB, Hammami M, Marzouk B (2009) Effects of growing region and maturity stages on oil yield and fatty acid composition of coriander (Coriandrum sativum L.) fruit. Scientia Horticulturae 120(4):525–531. https://doi.org/10.1016/j.scienta.2008.11.033
Ki Ichihara, Suda Y (2003) Lipid biosynthesis in developing perilla seeds. Phytochemistry 63(2):139–143. https://doi.org/10.1016/s0031-9422(03)00110-9
Vigeolas H, van Dongen JT, Waldeck P, Huhn D, Geigenberger P (2003) Lipid storage metabolism is limited by the prevailing low oxygen concentrations within developing seeds of oilseed rape. Plant Physiol 133(4):2048–2060. https://doi.org/10.1104/pp.103.031963
Kondo H, Ozaki H, Itoh K, Kato A, Takeno K (2006) Flowering induced by 5-azacytidine, a DNA demethylating reagent in a short-day plant Perilla frutescens var. crispa. Physiol Plant 127(1):130–137. https://doi.org/10.1111/j.1399-3054.2005.00635.x
Matsubara K, Hori K, Ogiso-Tanaka E, Yano M (2014) Cloning of quantitative trait genes from rice reveals conservation and divergence of photoperiod flowering pathways in Arabidopsis and rice. Front Plant Sci 5:193. https://doi.org/10.3389/fpls.2014.00193
Weller JL, Ortega R (2015) Genetic control of flowering time in legumes. Front Plant Sci 6:207. https://doi.org/10.3389/fpls.2015.00207
Song YH, Ito S, Imaizumi T (2013) Flowering time regulation: photoperiod- and temperature-sensing in leaves. Trends Plant Sci 18(10):575–583. https://doi.org/10.1016/j.tplants.2013.05.003
Tamaki S, Matsuo S, Wong HL, Yokoi S, Shimamoto K (2007) Hd3a protein is a mobile flowering signal in rice. Science 316(5827):1033–1036. https://doi.org/10.1126/science.1141753
Notaguchi M, Abe M, Kimura T, Daimon Y, Kobayashi T, Yamaguchi A, Tomita Y, Dohi K, Mori M, Araki T (2008) Long-distance, graft-transmissible action of Arabidopsis FLOWERING LOCUS T protein to promote flowering. Plant Cell Physiol 49(11):1645–1658. https://doi.org/10.1093/pcp/pcn154
Shim JS, Kubota A, Imaizumi T (2017) Circadian clock and photoperiodic flowering in Arabidopsis: CONSTANS is a hub for signal integration. Plant Physiol 173(1):5–15. https://doi.org/10.1104/pp.16.01327
Yamanaka N, Watanabe S, Toda K, Hayashi M, Fuchigami H, Takahashi R, Harada K (2005) Fine mapping of the FT1 locus for soybean flowering time using a residual heterozygous line derived from a recombinant inbred line. Theor Appl Genet 110(4):634–639. https://doi.org/10.1007/s00122-004-1886-3
Zhao XL, Shi ZY, Peng LT, Shen GZ, Zhang JL (2011) An atypical HLH protein OsLF in rice regulates flowering time and interacts with OsPIL13 and OsPIL15. N Biotechnol 28(6):788–797. https://doi.org/10.1016/j.nbt.2011.04.006
Nemoto Y, Kisaka M, Fuse T, Yano M, Ogihara Y (2003) Characterization and functional analysis of three wheat genes with homology to the CONSTANS flowering time gene in transgenic rice. Plant J 36(1):82–93
Kwak M, Velasco D, Gepts P (2008) Mapping homologous sequences for determinacy and photoperiod sensitivity in common bean (Phaseolus vulgaris). J Hered 99(3):283–291. https://doi.org/10.1093/jhered/esn005
Kim H-K, Cho Y-S, Yang J-W, Choi Y-W, Kang J-S, Lee Y-J, Son B-G (2010) Analysis of genetic variation of Perilla frutescens var. crispa germplasm using RAPD. J Life Sci 20(1):119–123. https://doi.org/10.5352/jls.2010.20.1.119
Lee MH, Yang KW, Ha TJ, Jung CS, Pae SB, Hwang CD, Park CH, Baek IY, Kim HK, Park SK (2011) Development of SCAR marker for identification of the Perilla species. Korean J Breed Sci 43(4):189–193
Kim Y, Kim AY, Jo A, Choi H, Cho SS, Choi C (2017) Development of user-friendly method to distinguish subspecies of the Korean medicinal herb Perilla frutescens using multiplex-PCR. Molecules 22(4):44. https://doi.org/10.3390/molecules22040665
Sa KJ, Choi SH, Ueno M, Park K-C, Park YJ, Ma KH, Lee JK (2013) Identification of genetic variations of cultivated and weedy types of Perilla species in Korea and Japan using morphological and SSR markers. Genes Genom 35(5):649–659. https://doi.org/10.1007/s13258-013-0117-1
Sa KJ, Choi SH, Ueno M, Lee JK (2015) Genetic diversity and population structure in cultivated and weedy types of Perilla in East Asia and other countries as revealed by SSR markers. Hortic Environ Biotechnol 56(4):524–534. https://doi.org/10.1007/s13580-015-0039-8
Sa KJ, Choi IY, Park KC, Lee JK (2018) Genetic diversity and population structure among accessions of Perilla frutescens (L.) Britton in East Asia using new developed microsatellite markers. Genes Genomics 40(12):1319–1329. https://doi.org/10.1007/s13258-018-0727-8
Woo SY, Sa KJ, Lee JK (2016) Collection and evaluation of genetic variation of perilla accessions in the Jeju Island. Plant Breeding and Biotechnology 4(1):87–98. https://doi.org/10.9787/pbb.2016.4.1.87
Liu L, Qu C, Wittkop B, Yi B, Xiao Y, He Y, Snowdon RJ, Li J (2013) A high-density SNP map for accurate mapping of seed fibre QTL in Brassica napus L. PLoS ONE 8(12):e83052. https://doi.org/10.1371/journal.pone.0083052
Eun MH, Han JH, Yoon JB, Lee J (2016) QTL mapping of resistance to the Cucumber mosaic virus P1 strain in pepper using a genotyping-by-sequencing analysis. Hortic Environ Biotechnol 57(6):589–597. https://doi.org/10.1007/s13580-016-0128-3
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7(2):e32253. https://doi.org/10.1371/journal.pone.0032253
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome J 5(3):92. https://doi.org/10.3835/plantgenome2012.05.0005
Deschamps S, Llaca V, May GD (2012) Genotyping-by-sequencing in plants. Biology (Basel) 1(3):460–483. https://doi.org/10.3390/biology1030460
Bielenberg DG, Rauh B, Fan S, Gasic K, Abbott AG, Reighard GL, Okie WR, Wells CE (2015) Genotyping by sequencing for SNP-based linkage map construction and QTL analysis of chilling requirement and bloom date in Peach [Prunus persica (L.) Batsch]. PLoS ONE 10(10):e0139406. https://doi.org/10.1371/journal.pone.0139406
Verma S, Gupta S, Bandhiwal N, Kumar T, Bharadwaj C, Bhatia S (2015) High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using Genotyping-by-Sequencing (GBS). Sci Rep 5:17512. https://doi.org/10.1038/srep17512
Hussain W, Baenziger PS, Belamkar V, Guttieri MJ, Venegas JP, Easterly A, Sallam A, Poland J (2017) Genotyping-by-Sequencing derived high-density linkage map and its application to QTL mapping of flag leaf traits in bread wheat. Sci Rep 7(1):16394. https://doi.org/10.1038/s41598-017-16006-z
Li L, Zhao S, Su J, Fan S, Pang C, Wei H, Wang H, Gu L, Zhang C, Liu G, Yu D, Liu Q, Zhang X, Yu S (2017) High-density genetic linkage map construction by F2 populations and QTL analysis of early-maturity traits in upland cotton (Gossypium hirsutum L.). PLoS One 12(8):e0182918. https://doi.org/10.1371/journal.pone.0182918
Lee MH, Oh KW, Kim MS, Kim SU, Kim JI, Oh EY, Pae SB, Yeo US, Kim T-H, Lee JH, Jung CS, Kwak DY, Kim YC (2018) Detection of QTLs in an interspecific cross between Perilla citriodora × P hirtella mapping population. Korean J Breeding Sci 50(1):13–20. https://doi.org/10.9787/kjbs.2018.50.1.13
Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF (2006) A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc 1(5):2320–2325. https://doi.org/10.1038/nprot.2006.384
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379. https://doi.org/10.1371/journal.pone.0019379
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17(1):3. https://doi.org/10.14806/ej.17.1.200
Cox MP, Peterson DA, Biggs PJ (2010) SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform 11:485. https://doi.org/10.1186/1471-2105-11-485
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Kim JE, Oh SK, Lee JH, Lee BM, Jo SH (2014) Genome-wide SNP calling using next generation sequencing data in tomato. Mol Cells 37(1):36–42. https://doi.org/10.14348/molcells.2014.2241
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283. https://doi.org/10.1016/j.cj.2015.01.001
Lin S, Kernighan BW (1973) An effective heuristic algorithm for the Traveling-Salesman problem. Oper Res 21(2):498–516
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugenic 12:172–175
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93(1):77–78
Wang S, Basten CJ, Zeng ZB (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm)
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142(1–2):169–196. https://doi.org/10.1007/s10681-005-1681-5
Stuber CW, Edwards MD, Wendel JF (1987) Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits 1. Crop Sci 27(4):639–648. https://doi.org/10.2135/cropsci1987.0011183x002700040006x
Blumel M, Dally N, Jung C (2015) Flowering time regulation in crops-what did we learn from Arabidopsis? Curr Opin Biotechnol 32:121–129. https://doi.org/10.1016/j.copbio.2014.11.023
Kim WJ, Ryu J, Im J, Kim SH, Kang SY, Lee JH, Jo SH, Ha BK (2018) Molecular characterization of proton beam-induced mutations in soybean using genotyping-by-sequencing. Mol Genet Genomics 293(5):1169–1180. https://doi.org/10.1007/s00438-018-1448-z
SuaÂrez-LoÂpez P, Wheatley K, Robson F, Onouchi H, Valverde F, George C (2001) CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 410(6832):1116–1120. https://doi.org/10.1038/35074138
Imaizumi T, Schultz TF, Harmon FG, Ho LA, Kay SA (2005) FKF1 F-box protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science 309(5732):293–297. https://doi.org/10.1126/science.1110586
Sawa M, Nusinow DA, Kay SA, Imaizumi T (2007) FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science 318(5848):261–265. https://doi.org/10.1126/science.1146994
Fornara F, Panigrahi KC, Gissot L, Sauerbrunn N, Ruhl M, Jarillo JA, Coupland G (2009) Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev Cell 17(1):75–86. https://doi.org/10.1016/j.devcel.2009.06.015
Kim Y, Lim J, Yeom M, Kim H, Kim J, Wang L, Kim WY, Somers DE, Nam HG (2013) ELF4 regulates GIGANTEA chromatin access through subnuclear sequestration. Cell Rep 3(3):671–677. https://doi.org/10.1016/j.celrep.2013.02.021
Cao D, Li Y, Lu S, Wang J, Nan H, Li X, Shi D, Fang C, Zhai H, Yuan X, Anai T, Xia Z, Liu B, Kong F (2015) GmCOL1a and GmCOL1b function as flowering repressors in soybean under long-day conditions. Plant Cell Physiol 56(12):2409–2422. https://doi.org/10.1093/pcp/pcv152
Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, Yano M (2002) Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43(10):1096–1105
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12(12):2473–2484
Cao D, Takeshima R, Zhao C, Liu B, Jun A, Kong F (2017) Molecular mechanisms of flowering under long days and stem growth habit in soybean. J Exp Bot 68(8):1873–1884. https://doi.org/10.1093/jxb/erw394
Acknowledgements
This work was supported by a Grant from the National Agricultural Genome Project (Nos. PJ01040803, PJ01335503), Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Contributions
YJ, BM, SH, and JH conceived and designed the experiments; YJ and BM conducted the SNP analysis, linkage map construction, and data analysis; MN constructed the GBS library; KW and MH developed the mapping population and evaluated the plant phenotypes; TH provided the draft genome and participated in discussions about the experiments; YJ, SH and JH wrote the manuscript; and SH and JH reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (PNG 5004 kb)
Fig. S1 Frequency distributions. The frequency distributions of three flowering-related traits in the 96 studied F2 populations of the parental lines P. citriodora x P. hiretella. The values corresponding to the parental lines (PLs) are shown by the vertical line. A: days to visible flower bud; B: days to flowering; C: days to maturity.
Rights and permissions
About this article

{kind=link}
Cite this article
Kang, YJ., Lee, BM., Nam, M. et al. Identification of quantitative trait loci associated with flowering time in perilla using genotyping-by-sequencing. Mol Biol Rep 46, 4397–4407 (2019). https://doi.org/10.1007/s11033-019-04894-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-04894-5