
Abstract
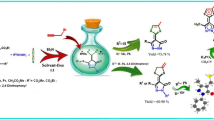
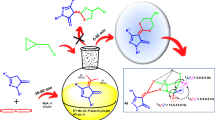
In this study, a one-pot reaction between β-keto esters or dialkyl acetylenedicarboxylates with hydrazines, carbon disulfide, and dialkyl acetylenedicarboxylates in the presence of triethylamine is reported. This reaction proceeded at room temperature and was completed within 6 h to produce functionalized pyrazolone-1,4-dithiafulvene hybrids in good yields.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pyrazolones represent important structural motifs in heterocyclic chemistry and are found in many biologically active molecules used in the pharmaceutical and agrochemical industries. Pyrazolones show anti-tuberculosis [1], anti-viral [2], anti-hypertension [3], anti-oxidation [4], neuroprotection [5], anti-diabetic [6], anti-inflammatory [7], and anti-cancer [8] activities. They are also used as ligands [9] in complexes with catalytic activity. Some pyrazolones are used as wool, cotton, and silk dyes [10]. In addition, derivatives of sulfur heterocycles show significant biological and pharmaceutical activities [11]. 1,3-Dithiol-2-ylidenes derivatives have attracted much attention due to their excellent electron donation characteristics as a component in electronic materials [12, 13]. Also, sulfur-containing heterocycles such as 1,3-dithiole derivatives have been considered potential new substances due to their superconducting, optical, and electrical switching capabilities [14].
Various methods for preparing 1,3-dithiole derivatives have been published [15,16,17,18,19]. Among the dithiols, 1,4-dithiafulvenes bearing ester groups have attracted much attention as building blocks of electronic materials [20, 21]. The most common methods reported for the synthesis of 1,4-dithiafulvenes containing ester groups are the use of Wittig reaction between aldehydes or ketones with phosphonium salts [22,23,24]. These reactions are usually carried out at − 78 °C under argon atmosphere and in the presence of strong base such as butyl lithium. On the other hand, preparation of phosphonium salts also includes several steps [25]. In this study, we attempted to prepare 1,4-dithiafulvenes containing ester groups using ketene dithioacetal intermediates under easier conditions.
Ketene dithioacetals are used as efficient intermediates in the synthesis of 1,3-dithiol derivatives. Ketene dithioacetals can generate from the reaction between carbon nucleophile and carbon disulfide [26]. The reaction between ketene dithioacetals and dual electrophilic species such as dihaloalkanes [27, 28], or α-halo carbonyl compounds produce sulfur-containing heterocycles with two sulfur atoms [29, 30].
Laboratory studies show that the biological activities of bioactive molecules are usually recovered if two or more bioactive units are grouped in a single molecule [31]. Therefore, hybrid molecules of various heterocycles with pyrazolones contain more effective biological activities [32]. Edaravone (I) has practical medicinal effects on a variety of diseases, including cardiovascular diseases [33], and Lanoconazole (II) shows significant antifungal activity [34]. Besides that, the 1,4-dithiafulvene unit (III) has a strong electron-donating property [21,22,23,24, 35] and is frequently used as a donor unit in donor–acceptor systems (Fig. 1).

Structures of pyrazolone and sulfur-containing molecules applied in medicine (I, II) and 1,4-dithiafulvene unit (III) applied in the material industry
Due to potential of sulfur-containing heterocycles and pyrazolones as mentioned above, we became interested in the synthesis of hybrid molecules containing pyrazolones and 1,4-dithiafulvenes. Following our research on the one-pot synthesis of new heterocyclic compounds [36,37,38,39] herein, we report the facile one-pot synthesis of pyrazolone-1,4-dithiafulvene hybrids 5 from the reactions between β-keto esters 1 or dialkyl acetylenedicarboxylates 2 with hydrazines 3, carbon disulfide, and dialkyl acetylenedicarboxylates 4 (Scheme 1).

One-pot reaction for the synthesis of pyrazolone-1,4-dithiafulvene hybrids 5
Results and discussion
Synthesis and optimization of reaction conditions
The one-pot reaction between ethyl acetoacetate 1a, phenyl hydrazine 3a, carbon disulfide, and dimethylacetylenedicarboxylate 4a was selected as a model reaction to produce pyrazolone-1,4-dithiafulvene hybrid 5a (Table 1). At first, the reaction was carried out in the water in the presence of two equimolar of Et3N at room temperature. The progress of the reaction was monitored by TLC. After compilation of the reaction, product 5a was separate as orange powder by filtration. The reaction yield was 25%. To optimize the reaction conditions, the reaction was carried out in the presence of various bases and solvents, and the results are collected in Table 1. As illustrated in Table 1, the reaction was not done in the presence of KOH, K2CO3, and pyridine in water, and the reaction yield in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) was negligible (Table 1, entries 1–5). Therefore, the Et3N was selected as the appropriate base for this reaction. The use of CH2Cl2 and EtOH as the solvent could not increase the reaction yield (Table 1, entries 6, 7). When the reaction was performed in THF or DMSO, the reaction yield increased, but the increase of reaction yield in acetonitrile was more significant (Table 1, entries 8–10). Further studies investigating the effect of temperature on the reaction yield showed that when the reaction was carried out in refluxing acetonitrile, the reaction yield was reduced because of the generation of complex by-products (Table 1, entry 11). Therefore, it is found that room temperature is the optimum temperature for the synthesis of pyrazolone-1,4-dithiafulvene hybrid 5a. In addition, the study of the effect of the amount of base on the reaction yield showed that the two equimolar of the base is the optimum amount of base for this reaction (Table 1, entries 10, 12–14).
In this reaction, no detectable by-products were formed. In addition to the desired product, small amounts of acetonitrile-soluble dark materials were formed which were separated from the main product by filtration. To evaluate the scalability of the reaction, the model reaction was performed at double and quadruple scale in optimal conditions and no significant change in the reaction yield was observed.
Characterization of products
The structure of 5a was confirmed by FT-IR, 1H NMR, 13C NMR, Mass, and elemental analysis data. In the IR spectra of 5a, the peaks related to the stretching vibration of the ester carbonyl groups and S-C bonds appear in 1735 and 755 cm−1, respectively. In the 1H NMR spectrum of 5a, methyl protons of pyrazolone moiety appear at δ = 2.46 ppm. Two methyl groups of ester moieties appear at δ = 3.95 and 3.96 ppm. The aromatic protons of 5a showed two triplets at δ = 7.16 ppm (3JHH = 8.4 Hz) and δ = 7.40 ppm (3JHH = 8.2 Hz), and a doublet at δ = 7.99 ppm (3JHH = 8.5 Hz). In addition, in the 13C NMR spectra of 5a, two ester CO signals overlapped at 160.4 ppm, and the 13C NMR spectra of 5a revealed 14 distinct signals, indicating that the hypothesized structure is correct. The mass spectrum of compound 5a showed the molecular ion peak at 390 m/z, which was expected (See experimental section).
To confirm the generalizability of the reaction, compounds 5a-i were synthesized through the one-pot reaction between β-keto esters 1 or acetylenic esters 2, with hydrazines 3, CS2, and dialkyl acetylenedicarboxylates 4 in the presence of Et3N in CH3CN. The reactions proceeded well, and the products 5b-i were obtained with a good yield within 6 h (See Table 2).
The proposed mechanism for this reaction is depicted in Scheme 2. Initially, the pyrazolone derivatives I were generated from the reaction of β-keto esters 1 or acetylenic esters 2, with hydrazine derivatives 3 [40, 41]. Subsequently, the tautomeric intermediates II and III were obtained by adding the in situ generated pyrazolone derivative I to CS2 in the presence of Et3N [42]. Following the Michel addition of sulfur anion of II or III to triple bound of acetylenic esters 4, resulting in the production of intermediate IV. Cyclization of intermediate IV through the second Michel addition, resulting in intermediate V [43]. Finally, intermediate V is then oxidized in the presence of air to produce product 5 spontaneously (Scheme 2).

The proposed mechanism for the synthesis of pyrazolone-1,4-dithiafulvene hybrids 5
To investigate the effect of atmospheric oxygen on the formation of the product 5a, the model reaction was performed under an argon atmosphere. In this case, TLC analysis showed that product 5a was not formed. This shows that atmospheric oxygen acts as an oxidant in this reaction.
Conclusion
In conclusion, we successfully presented a mild, facile, and one-pot method for the synthesis of new pyrazolone-1,4-dithiafulvene hybrids by using readily available starting materials through a one-pot reaction between β-keto esters or dialkyl acetylenedicarboxylates with hydrazines, carbon disulfide, and dialkyl acetylenedicarboxylates in good yields. A screening of the reaction conditions demonstrated that, performing this reaction at room temperature and in acetonitrile in the presence of two equivalents of triethylamine as a base, are the best ones. The good yields and the ease of workup procedure make it an appealing, practical, and acceptable one-pot method for producing functionalized pyrazolone-1,4-dithiafulvene hybrids.
Experimental section
General information
Dialkyl acetylendicarboxylate, hydrazines, carbon disulfide, β-keto esters, and all solvents were obtained from Merck (Germany) and were used without further purification. RF values refer to thin-layer chromatography (TLC) performed on silica gel 60 F254 aluminum-backed silica plates (Merck). Melting points were measured with a Stuart SMP-3 apparatus. IR spectra of products were measured with an FTIR Perkin Elmer RXI. NMR spectra were reported on a Varian Inova 500 MHz (500 MHz for 1H and 125 MHz for 13C) with CDCl3 as the solvent. Chemical shifts are given in ppm (δ) relative to internal TMS, and coupling constants (J) are reported in Hertz (Hz). Mass spectra were recorded with an Agilent 5977A Series MSD spectrometer operating at an ionization potential of 70 eV.
General procedure for the synthesis of products (5a-i), exemplified by 5a
A mixture of ethyl acetoacetate (1 mmol) with phenyl hydrazine (1 mmol) and Et3N (2 mmol) in CH3CN (5 mL) was stirred for 2 h at room temperature. After that, carbon disulfide (1.2 mmol) was added, and the mixture was stirred for 30 min. Then, dropwise additions of DMAD (1 mmol) were made, and the mixture was stirred for 6 h. Following that, the solvent was removed under reduced pressure, and the residue was washed with water to yield the pure product. The product was recrystallized in ethanol to achieve higher purity samples.
Dimethyl 2-(3-methyl-5-oxo-1-phenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5a)
Orange powder; yield: 0.25 g (64%); m.p. 200–201 ◦C. TLC RF = 0.65 (ethyl acetate/n-hexane = 3:7), IR (KBr, υ, cm−1): 1737 (CO2Me), 1526 (C=CS2), 1252 (C−O), 754 (S−C) cm−1. MS (EI): m/z (%) = 390 (M+, 100), 331 (40), 257 (27), 207 (17), 91 (17). 1H NMR (500 MHz, CDCl3): δ = 2.46 (s, 3H, N=CCH3), 3.95 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 7.16 (t, 1H, 3JHH = 7.4 Hz, CHpara), 7.4 (t, 2H, 3JHH = 7.2 Hz, CHmeta), 7.99 (d, 2H, 3JHH = 8.5 Hz, CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 16.6 (N=C–CH3), 54.1 and 54.2 (2OCH3), 112.9 (C=CS2), 118.7 (2CHortho), 124.8 (CHpara), 129.0 (2CHmeta), 131.2 and 137.5 (2 = C–CO2CH3), 138.7 (Cipso), 144.4 (C=N), 158.9 (S–C–S), 159.5 (N–C=O), 160.4 (2CO2CH3) ppm.
Diethyl 2-(3-methyl-5-oxo-1-phenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5b)
Orange powder; yield: 0.24 g (57%); m.p. 198–199 ◦C. TLC RF = 0.55 (ethyl acetate/n-hexane = 3:7), IR (KBr, υ, cm−1): 1735 (CO2Et), 1524 (C=CS2), 1238 (C−O), 755 (S−C) cm−1. MS (EI): m/z (%) = 418 (M+, 100), 345 (12), 215 (25), 185 (14), 91 (27), 28 (16). 1H NMR (500 MHz, CDCl3): δ = 1.40 (t, 6H, 3JHH = 7.1 Hz, 2CO2CH2CH3), 2.48 (s, 3H, N=CCH3), 4.41 (q, 4H, 3JHH = 7.1 Hz, 2CO2CH2CH3), 7.17 (t, 1H, 3JHH = 7.3 Hz, CHpara), 7.40 (t, 2H, 3JHH = 7.4 Hz, 2CHmeta), 7.99 (d, 2H, 3JHH = 8.0 Hz, 2CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 14.1 (2OCH2CH3), 16.6 (N=C–CH3), 63.8 (2OCH2), 112.7 (C=CS2), 118.6 (2CHortho), 124.8 (CHpara), 129.0 (2CHmeta), 131.4 and 137.5 (2=C–CO2CH3), 138.7 (Cipso), 144.5 (C=N), 158.5 (S–C–S), 158.6 (N–C=O), 159.1 (2CO2CH2CH3) ppm.
Dimethyl 2-(5-oxo-1,3-diphenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5c)
Orange powder; yield: 0.24 g (53%); m.p. 207–209 ◦C. TLC RF = 0.6 (ethyl acetate/n-hexane = 3:7), IR (KBr, υ, cm−1): 1738 (CO2Me), 1501 (C=CS2), 1258 (C−O), 756 (S−C) cm−1. MS (EI): m/z (%) = 394 (M+-CO2Me, 100), 366 (11), 335 (12), 281 (13), 261 (17), 207 (28), 145 (39), 91 (13). 1H NMR (500 MHz, CDCl3): δ = 3.87 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 7.20 (t, 1H, 3JHH = 7.3 Hz, CHpara), 7.42 (t, 2H, 3JHH = 7.8 Hz, 2CHmeta), 7.54–7.60 (m, 5H, 5CH-Ar), 8.07 (d, 2H, 3JHH = 8.2 Hz, 2CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 53.7 and 53.8 (2OCH3), 111.1 (C=CS2), 118.8 (2CHortho), 124.9 (CHpara), 128.7 (4CHmeta), 129.1 (2CHortho), 130.1 (CHpara), 131.4 (=C–CO2CH3), 132.4 (Cipso), 135.9 (=C–CO2CH3), 138.4 (Cipso), 147.2 (C=N), 159.0 (S–C–S), 159.2 (N–C=O), 162.0 and 162.1 (2CO2CH3) ppm.
Diethyl 2-(5-oxo-1,3-diphenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5d)
Orange powder; yield: 0.3 g (63%); m.p. 184–185 ◦C. TLC RF = 0.55 (ethyl acetate/n-hexane = 3:7), IR (KBr, υ, cm−1): 1729 (CO2Et), 1494 (C=CS2), 1244 (C−O), 756 (S−C) cm−1. MS (EI): m/z (%) = 480 (M+, 100), 407 (20), 310 (5), 277 (32), 247 (16), 145 (40), 91 (36). 1H NMR (500 MHz, CDCl3): δ = 1.32 (t, 3H, 3JHH = 7.1 Hz, CO2CH2CH3), 1.39 (t, 3H, 3JHH = 7.0 Hz, CO2CH2CH3), 4.33 (q, 2H, 3JHH = 7.1 Hz, OCH2), 4.39 (q, 2H, 3JHH = 7.0 Hz, OCH2), 7.20 (t, 1H, 3JHH = 7.3 Hz, CHpara), 7.42 (t, 2H, 3JHH = 7.8 Hz, 2CHmeta), 7.55–7.61 (m, 5H, 5CH-Ar), 8.08 (d, 2H, 3JHH = 8.2 Hz, 2CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 14.0 and 14.1 (2OCH2CH3), 63.4 and 63.5 (2OCH2), 111.2 (C=CS2), 119.0 (2CHortho), 125.1 (CHpara), 129.0 (4CHmeta), 129.3 (2CHortho), 130.3 (CHpara), 131.7 (=C–CO2CH3), 133.0 (Cipso), 136.1 (=C–CO2CH3), 138.7 (Cipso), 147.4 (C=N), 158.9 (S–C–S), 159.2 (N–C=O), 162.4 and 162.8 (2CO2CH3) ppm.
Dimethyl 2-(3-(methoxycarbonyl)-5-oxo-1-phenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5e)
Yellow powder; yield: 0.22 g (51%); m.p. 246–248 ◦C. TLC RF = 0.55 (ethyl acetate/n-hexane = 4:6), IR (KBr, υ, cm−1): 1753 and 1712 (CO2Me), 1487 (C=CS2), 1226 (C−O), 764 (S−C) cm−1. MS (EI): m/z (%) = 434 (M+, 100), 406 (12), 375 (11), 347 (21), 315 (15), 273 (11), 77 (19). 1H NMR (500 MHz, CDCl3): δ = 3.97 (s, 6H, 2OCH3), 4.03 (s, 3H, OCH3), 7.27 (t, 1H, 3JHH = 7.0 Hz, CHpara), 7.45 (t, 2H, 3JHH = 7.0 Hz, CHmeta), 8.02 (d, 2H, 3JHH = 8.0 Hz, CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 53.0, 54.0, and 54.1 (3OCH3), 108.7 (C=CS2), 120.2 (2CHortho), 126.4 (CHpara), 129.1 (2CHmeta), 135.3 (=C–CO2CH3), 135.5 (Cipso), 137.1 (=C–CO2CH3), 138.0 (C=N), 159.3 (S–C–S), 159.4 (N–C=O), 162.7 (2CO2CH3), 167.6 (CO2CH3) ppm.
Diethyl 2-(3-(methoxycarbonyl)-5-oxo-1-phenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5f)
Orange powder; yield: 0.25 g (54%); m.p. 165–167 ◦C. TLC RF = 0.5 (ethyl acetate/n-hexane = 4:6), IR (KBr, υ, cm−1): 1731 (CO2Et), 1491 (C=CS2), 1241 (C−O), 757 (S−C) cm−1. MS (EI): m/z (%) = 390 (M+-CO2Et, 74), 281 (42), 253 (13), 207 (100), 133 (10), 77 (14). 1H NMR (500 MHz, CDCl3): δ = 1.39 (t, 6H, 3JHH = 7.1 Hz, 2CO2CH2CH3), 4.03 (s, 3H, OCH3), 4.43 (q, 4H, 3JHH = 7.1 Hz, 2OCH2), 7.20 (t, 1H, 3JHH = 7.3 Hz, CHpara), 7.44 (t, 2H, 3JHH = 7.3 Hz, 2CHmeta), 8.01 (d, 2H, 3JHH = 8.6 Hz, 2CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 14.1 (2OCH2CH3), 53.0 (OCH3), 63.7 and 63.8 (2OCH2), 108.5 (C=CS2), 120.3 (2CHortho), 126.4 (CHpara), 129.1 (2CHmeta), 135.3 (=C–CO2CH3), 135.9 (Cipso), 137.0 (=C–CO2CH3), 138.0 (C=N), 159.1 (S–C–S), 162.3 (N–C=O), 162.7 (2CO2CH3), 167.9 (CO2CH3) ppm.
Dimethyl 2-(3-(ethoxycarbonyl)-5-oxo-1-phenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5 g)
Yellow powder; yield: 0.22 g (50%); m.p. 223–224 ◦C. TLC RF = 0.5 (ethyl acetate/n-hexane = 4:6), IR (KBr, υ, cm−1): 1717 (CO2Me), 1491 (C=CS2), 1227 (C−O), 759 (S−C) cm−1. MS (EI): m/z (%) = 448 (M+, 100), 347 (23), 284 (12), 257 (25), 207 (23), 77 (17). 1H NMR (500 MHz, CDCl3): δ = 1.49 (t, 3H, 3JHH = 7.1 Hz, CH3), 3.97 and 3.98 (2 s, 6H, 2OCH3), 4.51 (q, 2H, 3JHH = 7.1 Hz, CH2), 7.26 (Overlapped with solvent peak, 1H, CHpara), 7.45 (t, 2H, 3JHH = 8.6 Hz, 2CHmeta), 8.03 (d, 2H, 3JHH = 8.6 Hz, 2CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 14.4 (OCH2CH3), 54.0 and 54.1 (2OCH3), 62.4 (OCH2), 109.1 (C=CS2), 120.3 (2CHortho), 126.4 (CHpara), 129.1 (2CHmeta), 135.5 (=C–CO2CH3), 135.7 (Cipso), 137.0 (=C–CO2CH3), 138.0 (C=N), 159.4 (S–C–S), 159.5 (N–C=O), 162.4 and 162.5 (2CO2CH3 and CO2C2H5) ppm.
Diethyl 2-(3-(ethoxycarbonyl)-5-oxo-1-phenyl-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5 h)
Yellow powder; yield: 0.24 g (51%); m.p. 158–159 ◦C. TLC RF = 0.65 (ethyl acetate/n-hexane = 4:6), IR (KBr, υ, cm−1): 1725 (CO2Et), 1494 (C=CS2), 1254 (C−O), 756 (S−C) cm−1. MS (EI): m/z (%) = 404 (M+-CO2Et, 58), 312 (100), 281 (18), 212 (24), 207 (45), 110 (11), 77 (7). 1H NMR (500 MHz, CDCl3): δ = 1.40 (t, 3H, 3JHH = 6.9 Hz, OCH2CH3), 1.41 (t, 3H, 3JHH = 6.9 Hz, OCH2CH3), 1.49 (t, 3H, 3JHH = 7.1 Hz, OCH2CH3), 4.40–4.45 (m, 4H, 2OCH2), 4.51 (q, 2H, 3JHH = 7.1 Hz, OCH2), 7.26 (Overlapped with solvent peak, 1H, CHpara), 7.44 (t, 2H, 3JHH = 8.2 Hz, 2CHmeta), 8.03 (d, 2H, 3JHH = 8.1 Hz, 2CHortho) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 14.1 (2OCH2CH3), 14.4 (OCH2CH3), 62.3, 63.6, and 63.7 (3OCH2), 108.6 (C=CS2), 120.3 (2CHortho), 126.3 (CHpara), 129.1 (2CHmeta), 135.7 (=C–CO2CH3), 135.9 (Cipso), 136.9 (=C–CO2CH3), 138.1 (C=N), 159.0 (S–C–S), 159.1 (N–C=O), 162.4 (2CO2C2H5), 167.8 (CO2 C2H5) ppm.
Diethyl 2-(3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene)-1,3-dithiole-4,5-dicarboxylate (5i)
Orange powder; yield: 0.18 g (52%); m.p. 187–190 ◦C. TLC RF = 0.5 (ethyl acetate/n-hexane = 6:4), IR (KBr, υ, cm−1): 3462 (N–H), 1742 and 1736 (CO2Et), 1507 (C=CS2), 1287 (C−O), 767 (S−C) cm−1. MS (EI): m/z (%) = 342 (M+, 100), 297 (5), 269 (9), 242 (35), 185 (16), 140 (29), 83(16). 1H NMR (500 MHz, CDCl3): δ = 1.35–1.37 (m, 6H, 2OCH2CH3), 2.35 (s, 3H, CH3), 4.37–4.39 (m, 4H, 2OCH2CH3), 9.67 (s, 1H, NH) ppm. 13C NMR (125.59 MHz, CDCl3): δ = 14.0 and 14.1 (2OCH2CH3), 16.6 (N=C–CH3), 63.6 and 63.7 (2OCH2), 111.5 (C=CS2), 131.0 and 137.4 (2=C–CO2CH3), 144.9 (C=N), 158.6 (S–C–S), 159.2 (N–C=O), 160.6 and 165.5 (2CO2C2H5) ppm.
References
Sweeney NL, Lipker L, Hanson AM, Bohl JC, Engel KE, Kalous KS, Stemper ME, Sem DS, Schwan WR (2017) Docking into mycobacterium tuberculosis thioredoxin reductase protein yields pyrazolone lead molecules for methicillin-resistant staphylococcus aureus. Antibiotics 6:1–11. https://doi.org/10.3390/antibiotics6010004
Ramajayam R, Tan KP, Liu HG, Liang PH (2010) Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg Med Chem 18:7849–7854. https://doi.org/10.1016/j.bmc.2010.09.050
Hassan MQ, Akhtar MS, Afzal O, Hussain I, Akhtar M, Haque SE, Najmi AK (2020) Edaravone and benidipine protect myocardial damage by regulating mitochondrial stress, apoptosis signalling and cardiac biomarkers against doxorubicin-induced cardiotoxicity. Clin Exp Hypertens 42:381–392. https://doi.org/10.1080/10641963.2019.1676770
Bailly C (2019) Potential use of edaravone to reduce specific side effects of chemo-, radio-and immuno-therapy of cancers. Int Immunopharmacol 77:1–8. https://doi.org/10.1016/j.intimp.2019.105967
Bhandari R, Kuhad A, Kuhad A (2018) Edaravone: a new hope for deadly amyotrophic lateral sclerosis. Drugs Today 54:349–360. https://doi.org/10.1358/dot.2018.54.6.2828189
Haroun M (2019) Novel hybrids of pyrazolidinedione and benzothiazole as TZD analogues. Rationale design, synthesis and in vivo anti-diabetic evaluation. Med Chem 15:624–633. https://doi.org/10.2174/1573406415666190515093657
Abdelgawad MA, Labib MB, Ali WAM, Kamel G, Azouz AA, El-Nahass ES (2018) Design, synthesis, analgesic, anti-inflammatory activity of novel pyrazolones possessing aminosulfonyl pharmacophore as inhibitors of COX-2/5-LOX enzymes: Histopathological and docking studies. Bioorg Chem 78:103–114. https://doi.org/10.1016/j.bioorg.2018.03.011
Sun X, Zhang L, Gao M, Que X, Zhou C, Zhu D, Cai Y (2019) Nanoformulation of a novel pyrano [2,3-c] pyrazole heterocyclic compound AMDPC exhibits anti-cancer activity via blocking the cell cycle through a P53-independent pathway. Molecules 24:1–11. https://doi.org/10.3390/molecules24030624
Akcha S, Gómez-Ruiz S, Kellou-Tairi S, Lezama L, Pérez FB, Benali-Baitich O (2018) Synthesis, characterization, solution equilibria, DFT study, DNA binding affinity and cytotoxic properties of a cobalt(II) complex with a 5-pyrazolone ligand. Inorg Chim Acta 482:738–748. https://doi.org/10.1016/j.ica.2018.06.051
Rizk HF, Ibrahim SA, El-Borai MA (2017) Synthesis, dyeing performance on polyester fiber and antimicrobial studies of some novel pyrazolotriazine and pyrazolyl pyrazolone azo dyes. Arab J Chem 10:S3303–S3309. https://doi.org/10.1016/j.arabjc.2014.01.008
Pathania S, Narang RK, Rawal RK (2019) Role of sulphur-heterocycles in medicinal chemistry: an update. Eur J Med Chem 180:486–508. https://doi.org/10.1016/j.ejmech.2019.07.043
Segura JL, Martin N (2001) New concepts in tetrathiafulvalene chemistry. Angew Chem Int Ed 40:1372–1409. https://doi.org/10.1002/1521-3773(20010417)40:83.0.CO;2-I
Bryce MR (1995) Current trends in tetrathiafulvalene chemistry: towards increased dimensionality. J Mater Chem 5:1481–1496. https://doi.org/10.1039/JM9950501481
Marcos CF, Polo C, Rakitin OA, Rees CW, Torroba T (1997) One-pot synthesis and chemistry of bis [1,2] dithiolopyrroles. Chem Commun 9:879–880. https://doi.org/10.1039/A701340J
Ahadi S, Hosseini Gh, Bazgir A (2012) Synthesis of oxoindolin-3-ylidene-1,3-dithioles. J Iran Chem Soc 9:333–338. https://doi.org/10.1007/s13738-011-0028-5
Bazgir A, Astaraki AM (2011) Simple and efficient synthesis of 1,3-dithioles with pyrimidinylidene or pyrazolylidene substituents. Phosphorus Sulfur Silicon 186:1916–1921. https://doi.org/10.1080/10426507.2010.551617
Narayanan K, Shanmugam M, Vasuki G, Kabilan S (2014) Synthesis, spectral, crystal and theoretical studies of some novel 4-heterocyclic substituted pyrazolones. J Mol Struct 1056–1057:70–78. https://doi.org/10.1016/j.molstruc.2013.10.018
Xu CF, Liu YX, Cao RZ, Liu LZ (2002) The synthesis of 2-arylidene-1,3-dithioles containing phosphonyl group. Synth Commun 32:535–538. https://doi.org/10.1081/SCC-120002398
Konstantinova LS, Lysov KA, Amelichev SA, Obruchnikova NV, Rakitin OA (2009) A one-pot synthesis and 1,3-dipolar cycloaddition of [1,2]dithiolo[4,3-b]indole-3(4H)-thiones. Tetrahedron 65:2178–2183. https://doi.org/10.1016/j.tet.2009.01.069
Lissau H, Jevric M, Madsen AØ, Nielsen MB (2015) Synthesis of dithiafulvene-quinone donor-acceptor systems: isolation of a Michael adduct. Acta Cryst C71:452–455. https://doi.org/10.1107/S2053229615008578
Mansø M, Kilde MD, Singh SK, Erhart P, Moth-Poulsen K, Nielsen MB (2019) Dithiafulvene derivatized donor–acceptor norbornadienes with redshifted absorption. Phys Chem Chem Phys 21:3092–3097. https://doi.org/10.1039/C8CP07744D
Åxman Petersen M, Zhu L, Jensen SH, Andersson AS, Kadziola A, Kilså K, Brøndsted Nielsen M (2007) Photoswitches containing a dithiafulvene electron donor. Adv Funct Mater 17:797–804. https://doi.org/10.1002/adfm.200600888
Sune Andersson A, Qvortrup K, Rossel Torbensen E, Mayer JP, Gisselbrecht JP, Boudon C, Gross M, Kadziola A, Kilså K, Brøndsted Nielsen M (2005) Synthesis and characterization of extended tetrathiafulvalenes with Di-, Tri-, and tetraethynylethene cores. Eur J Org Chem 17:3660–3671. https://doi.org/10.1002/ejoc.200500287
Nielsen MB, Petersen JC, Thorup N, Jessing M, Andersson AS, Jepsen AS, Gisselbrecht JP, Boudon C, Gross M (2005) Acetylenic dithiafulvene derived donor–π–acceptor dyads: synthesis, electrochemistry and non-linear optical properties. J Mater Chem 15:2599–2605. https://doi.org/10.1039/B504124D
Bryce M R, Coffin M. A, Clegg W (1992) New vinylogous tetrathiafulvalene .pi.-electron donors with peripheral alkylseleno substitution. J. Org. Chem 57(6): 1696–1699. https://doi.org/10.1021/jo00032a018
Hamrouni K, Saied T, El Abed N, Ben Hadj Ahmed S, Boujlel K, Ben Khoud ML (2015) Electrogenerated base-promoted synthesis and antimicrobial activity of 2-(1,3-dithian-2-ylidene)-2-arylacetonitrile and 2-(1,3-dithiolan-2-ylidene)-2-arylacetonitrile. J Sulphur Chem 36:196–206. https://doi.org/10.1080/17415993.2015.1005620
Ahluwalia VK, Dudeja S (2001) A convenient synthesis of 1,3-disubstituted-4-(1′,3′-dithiolane/ dithiane-2′-ylidene)-2-pyrazolin-5-ones. Synth Commun 31:3175–3181. https://doi.org/10.1081/SCC-100105894
Khalil AKh, Hassan MA, Mohamed MM, El-Sayed AM (2005) Phase-transfer catalyzed alkylation and cycloalkylation of 3-substituted-1H-pyrazol-2-in-5-ones in the absence or presence of carbon disulphide. Phosphorus Sulfur Silicon 180:479–496. https://doi.org/10.1080/104265090517208
El-Saraf GA, El-Sayed AM, El-Saghier A (2003) One-pot PTC synthesis of polyfused pyrazoles. Heteroatom Chem 14:211–217. https://doi.org/10.1002/hc.10129
Sabahi-Agabager L, Akhavan S, Nasiri F (2022) A facile one-pot, solvent-free synthesis of new pyrazolone-1,3-dithiolan hybrids through the reaction between 2-pyrazoline-5-ones, CS2, and α-chloroacetaldehyde. J Sulphur Chem 43:391–401. https://doi.org/10.1080/17415993.2022.2055432
Mishra S, Singh P (2016) Hybrid molecules: the privileged scaffolds for various pharmaceuticals. Eur J Med Chem 124:500–536. https://doi.org/10.1016/j.ejmech.2016.08.039
Guha C, Sepay N, Mallik S, Mallik AK (2018) Facile synthesis of a new class of pyrazolone attached chromene derivatives showing good binding with β-Lactoglobulin. ChemistrySelect 3:5138–5142. https://doi.org/10.1002/slct.201800702
Zhao Z, Dai X, Li C, Wang X, Tian J, Feng Y, Xie J, Ma C, Nie Z, Fan P, Qian M (2020) Pyrazolone structural motif in medicinal chemistry: retrospect and prospect. Eur J Med Chem 186:111893. https://doi.org/10.1016/j.ejmech.2019.111893
Niwano Y, Ohmi T, Seo A, Kodama H, Koga H, Sakai A (2003) Lanoconazole and its related optically active compound NND-502: novel antifungal imidazoles with a ketene dithioacetal structure. Curr Med Chem Anti-Infect Agents 2:147–160. https://doi.org/10.2174/1568012033483097
Qvortrup K, Sune Andersson A, Mayer JP, Sofie Jepsen A (2004) Cross-coupling reactions with acetylenic dithiafulvenes. Synlett 15:2818–2820. https://doi.org/10.1055/s-2004-835641
Aghaalizadeh T, Nasiri F (2018) Regioselective four-component synthesis of new tetrazolo [1, 5-a] quinoline-based 2-amino-1, 4-dihydropyridine and pyridin-2(1H)-one derivatives using nano-ZnO catalysis. Mol Divers 22:907–917. https://doi.org/10.1007/s11030-018-9844-1
Nasiri F, Nazari P (2018) One-pot solvent-free three-component reaction between primary amines, carbon disulfide, and 5-alkylidene rhodanines: a convenient synthesis of asymmetric birhodanines. Mol Divers 22:601–608. https://doi.org/10.1007/s11030-018-9816-5
Aghaalizadeh T, Nasiri F, Sabahi-Agabager L (2019) Synthesis of new 2-amino-4H-thiopyran derivatives via the one-pot reaction between 3,4-dihydro-3-((benzylamino) methylene)-4-thioxochromen-2-ones and alkyl-2-cyanoacetates in the presence of nano-ZnO catalysis. J Sulfur Chem 40:42–51. https://doi.org/10.1080/17415993.2018.1502293
Sabahi-Agabager L, Nasiri F (2020) One-pot, solvent-free facile stereoselective synthesis of rhodanine-furan hybrids from renewable resources. J Sulfur Chem 41:170–181. https://doi.org/10.1080/17415993.2019.1702196
Deruiter J, Carter DA, Arledge WS, Sullivan PJ (1987) Synthesis and reactions of 4-isopropylidene-1-aryl-3-methyl-2-pyrazolin-5-ones. J Heterocycl Chem 24:149–153. https://doi.org/10.1002/jhet.5570240128
Ghonchepour E, Islami MR, Mostafavi H, Momeni Tikdari A (2018) Three-component reaction for an efficient synthesis of 5-hydroxy-1-phenyl-1H-pyrazoles containing a stable phosphorus ylide moiety. Phosph Sulfur Silicon Relat Elem 193:459–463. https://doi.org/10.1080/10426507.2018.1437619
Yavari I, Sheykhahmadi J, Saffarian H, Halvagar MR (2019) Nef-isocyanide-Perkow access to novel pyrazolone derivations containing a cyclic ketene dithioacetal moiety. Synth Commun 49:1–7. https://doi.org/10.1080/00397911.2018.1560474
Habibi A, Valizadeh Y, Alizadeh A, Rudbari HA, Nardo VM (2014) Regioselective synthesis of novel ketene dithioacetals. J Sulfur Chem 35:362–372. https://doi.org/10.1080/17415993.2013.879871
Funding
This work was supported by the University of Mohaghegh Ardabili, Ardabil, Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest was reported by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Salehzadeh, J., Nasiri, F. A facile one-pot synthesis of new functionalized pyrazolone-1,4-dithiafulvene hybrids. Mol Divers 28, 19–28 (2024). https://doi.org/10.1007/s11030-022-10473-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10473-x