
Abstract
Myosin heavy chain gene 7 (MYH7), a sarcomeric gene encoding the myosin heavy chain (myosin-7), has attracted considerable interest as a result of its fundamental functions in cardiac and skeletal muscle contraction and numerous nucleotide variations of MYH7 are closely related to cardiomyopathy and skeletal muscle myopathy. These disorders display significantly inter- and intra-familial variability, sometimes developing complex phenotypes, including both cardiomyopathy and skeletal myopathy. Here, we review the current understanding on MYH7 with the aim to better clarify how mutations in MYH7 affect the structure and physiologic function of sarcomere, thus resulting in cardiomyopathy and skeletal muscle myopathy. Importantly, the latest advances on diagnosis, research models in vivo and in vitro and therapy for precise clinical application have made great progress and have epoch-making significance. All the great advance is discussed here.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Myosin which makes up the backbone of the sarcomere thick filament, plays a key role in the process of muscle cell contraction. Emerging evidence proved that hereditary myosin myopathies are caused by mutations in skeletal muscle myosin heavy chain (MYH) gene family, including MYH1, MYH2, MYH3, MYH4, MYH8, and MYH13 on chromosome 17 expressed in skeletal muscles, as well as MYH6 and MYH7 on chromosome 14, encoding two main types of cardiac muscle, alpha isoform and beta isoform, respectively [1]. Myosin is highly sensitive to the mutation of MYH gene family [2]. Consistent with the locations and predominate muscles of myosin, pathogenic variants in the respective genes are associated with distinctive phenotypes of cardiac and skeletal myopathies.
Myosin heavy chain (myosin-7), the same substance with cardiac muscle beta isoform as mentioned above, is a slow ATPase myosin and is encoded by MYH7 gene. Myosin-7 is located in both ventricular muscle fibers and slow/type 1 skeletal muscle fibers. Therefore, pathogenic mutations in MYH7 gene could cause cardiomyopathy, skeletal muscle myopathy, and both of them. Known human cardiomyopathy includes hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restricted cardiomyopathy (RCM), left ventricular non-compaction cardiomyopathy (LVNC), and other less common congenital cardiomyopathies. The common skeletal muscle myopathy includes myosin storage myopathy (MSM), Laing distal myopathy (LDM), and congenital fiber-type disproportion (CFTD). Compound MYH7 mutations increase the severity of disease and the risk of sudden cardiac death (SCD). The variable clinical phenotypes and coexisting multiple mutations increase the difficulty and complexity of clinical work.
In this review, we summarized the structure of MYH7 and provide a brief overview of the relationship between the mutations in MYH7 and its disorders, as well as updating the latest research progress on diagnose methods and target therapy.
Structure
The structure and sequence of MYH7 (MIM:160760) have been completely discovered in 1990 [3], which is 22,883 bp long and located on chromosome 14q11.2, 3.6 kb upstream from the MYH6 (MIM:160710) in a head-to-tail tandem fashion [4, 5]. MYH7 is composed of 39 introns and 40 exons, including 38 coding. The 5-prime untranslated region, 86 bp long, is split by 2 introns and the 3-prime untranslated region (UTR) is 114 bp long. The translation start codon (ATG) is located in nucleotide position 9 of the third exon. Three Alu repeats (–GATC–) were identified within the gene and the fourth one is in the 3-prime flanking intergenic region [3] (Fig. 1a).

a Schematic of MYH7 gene, mRNA and protein, its interactomes. MYH7 is composed of 39 introns and 40 exons, whose 38 are translated into a 223,097 Da peptide called myosin-7, containing 1935 amino acids. The exons are drawn as black boxes. Locations of the TATA box, translation start codon (ATG), and poly (A) signal (AATAAA), two “active thiols” (SHl, SH2), and the S2-hinge, S2:LMM are illustrated, in addition to promoter indicated by a triangle and 4 Alu repeats by asterisks in MYH7. b Representation of S1 constructed by Swiss Model. Using amino acid sequence of human myosin-7 as target (sequence identity: 78.88%), the N-terminal 25 kDa, central 50 kDa which are functionally divided into upper 50 kDa and lower 50 kDa domains, and C-terminal 20 kDa, are indicated with corresponding colors. Loop1 connecting the 25 kDa and 50 kDa, loop 2 connecting the 50 kDa and 20 kDa, and the cleft formed by numerous α-helix surrounding a 7-stranded β-sheet core are illustrated. (https://swissmodel.expasy.org/). c The proteolytic cleavage form of S1. It contains the N-terminal 25 kDa, central 50 kDa, and C-terminal 20 kDa. The locations of loop1 and loop2 are indicated. SH3-like domain Src homology 3-like domain, IQ motif isoleucine–glutamine motif, HMM heavy meromyosin, LMM light meromyosin, S1 subfragment 1, S2 subfragment 2, ELC essential light chain subunit, RLC regulatory light chain subunit, Unc-45 uncoordinated mutant number-45, MyBP-C myosin binding protein-C, MyBP-H myosin binding protein-H, AMPD1 adenosine monophosphate deaminase 1, MuRF1 muscle RING dinger protein1, MuRF3 muscle RING dinger protein3, Interactome interaction partner
Myosin-7 (P12833), a protein of 1935 amino acids and a chemical mass of 223,097 Da, is encoded by the MYH7 gene. It is also known as heart muscle beta-myosin heavy chain, myosin heavy chain 7, myosin heavy chain slow isoform. According to Geeves et al. [6], myosin-7 is composed of a Src homology 3-like (SH3-like) domain, a head motor domain, a lever arm, and a coiled-coil “tail.” Proteolytic cleavage yields myosin-7 into two parts: heavy meromyosin (HMM) and light meromyosin (LMM). HMM can be further digested into subfragment 1 (S1) and subfragment 2 (S2). S1 contains the SH3-like domain in N-terminus, the motor head domain containing a converter segment in the C-terminus via a relay helix, and the lever arm compounded from two isoleucine–glutamine (IQ) motifs. The two IQ motifs extend to the “tail” structure of myosin molecular, containing S2 and LMM. S2 and LMM contain the NH2- and COOH-terminal regions of the α-helical rod domain, respectively, and these two α-helical heavy chains dimerize to form a coiled-coil in the “tail” (Fig. 1a). The motor head region has the actin binding site and the ATP binding site. And the region is composed of a 7-stranded β-sheet core surrounding by numerous α-helices, which forms a cleft stretching between these two binding sites. Furthermore, S1 can be cleaved to 3 subdomains by trypsin, the central 50 kDa, the N-terminal 25 kDa, and the C-terminal 20 kDa [7]. The N-terminal 25kD contains the SH3-like domain. The central 50 kDa domain is further functionally divided into upper and lower 50 kDa regions [8]. The ATP binding site is located in the cleft which is formed between the upper and lower 50 kDa subregions, and the actin binding site is in the central 50 kDa domain, mostly the lower 50 kDa subregion [9, 10] (Fig. 1b). Moreover, these two binding procedures are implemented in a well-defined coupling process in which when the ATP binds to the head region, the actin binding cleft is controlled to open, which is the pre-requisite for functioning effectively [11]. In addition to two active thiols (SH1 and SH2), the ATP and actin binding sites, are all well conserved, and there are also several variable subregions, including N-terminus of S1, the hypervariable loop1 and loop2, the S2-hinge and the S2:LMM junction. Loop1, which connects the 25 kDa and 50 kDa, sets above the nucleotide binding pocket, and is suggested to be essential in modulating the ATPase kinetics. Loop 2, connecting the 50 kDa and 20 kDa, involves in actin binding step [12]. The S2 hinge and the S2:LMM junction are located in the linking area between the head and the rod, and exert to mediate the flexibility between them [3] (Fig. 1c).
In addition to ATP and actin binding sites, myosin-7 also react with several interaction partners (interactomes) in stable or transient forms to properly complete its functions, including essential light chain subunit (ELC) and regulatory light chain subunit (RLC) binding to the NH2- and COOH-terminal IQ motifs of lever arm, respectively, myosin binding protein-C (MyBP-C) and MyBP-H binding to the coiled-coil tail containing both S2 and LMM [13, 14], myomesin and M-protein binding to the LMM [15, 16], titin binding to S1 and LMM [17, 18], nonerythroid 4.1 protein, MuRF1 and MuRF3 binding to HMM [19, 20], AMPD1 binding to S2 [19], and Unc-45 binding to the head domain [21] (Fig. 1a). There are also multiple posttranslational modifications (PTMs) in myosin-7, most of which are phosphorylation sites, followed by acetylation, ubiquitylation, and a limited O-glycosylation and methylation sites (Fig. 2).

Sites of PTMs in myosin-7. The many PTMs published in papers, which affect myosin-7 are represented by variously colored circles, and the PTMs with more than 5 references are shown in detail, required from PhosphatePlus database (https://www.phosphosite.org/homeAction). PTMs posttranslational modifications, Other O-glycosylation, and methylation sites
In striated muscles, two myosin heavy chain subunits (myosin-7), along with 4 light chain subunits, 2 of which are ELC subunits and the others are RLC subunits, compose the sarcomeric myosin, the main component of thick myosin filament. A number of highly ordered bipolar thick myosin filaments, together with thin actin filaments, in addition to several accessory proteins, comprises the sarcomere, the fundamental contractile unit of striated muscle containing both cardiac and skeletal muscle. Thousands of sarcomeres make up myofibrils, which come together to form myofibers that give rise to mature muscles (Fig. 3). Consequently, the thick filaments slide past the thin filaments orderly, consuming ATP and phosphate and driving the contraction of muscles.

The composition of striated muscle, the schematic drawing of sarcomere and thick myosin heavy chain. The thick myosin filaments and thin actin filaments, together with the binding proteins of myosin are represented in corresponding colors. The area between two adjacent Z lines is called a sarcomere, which consists of a dark band chiefly containing thick myosin filaments (myosin-7, MyBP-C, etc.) anchored to M line in center and two 1/2 light bands only containing part of thin actin filaments anchored to Z line in lateral sides. The M line, which contains myomesin and M-protein et al., is the center of the dark band and the H band is the relatively bright region in dark band as a result of consisting of only thick filaments. The schematic illustration of a thick myosin heavy chain composed of two myosin-7 and 4 light chain subunits (2 ELC and 2 RLC) is indicated in different colors
MYH7 and inherited cardiomyopathies
Sarcomere is the basic unit of the contraction in cardiac muscles. Genetic mutation in genes coding sarcomeric proteins can exactly cause the impairment of the integrity of structure or function of sarcomere. Those disorders can hinder myocardial contraction greatly and are predominant with family clustering, thus classified as inherited cardiomyopathy. The two majors of MYH7-related inherited cardiomyopathy include HCM and DCM. RCM, LVNC, congenital heart defects (CHD), arrhythmia, etc., can be affected as well.
MYH7 and hypertrophic cardiomyopathy
HCM (MIM#192,600), the most common family cardiovascular disease, with a prevalence of at least 1:500 in global population [22, 23], is characterized by significant ventricular hypertrophy, usually asymmetric and frequently involving interventricular septum, with disorganized myocytes and diastolic dysfunction but without elevated loading conditions. The symptoms between inter- and intra-family vary exceedingly from benign to malignant kinds with a considerable risk of heart failure and SCD in younger adults and athletes [24]. Majority of HCM are single-gene hereditary, in an autosomal dominant mode or a de novo mutation fashion displaying family HCM or sporadic HCM, and more than 1500 mutations involved in at least 11 cardiac sarcomeric genes have been identified [25]. The missense variant p.R403Q in MYH7 is the firstly found pathogenic gene related to family HCM [5, 26]. Gradually, other genes, which are mostly sarcomeric protein encoding genes, have been successively found and MYH7 ranks the second frequently pathogenic gene in HCM followed MyBP-C3 [27, 28]. Besides, MYH7 variants are highly related to evolute toward impaired systolic function and end-stage HCM [29]. The disease-relevant missense variants are enriched in S1 and S2 [9], and it is supported by a recent study, which showed the converter domain and residues in myosin mesa, a single flat surface on myosin head, are the common sites of MYH7-associated HCM mutations [30]. Moreover, patients with variants in these enriched regions tend to develop an earlier-onset disease compared with HCM patients carrying mutations in elsewhere of MYH7 [30]. Furthermore, mutants in converter region are associated with adverse prognosis and overlapping phenotypes of other cardiomyopathies [31]. The phenotypic diversity and the relationship between variation and clinical characteristics are described herein after.
MYH7 mutations
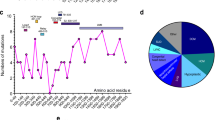
The groupwork enrolling the American College of Medical Genetics and Genomics (ACMG), the Association for Molecular Pathology (AMP), and the College of American Pathologists (CAP), recommended a guideline, giving five classifications of variants transmitted in Mendelian inherited pattern in addition to mitochondrial variants, “pathogenic (P),” “likely pathogenic (LP),” “uncertain significance (VUS),” “likely benign,” and “benign” [32]. This classification framework has been applied universally in MYH7-associated cardiomyopathies. The major known “P” and “LP” mutations of MYH7 associated with HCM are missense variants [33], with a small proportion of nonsense, frameshift, and splice variants which are predicted to produce loss-of-function (LOF) proteins or unstable transcripts. These missense mutations have been reported in numerous cases and recoded in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) with variable degree of confidence (Table 1), and they occur more common in the head and neck than the tail of MYH7 [34]. Despite the knowledge of LOF variants in MYH7 is still incomplete, reports of its potential pathogenicity have sprung up. To date, the ClinVar records a total of 25 LOF variants in MYH7 tabbed as “P” or “LP,” among which 7 are considered to be related with HCM (Table 2). Additionally, two protein-truncating variants in MYH7 were found. c.3562_3574delACTGCCGCGGCCC/p.T1188Cfs*22 is a sporadic 13 base pairs deletion variant in the tail domain and is predicted to produce a truncated protein resulting in dysfunction to dimerize to coiled-coil tail, manifesting both HCM and RCM [35]. And p.Lys1173ArgfsTer41 is a frameshift variant found in 2 HCM carriers and is considered to lead to a truncated protein [36].
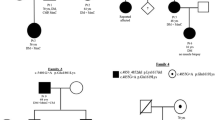
Furthermore, several founder mutations which are not common in MYH7 have been discovered in specific communities and nations, where they account for a sizable proportion of HCM instances. The missense variant p.G584R found in two families in 1993 with a putative Portuguese ancestor is the first mutation believed to have a founder impact [37]. The missense variants p.A797T and p.R403W, accounting for 25% and 5% HCM cases of a subpopulations in Western and Eastern Cape provinces in South Africa, respectively, in addition to p.R249Q, p.R719Q, and p.Glu499Lys accounting for 7.5% altogether, are assumed to exert founder effects, originating from mixed ancestors [38]. The variant p.N1918K is the first founder mutation found in Dutch, causing a variable phenotype, with a relatively early-onset age in period of childhood but with a generally benign outcome [39]. The missense variant p.A797T is a founder mutation, accounting for 25% probands in the panel of South Africa, resulting in a poor outcome and a high risk of SCD [40]. The missense variant p.E894G has been found in 6 unrelated families worldwide [41,42,43,44], a novel missense variant p.R652K, a significant methylation site, was found in Spain [45], and the deletion variant p.K847del found in Manaus [46]. Whether they are founder mutations is still an unsolved issue. Therefore, these founder genes are more frequently sequenced in individual communities and the existence of them would play a key role in saving cost and time in the program of molecular diagnose.
Mutation of MYH7 in pediatric cardiomyopathy
Variants found in childhood are largely associated with early-onset age, complicated manifestations, and high risk of adverse cardiovascular events [47, 48]. A hebetic girl, identified a “P” missense variant p.R719W, experienced combined symptoms, including non-obstructive HCM, RCM, complete left bundle branch block and intermittent third-degree atrioventricular block [49]. A 7-year-old boy with a “P” missense variant p.R453C, presented HCM and WPW with increased likelihood of SCD syndrome [50]. A 9-month female newborn carrying a “LP” missense variant p.Y386C suffered SCD with the diagnose of RCM companied with coronary artery bridging [51]. Moreover, childhood with MYH7 variants-related CHD would manifest more complexly, displaying a couple forms of CHD and other cardiovascular diseases simultaneously, including ventricular septal defect (VSD), Ebstein anomaly (EA), hypoplastic left heart syndrome, Taussig–Bing type double-outlet right ventricle, LVNC, arrhythmias, and so forth [52]. The complex clinical phenotypes and adverse cardiovascular events are associated with the onset of disease in childhood.
Compound MYH7 mutations
Evidence has proved that compound variants with more than one allelic mutation often develop a more complex and severe phenotypes. In comparison to compound heterozygous diallelic mutations with double mutations in two alleles, the clinical manifestation, severity of disease, and prognosis of monoallelic double mutations illustrated in a cis-manner in MYH7 are thought to be better [53, 54]. Cumulative effect is thought to exist when another mutation occurs, presenting a more severe clinical phenotype, such as an earlier-onset age, a higher chance of SCD, and a worse prognosis in MYH7-related HCM, than that generated by each of the single one [55,56,57,58,59,60,61,62,63,64,65,66,67]. A mouse model reconfirmed the addictive effect that mouse with single missense variant p.Val606Met manifested a relatively benign phenotype, but when mouse model was introduced by dual “P” variants p.V606M & p.R453C or p.V606M & p.R719W, it developed a more hypertrophic phenotype [, 67, 68]. In the same family, the members carrying homozygotes variant p.R869G developed a more severe phenotype than it in heterozygous individuals, also indicating a dose-dependent effect [69]. All evidence indicate that compound variations are related with poor prognosis, thus comprehensive and precise genetic sequencing is necessary in patients with complex and severe clinical manifestations.
MYH7-associated pathomechanisms
The molecular pathogenic mechanisms of HCM have been detailed described in a review by Norbert Frey et al. [25], including impaired calcium cycling and calcium sensitivity, increased myocardial fibrosis, disturbed biomechanical stress sensing, and altered cardiac energy homeostasis. MYH7-associated pathomechanisms also have been gradually revealed, but it has not been completely elucidated. Increased actin-activated ATPase activity, higher average force generation and faster actin filament sliding velocity [70, 71], diastolic dysfunction [72] and impaired cardiac relaxation [73], abnormal Ca2+ response [74], cardiac fibrosis and remodeling [75], and a series of differences in gene transcription factors [76] were all observed in mouse models harboring p.R403Q, which is a known “P” variant. As mentioned above, the major “P” and “LP” mutations in MYH7-related HCM are missense variants, which encode stable proteins which are anticipated to be integrated into sarcomeres, disturb normal motor function, and trigger pathologic signals. The motor activity is predicted to be either enhanced [77] or reduced [78] in MYH7-related HCM. Currently, the gain-of-function pathophysiologic mechanism associated missense variants in MYH7 is widely accepted, which proposed the poison peptides produced by MYH7 incorporating into the sarcomere and perturbing the formation of proper and functional sarcomere, leading to elevated contractility and relayed relaxation. As a result, destabilization of interacting-heads motif (IHM) irregulates the balance of increased numbers of myosin in disordered relaxed state (DRX) and decreased number of it in super relaxed state (SRX), consequently excessing mitochondrial quantity, energetic consumption, metabolic stress, and remodeling of cardiomyocytes with hypertrophy [79]. The IHM has already been found in all human muscle myosins, and is definitely a conserved motif, playing an important role in conserving ATP consumption [80]. Myosin in DRX conformation consumes five times energy than it in SRX state [81, 82].
HCM patients with “P” and “LP” variants in MYH7 are sequenced that those variants are significantly enriched in the interacting region with IHM [83]. Two human-induced pluripotent stem cell-cardiomyocyte (iPSC-CM) models harboring missense variant p.P710R and p.R723C, respectively, and both of them are located in converter domain, observed prolonged myosin working time and slowed relaxation, emphasizing the key role of dysregulated SRX state in hypercontractility [84, 85]. The hypercontractility has also been verified in molecular level by using microscale thermophoresis technique based on four variants p.R249Q, p.H251N, p.D382Y, and p.R719W [86]. Mutations in S2 domain are assumed to interfere the normal interaction of this domain with C0-C2 domain of MyBP-C by decreasing the phosphorylation lever of MyBP-C, eventually inhibiting myosin transforming into the SRX state, thereby leading to sarcomeric hypercontractility, impaired full relaxation and increased energy consumption, in a vitro test using three known variants associated with HCM in this region, p.R870H, p.E924K, and p.E930del [87].
Furthermore, it has long been believed that the quantity of toxic peptides acts as a significant risk factor for the severity of disease. However, recently, this hypothesis has gradually rectified, as the variable genotype–phenotype relationship emerged, even exhibiting heterogeneity in a family with the same mutation. The latest studies about the mechanism of genotype–phenotype relationship of different mutations in heterozygous HCM patients highlight the allelic expression imbalance of mutant and wildtype mRNA in cell level, in a stochastic switch on–off, burst-like transcription pattern, resulting in imbalance of proteins eventually, which generate distinct contractile force from cell to cell, leading to different force generation in myofibril level and developing to cardiac hypertrophy in different degrees [88,89,90]. A pair of monozygotic twins with the same “P” variant p.G768R demonstrated different clinical manifestations and tissue characteristics increase the credibility of the mechanism [91]. Another possible intrinsic process involved in the allelic imbalance is presumed that different mutations in coding regions of MYH7 could alter the secondary structure of mRNA, affecting its stability and lifetime and leading to allelic imbalance, which was proposed from an experiment in vivo according to a “P” variant p.R723G [92].
Besides, the variable clinical phenotypes demonstrated by patients from SCD to lifetime survival, and even asymptomatic, with the same variant, indicated diverse additional mechanisms should have taken part in the regulation of allelic imbalance, including environment factors, epigenetic factors, etc. [40, 93,94,95,96,97,98]. The considerable phenotypic heterogeneity of HCM has been explained by a number of moderating factors, including lifestyle [99], gender [100], genetic background [, 94, 101], and so on. Additionally, the discrepancy of level between the protein/gene and their regulatory factors, such as microRNAs, probably plays a conceivable role in diversity of genotype–phenotype [102]. MyBP-H is validated to be a modifier gene in HCM patients with the “P”/“LP” missense variant p.A797T [103]. The genetic polymorphism of renin–angiotensin–aldosterone system is predicted as a modifier factor for the penetrance and severe degree of HCM [104, 105]. The conceivable pathogenetic mechanisms correlated in patients carrying a same variant, suffered from HCM to DCM, and even heart failure, are proposed to include impaired energy generation, dose addictive effect of the poison proteins, environmental factors, the modifier factors of gene, and so on [106, 107]. Virus infection is also considered to deteriorate the condition of a patient with HCM [108]. The factors involved in the diversity of genotype–phenotype needs further exploration for clarifying the mechanism of clinical phenotypic variability.
Several assumptions for the mutations in specific regions of MYH7 have risen. The mutations in the promoter region of the gene are presumed to perturb the formation of triple-stranded G-quadruplex, which is enriched in this region and nears a variety of transcription factors, affecting the process of protein expression [109]. And the mutations located in the promoter domain are predicted to be a hazard to develop HCM, nevertheless no evidence of disease-causing effect for mutations in the introns and 3-prime UTR has been found [110]. Mutations located close to the SH1 or SH2 cysteine are speculated to generate disulfide crosslinking, resulting in non-functional proteins [111]. More researches about the specific regions-related pathogenic mechanism and clinical characteristic are needed.
Diagnosis tools for MYH7-associated HCM
Genetic testing, predominantly whole-exome sequencing (WES), next genetic sequencing (NGS), has been widely used in HCM. For the past 20 years, the estimated prevalence of MYH7-related HCM was about 0.2% in adult [112]. Moreover, with the widespread application of genetic testing, in addition to taking the analysis of family information, sex, specific ethnic, and locational backgrounds into account, the detectable rate in general population is elevated to 1:250 [113]. The most common sequence variants in human are single-nucleotide polymorphisms (SNPs) [114]. Primer extension technique by labeling the dideoxynucleotides (ddNTPs) is proposed an appropriate tool to detect disease-specific SNPs with individual mutation [115]. Recently, a novel platform using ferrocene-labeled oligonucleotides was validated on the basis of primer elongation to electrochemically detect SNPs in MYH7, just depending 10 μL fingerprick blood sample, which facilitates the efficiency of detecting SNP [116]. Those novel detection techniques are important impetus for genetic testing and worth further developing.
Although the category provides convenience for clinical experts to make good judgment and offer useful advice for patients and their families, variants of VUS still cause misjudgment and delayed judgment, leading to delay in treatment. A new PE-MYH7-ACMG tactics which adds the phenotype-enhanced criteria (PE-ACMG) using the HCM Genotype Predictor Score (HGPS) on the basis of the MYH7-specific ACMG guidelines is considered as available criteria for the designation of VUS in order to better use genetic testing, with a considerably reduced VUS of 30 to 16 in a cohort of Australia and 49 to 27 in Mayo Clinic [117]. Moreover, the modified ClinGen’s guideline confirms a professional guidance for clinical experts to give more accurate classification diagnose of the variants than ACMG/AMP framework, with increased variants of 65% contrast to 54% and a decreased VUS number of 30% compared to 42% [118]. While about 181 missense variants have been recorded in ClinVar database classified as “P” or “LP” variants, REVEL score sets a threshold with 0.05 REVEL that warrants a new predicting index for recognizing deleterious variants of MYH7 associated with HCM [119]. Diagnose criteria mentioned above provide a new efficient idea to identify VUS for early treatment.
Researchers also devoted to pursue the difference methods for MYH7 and other sarcomere encoding gene-associated HCM to simplify the process of diagnosis. In spite of different onset age, the degree of cardiac hypertrophy and prognosis in adult, MyBP-C3 and MYH7 variant carriers are observed with no significant difference not only in the echocardiographic parameters reflecting the degree of myocardial deformation, of both right and left ventricle, but also CMR imaging [46, 120,121,122,123,124]. Nevertheless, Radiomic Analysis of Native T1 Mapping Images makes it possible to distinguish these two genotypes with subtle clinical phenotypic difference, and importantly, it could be a potential tool to predict and provide prescient treatment [125]. The level of circulating miR-499a-5p in the plasma of patients with “P” or “LP” MYH7 mutation is considered to be a potential biomarker as well, with higher level than both non-HCM patients and MyBP-C3-related HCM patients [126]. Whether there is significant clinical target difference to distinguish MYH7 and MyBP-C3 needs large-scale study in future.
Clinical manifestations and auxiliary examinations also provide useful information for physicians to evaluate the severity of disease and prognosis of patients, and to offer proper and timely treatment. Patients with the variants located in the enriched mutation region suffered from higher incidence of AF than those with not-enriched region. Moreover, patients with mutations are in higher risk of SCD when they suffered AF at an early age [127]. Pediatric patients with MYH7-related HCM always suffered from severe phenotype and a higher risk of SCD, so an implantable cardioverter defibrillator (ICD) placement is suggested to be a feasible intervention treatment and should be adopted in early stage [128]. Due to the adverse effect of this gene mutations, left ventricular global longitudinal strain, using 3D speckle tracking imaging technique is considered as a valuable parameter to predict adverse cardiovascular event of HCM patients carrying MYH7 mutations [129]. The changes of parameters in ECG appear earlier than that in echocardiography, divulging the importance of using ECG to assist in diagnosing the mutations carriers in early stage and screen condition progression [130, 131]. Two cases of patients carrying variant p.Leu517Arg and p.Arg858Leu, respectively, both suffered from cardiac arrest caused by ventricular fibrillation before demonstrating HCM, and a case with identified “P” / “LP” p.A1379T variant presented AF and atrial fibrosis as the first clinical manifestation, all emphasizing the significance of ECG [132, 133]. All cases above proved that highly efficient use of clinical manifestation and auxiliary inspection could facilitate the diagnosis and treatment.
Besides, researchers identified a promising hallmark for inherited cardiomyopathy, including genetic HCM and DCM, that the shortened telomere is an abnormal feature of CMs, which is illustrated in iPSC-CM models in vitro harboring mutations associated with both HCM and DCM, significantly decreased by 26% and 40%, respectively [134]. The shortening of telomeres is proposed to be essential in developing into the dysfunction of mitochondria, which is the important pathogenic link in HCM and DCM [135]. Although there is no difference between MYH7 and other genetic variants, the important role of telomere is undoubtable and needs to be further elucidated.
MYH7-targeting therapy for HCM
Therapies according HCM have been systematically reviewed by Ali J. Marian, M.D et al. [136]. Nowadays, many novel ideas about MYH7 gene or base targeting therapy for HCM patients have been put on. Experimental data from a cell and mice model indicated that YTHDF2, which is a m6A reader protein, plays a protective role in the regulation of cardiac hypertrophy and heart failure, expression upregulating in a self-regulation mechanism, by interacting with m6A site of MYH7 mRNA via its YTH domain, and promoting its degradation to alleviate cardiomyocyte hypertrophy [137]. As the switch from fetal dominant MYH7 phenotype to MYH6 is considered to be completed during the maturation period, the MYH7 gene targeting therapy to delete the re-expressed MYH7 is supposed to be effective after its transformation [138], and it is proved available by an iPSC model harboring MYH7/MYH6 mutations [58]. An mice experiment both in vitro and in vivo provided a feasible targeting therapeutic option, selective knocking down of rs7157716, which is a common SNP with high heterozygosity using antisense oligonucleotides technique [139]. And another group identified it in a further human-cell model, which also studied the short hairpin RNA method in this hiPSC-CM model [140]. Moreover, cationic porphyrins are speculated to destabilize the G-quadruplex as said before, by binding to the structure, which proposes a sight in drug designation [141]. The telomere shortening as previously stated is also a challenging drug target. For a fetus suffered from high risk of familial HCM accompanied by diastolic dysfunction, intrauterine treatment using beta-receptor blockers is recommended as an optional treatment [142]. Although more and more new notions emerged and have been validated to be feasible and effective, MYH7 gene therapy has not been used clinically. Whether those therapies are practically available needs further research.
Models for studying MYH7-associated HCM
A number of animal models have been used for studying the pathogenic mechanism of MYH7-associated HCM. The first animal model is a transgenic mouse model according to a missense variant, p. R403Q, the first found mutation related to HCM, also the most common used [143]. For a long time, researchers preferred mice model to imitate human HCM cases and investigate the possible pathogenic mechanisms. However, a significant limitation has also merged that human express slow beta-myosin heavy chain but mouse models encode fast alpha-myosin, which do not result in the typical phenotype found in human with HCM [144]. Therefore, several alternative models have been found. Transgenic rabbit carrying the variant p.R403Q precedes the understanding of molecular mechanism in human HCM [145]. The genetic editing pig model with the knock-in orthologous “P” point variant p.R723G based on somatic cell nuclear transfer technique provided a more suitable large animal model to investigate the pathogenic mechanism of human HCM than mouse model, with more similar cardiovascular physiology to human [146]. Another group proved that zebrafish is also an alternative animal model for the study of cardiomyopathy caused by the mutant of MYH7, and confirmed that the inhibition of mTOR and MAPK signal pathway had a therapeutic effect using the zebrafish homolog of human MYH7-based cardiomyopathy model [147]. A human orthologous “VUS” variant p.E1883K has been found in a cat suffered from HCM, making it possible that the cat with HCM is an optional model as well [148]. Those new animal models are all alternative to study the molecular pathogenic mechanism and therapeutic tools in future.
Meanwhile, a number of iPSC-CM HCM models, harboring a number of well-known HCM-causing heterozygote variants, including “P” p.R403L, p.R719Q, p.Ala355Thr, p.R723G, p.R663H, p.E1356K, p.R1712Q, p.R723C, and “VUS” p.M659I and p.E1462K, using CRISPR/Cas-9 editing protocol, could be an useful tool for the future study of the molecular mechanism of HCM [149,150,151,152,153,154,155,156,157,158,159]. Also, using the same editing tool, a homozygous knockout human embryonic stem cell (hESC) line of MYH7 gene has been produced [160]. Genome-editing technique combining with iPSC-CMs provides a practical platform to guide the understanding of mechanism and evaluation of precision drug. Isogenic genome-edited human pluripotent stem cell-cardiomyocytes (hPSC-CMs) using CRISPR/Cas-9 editing protocol, produced 11 isogenic variants centered on “P” p.R453C and comprehensively phenocopied the features of adult hypertrophic cardiomyocytes [161]. Ioannis Karakikes et al. produced a transcription activator-like effector nucleases (TALEN)-instructed knocking out iPSC-CMs line [162]. Those iPSC lines models provide an available, valuable, and validated opportunity for studying the pathogenic mechanisms and therapeutic tools of HCM in vitro in future.
MYH7 and other cardiomyopathies
Increasing evidence has revealed that MYH7 could also lead to many other kinds of cardiomyopathies except HCM, including DCM, RCM, LVNC, arrhythmogenic cardiomyopathy, and other types of CHD. Majority of them are identified in family cases, also predominantly in an autosomal dominant transmitted Mendel pattern, and also involving autosomal recessive, X-linked, and mitochondrial inherited mode [163,164,165,166].
DCM and RCM are two well-known cardiomyopathies. DCM (MIM#613,426) is a kind of relatively rare cardiomyopathy, about 1:2500 [167], with the characteristics of ventricular enlargement predominantly in left ventricular with systolic dysfunction [168]. MYH7 gene is ranked the third common pathogenic gene of idiopathic DCM [169] and most of them are non-truncating variants with a high penetrance in family, with a relatively high proportion of pediatric patients [170,171,172]. Fifty-nine “P” and “LP” missense variants are recorded in ClinVar with at least one submitter, among which 6 are with high confidence (Table 1), in addition to 3 LOF variants (Table 2). DCM is the leading reason for congestive heart failure and patients are at high risk of SCD [173, 174], predominantly in working people with early age [175]. Unlike HCM, MYH7 mutations associated with DCM are scattered throughout entire length of the gene without a significant enriched region, like IHM interacting residues. The biological mechanism in DCM is opposite to HCM, with a decreased sarcomeric contractility as a result of impaired ATPase activity and reduced velocity sliding along actin filaments [176], ultimately triggering the process of remodeling, which is tested in mouse models by knocking in the “P” missense variant p.S532P in actin binding domain and “P” p.F764L in converter domain which are recognized pathogenic MYH7 mutation in human DCM [177]. Same with HCM, digenetic mutations involving a variant in MYH7 gene and the other gene predispose to a severe phenotype of DCM, leading to an addictive effect [178,179,180]. Furthermore, the variable manifestations of DCM patients with a causative-variant in MYH7 are also presumed to attribute to environmental and genetic modifiers [166]. RCM, least common cardiomyopathy with unknown prevalence [181, 182], with the estimated 5% in pediatric cardiomyopathies [183, 184], is characterized by diastolic dysfunction but without impaired systolic function, in the condition of stiffed ventricular walls but not necessary for thickening, leading to lower the appropriate filling of ventricular [185, 186]. Idiopathic RCM is predominantly an inherited ailment [187]. Most of mutations in MYH7-related RCM are inherited in an autosomal dominant pattern and are missense variants [185]. There are 2 “P” and “LP” missense variants in ClinVar with more than one submitter, and 1 of them has high confidence with a number of references (Table 1). Unexpectedly, RCM is related with the worst prognosis among cardiomyopathy [188].
LVNC (MIM#613,426), resulting of the incomplete or arrested development and compaction of human myocardium during the 5th to 8th week of embryonic development [181], is the third common genetic cardiomyopathy, characterized by the presence of numerous thickened trabeculations and deep recesses, prominent in the left ventricular with a spongy like, with a risk of developing to HCM and DCM [189,190,191]. MYH7 is one of the pathogenic gene for LVNC [192]. Unlike HCM and DCM, except point mutation, truncating variants in MYH7 are thought to be pathogenic in LVNC [193], with a relatively high proportion of pediatric patients [194]. In addition to 7 “P” and “LP” missense variants (Table 1), 4 “LP” LOF mutants (Table 2) are documented in ClinVar. Patients in LVNC with MYH7 mutations are prone to have a low risk of adverse cardiovascular events [195], and the proportion of asymptomatic individuals accounts for a significant ratio, about 8% subjects meeting the criteria for LVNC in high-trained athletes from UK and France [196]. The risk of adverse cardiovascular event is also considered lower in LVNC with MYH7 mutations compared with patients carrying other gene variants [197]. The potential mechanism for the development of irregular trabeculations and deepened recesses is interpreted by the notion that variants in MYH7 may increase apical–basal polarization, resulting in the delamination of compact layer cardiomyocytes [198]. Moreover, the too-early isoform switch from MYH7 to MYH6 which are programed to be finished at birth may trigger pathological remodeling and abnormal sarcomere assembly, leading to impaired trabeculation and compaction of myocardium, possibly accompanied by specific modulator of gene expression, such as G-quadruplex resolvase RNA helicase associated with AU-rich element [199]. Like HCM, coexistence of digenic mutations is prone to develop an early-onset age, severe phenotype, and poor outcome [200]. Although LVNC is often exist individually, it sometimes coexists with several CHD [191], most common one of which is EA [201], a relatively rare kind of CHD, with a prevalence of 1:200,000 in births [202], characterized by a lower position of tricuspid and malformed leaflets [203], leading to an enlarged right atrium, tricuspid regurgitation, and eventually heart failure [204, 205]. Moreover, variants in MYH7 are increasingly identified as the disease-causative of the combination of LVNC and EA, sometimes incorporated with other CHD, also in a dominant autosomal pattern, and patients presenting variable manifestations too, significantly from asymptomatic, mild symptomatic to fetal [65, 206, 207]. Many other types of CHD and malformations also have been found in combination with LVNC, including bicuspid aortic valve, single umbilical artery [208, 209].
Besides, limited cases of rare CHD independently associated with MYH7 have been reported, such as double-chambered right ventricle and double-chambered left ventricle [205, 210]. Additionally, a single variant also could develop to a complicated clinical phenotype, such as a young female identified with a novel missense “VUS” variant p.F252S manifested both RCM and left ventricular hypertrophy [211]. The phenotypes of patients with those kinds of cardiomyopathies present variably and the conceivable mechanism of genotype–phenotype is still unclear [209, 212]. Meanwhile, pediatric patients suffered from a more severe degree of cardiomyopathies with more severe malformations or more coexist cardiomyopathies or poor prognosis [49, 209, 213]. Interestingly, MYH7 variants in pediatric patients with both DCM and LVNC are totally located in residues among 1 to 600 found in a large pediatric cohort [214]. A few cases of fetus with LVNC identified in the third trimester by using prenatal ultrasound technique, carrying mutations in MYH7, elucidate the importance of seeking the pathogenic gene and investigating the family disease history of suspicious fetus [215, 216].
Although the application of experimental models in these cardiomyopathies is relatively countless, human iPSC models and animal models also extended to this field, including the iPSC model according to systolic cardiomyopathy derived from “P”/“LP” variant p.E848G, and zebrafish model which is considered an available animal platform to investigate the pathogenic molecular mechanism of LVNC [198, 217]. Zebrafish has been found corresponding homologues according to 96% genes associated with DCM, therefore, zebrafish is a promising animal model for future research of cardiomyopathy [218]. To find more experimental models is necessary for better understanding the pathogenic mechanism and clinical application.
MYH7 and skeletal muscle myopathy
MYH7 and Laing distal myopathy
LDM (MIM#160,500), a predominantly autosomal dominant condition, as a result of heterozygous mutations enriched in C-terminus of MYH7 [219], which affects the anterior compartment of the legs, with the characteristics of progressively progressing distal weakness, results in the recognizable “hanging big toe” sign [220]. It is well recognized that the typical LDM phenotype exhibits an early-onset age but could also range from infancy to adult up to 45 years old [220,221,222,223], development of the foot dorsiflexors and big toe extensors, then a weakening of the proximal upper and lower limbs, cervical flexor muscles and finger extensor muscles, even respiratory and cardiovascular system diseases, moreover, with substantial variability in clinical presentations and frequent histological morphologic changes in different instances [220, 224,225,226,227]. However, the footdrop is valid not a specific feature, the weakening of finger extension is thought to be more specific [226]. CFTD, cores and minicores, dystrophic alterations, and moderate unspecified abnormalities are only a few examples of the variety in muscle histology in LDM [, 228, 229].
Often these LDM patients possess mutations in the mid-rod domain of MYH7 gene within 32–36 exons including p.E1508del, p.R1500P, p.Lys1617del, p.Ala1663Pro, p.Leu1706Pro, and p.Lys1729del, etc. [219, 224, 227], which interfere the normal process of tail forming coiled-coil, whereas in a few limited cases, the globular head region has also been linked to the condition, including p.Tre441Met, p.R783P, and p.V606M [230,231,232]. The “P” and “LP” missense variants (Table 3) and LOF variants (Table 2) are recorded in ClinVar. The phenotype spectrum is also significantly variable, and the case in point is deletion variant p.E1508del [224], which is a susceptible residue [233], and missense variant p. Leu1551Pro, located in the exon 34 of the MYH7 [234]. Furthermore, missense variant p.L1453P, located in exon 32, was found related to brain white matter lesions on imaging, but whether the mutation in MYH7 gene is to blame for the neurologic abnormalities requires further research [235]. Moreover, patients with LDM, having the fatty atrophies and substitutions of the proximal or paraspinal muscles could be more severe and illness progression more quickly compared with that of the lower thigh muscles [236]. All cases above broadened the phenotype spectrum of MYH7-related LDM.
There are also founder-effect mutations which have been found in several regions. Geographically restricted to the South of Spain, the missense “LP” variant p.R1560P was confirmed to be a novel founder mutation linked to LDM [237]. The deletion variant p.K1729del was assumed to be a founder mutation in Safor of Spanish, being brought into the population around the start of the seventeenth century, having an origination of Italian, according to the mathematical method [238].
A fly model harboring the known LDM variant p.K1729del imitated the morphological feature and impaired muscle function as seen in human LDM, using CRISPR/Cas-9 genome engineering protocol, indicated that the severity of disease is explained by the number of mutated alleles, illustrating a dose-dependent effect, and increasing the expression of protein Abba/Thin which are instrumental in maintaining the integrity of sarcomere could alleviate the phenotype [239]. It provides a potential treatment tool.
MYH7 and myosin storage myopathy
MSM (MIM#608358) was first listed in 2003 [240], the first skeletal muscle myopathy found to be caused by MYH7 gene, previously named as hyaline body myopathy due to sluggish myosin hyaline body accumulation seen in type 1 muscle fibers in subsarcolemmal tissues displayed in histopathologic features, perturbing the assembly of thick filaments. Clinical features include mainly early-onset age predominantly in infancy and childhood, prominent axial and proximal weakening, spinal stiffness, severe scoliosis, accompanied by or without respiratory and cardiac involvement. Variants of MYH7-related MSM are mostly in an autosomal dominant inherence pattern with mutations in the distal rod region corresponding to 37–40 exons of MYH7 gene, including missense variant p.Arg1845Trp, p.His1904Leu, p.Leu1793Pro, p.Glu1883Lys [240,241,242,243,244,245], in-frame deletion variant p.K1784del [246] and a missense variant p.X1936WfsX32 changing the TAG to tryptophan (W), which leads to the elongation of the C-terminus [244]. Additionally, countless occurrences of recessive inheritance have been documented, such as homozygous variant p.R1712W, heterozygous variant of truncating p.Gln1567*, and missense p.E1555G [247]. The clinical manifestations of MYH7-related MSM are incredibly varied from asymptomatic to severe weakness [241, 248,249,250,251,252]. The mechanism of pathology could be interpreted by mutations perturbing the process of proteins to assemble to proper, stable, and functional thick filaments, corresponding to variants enriched in distal rod [253, 254]. Different mutations interfere distinct steps in assembling process, including two α-helices properly folded into coiled-coils, then assembling to bundles of coiled-coils, and ultimately into thick filaments. An uncommon missense variant p.Ile457Arg is located in the head domain, performing pronounced thigh weakness as well as respiratory and cardiac impairment, indicating the correlation of variants location and its functional region [255]. The exact mechanism needs further exploration.
MYH7 and congenital myopathy with fiber-type disproportion
CFTD (MIM#255,310), is an uncommon myopathy, defined by the characteristic pattern with a predominance of slowly contracting type I fibers in skeletal muscles seen by histological analysis [256]. MYH7 is one of the pathogenic genes of CFTD, inherited in autosomal dominant, recessive or X-linked forms. Also, CFTD presents a variable range of clinical manifestations [257]. Moreover, the stop-loss variant p.X1936WfsX32 linked to CFTD is speculated to eventually develop to MSM later, as a result of the absence of stop signal and producing an elongated protein, leading to disturb the degradation of the protein, protein buildup, and accumulation in sarcomere, which caused the weakness of axial muscles, prominent neck muscle [244, 258].
MYH7 and other myopathies
MYH7 mutations were also identified in many other forms of myopathies, including usually scapuloperoneal myopathy, axial stiffness, drop-head syndrome, congenital core myopathy, and asymptotic hyperCKemia accompanied by or without hyaline bodies [259,260,261,262]. The variable phenotypes and muscle biopsy findings are postulated to have a relationship with Ca2+ regulatory process by interfering the charge of residuals, leading to assembly destabilization or structural alterations, eventually perturbing the normal structure and stability of myosin [227].
There are also a variety of animal models used to study the molecular pathogenic mechanism of human myopathy related with MYH7 gene, including nematode model with the ortholog (unc-54) of human MYH7, pig model carrying an in-frame insertion variant [263, 264].
MYH7 with clinical phenotypic diversity
The clinical phenotype of patients with mutations in MYH7 gene significantly vary from person to person, even with the same variant within a family, presenting either cardiomyopathy or skeletal muscle myopathy independently, also showing an overlapping complex form of both of them, possibly accompanied by other complications [265], such as nonsense “VUS” variant p.Q1916* [266], deletion variant p.Glu1508del [267], and missense variant p.E1801K [268], p.E1856K [269], p.R1820W [270], p.R783P [231], p.V606M [232], p.R249Q [271], p.Arg1820Gln [272], p.Glu1883Lys [245], p.Leu1467Val, p.Arg1588Pro [273], and so on. The existence of clinical phenotypic diversity in MYH7-related diseases adds difficulty to clinical practice, so it is very important to find out the regular pattern between mutation and phenotype, which needs more exploration in future.
MYH7 with tumorigenesis diseases
Additionally, MYH7 is also found in many tumorigenesis diseases. MYH7 has been found highly expressed in lung cancer, especially in cigarette smoking-associated lung adenocarcinoma patients, 12% of which experiencing MYH7 mutation. High expression of MYH7 is also considered to be related to cancer progression and poor prognosis, which indicated that MYH7 is a potential biomarker for smoking-related lung cancer and a promising targeting-therapy point [274]. MYH7 is ranked in the top ten hub gene of prostate cancer as well [275]. MYH7 is also found enriched in the biological processes of oral cancer [276]. A large-scale research held in China identified MYH7 mutations in Epstein–Barr virus-associated intrahepatic cholangiocarcinoma [277]. Both gene mutations and changes in expression can affect the occurrence and development of cancer. All the above results show the possible influence of MYH7 on tumorigenesis.
Conclusion
MYH7 is one of the most important sarcomere protein encoding genes, and its variants are disease-causative for a series of cardiomyopathy and skeletal myopathy, which sometimes exhibit clinical overlap. MYH7 can also affect a limited number of tumorigenesis diseases from expression change to base change. Better understanding of the structure of MYH7 and the functional regions of myosin-7 can improve the insight into its related disorders. Genetic testing provides and accelerates the accurate and early diagnosis of inherited diseases, especially in families. Deeper comprehension to the pathogenic mechanisms of cardiomyopathy and/or skeletal myopathy as well as the investigation of variable genotype–phenotype is necessary. It is essential to extend application of iPSC models and animal models in all forms of MYH7-associated diseases and establish more suitable animal models to study the disease mechanisms and morphological performances. There are still several urgent questions to be resolved. More precise and cost-effective sequencing technology are needed to distinguish the VUS and missing variants, and re-evaluation is necessary probably, in addition to more sophisticated management of pediatric patients. In conclusion, MYH7 is a potential biomarker to predict disease. Further work in the development of base- and gene-specific therapies are required for the pinpoint management of patients.
Data availability
Enquiries about data availability should be directed to the authors.
References
Oldfors A (2007) Hereditary myosin myopathies. Neuromuscul Disord 17:355–367. https://doi.org/10.1016/j.nmd.2007.02.008
Buvoli M, Hamady M, Leinwand LA, Knight R (2008) Bioinformatics assessment of β-myosin mutations reveals myosin’s high sensitivity to mutations. Trends Cardiovasc Med 18:141–149. https://doi.org/10.1016/j.tcm.2008.04.001
Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg H-P (1990) The complete sequence of the human β-myosin heavy chain gene and a comparative analysis of its product. Genomics 8:194–206. https://doi.org/10.1016/0888-7543(90)90272-v
Yamauchi-Takihara K, Sole MJ, Liew J, Ing D, Liew C-C (1989) Characterization of human cardiac myosin heavy chain genes. Proc Natl Acad Sci 86:3504–3508. https://doi.org/10.1073/pnas.86.10.3504
Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg H-P, McKenna W, Seidman CE, Seidman J (1990) A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell 62:999–1006. https://doi.org/10.1016/0092-8674(90)90274-i
Geeves MA, Holmes KC (1999) Structural mechanism of muscle contraction. Annu Rev Biochem 68:687–728. https://doi.org/10.1146/annurev.biochem.68.1.687
Mornet D, Pantel P, Audemard E, Kassab R (1979) The limited tryptic cleavage of chymotryptic S-1: an approach to the characterization of the actin site in myosin heads. Biochem Biophys Res Commun 89:925–932. https://doi.org/10.1016/0006-291x(79)91867-9
Rayment I, Rypniewski WR, Schmidt-Bäse K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261:50–58. https://doi.org/10.1126/science.8316857
Colegrave M, Peckham M (2014) Structural implications of β-cardiac myosin heavy chain mutations in human disease. Anat Rec 297:1670–1680. https://doi.org/10.1002/ar.22973
Mornet D, Bertrand R, Pantel P, Audemard E, Kassab R (1981) Proteolytic approach to structure and function of actin recognition site in myosin heads. Biochemistry 20:2110–2120. https://doi.org/10.1021/bi00511a007
Kiani FA, Fischer S (2016) ATP-dependent interplay between local and global conformational changes in the myosin motor. Cytoskeleton 73:643–651. https://doi.org/10.1002/cm.21333
Chaussepied P, Morales M (1988) Modifying preselected sites on proteins: the stretch of residues 633–642 of the myosin heavy chain is part of the actin-binding site. Proc Natl Acad Sci 85:7471–7475. https://doi.org/10.1073/pnas.85.20.7471
Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC (1993) The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J Cell Biol 123:619–626. https://doi.org/10.1083/jcb.123.3.619
Moos C, Offer G, Starr R, Bennett P (1975) Interaction of C-protein with myosin, myosin rod and light meromyosin. J Mol Biol 97:1–9. https://doi.org/10.1016/s0022-2836(75)80017-9
Obermann WM, Gautel M, Weber K, Fürst DO (1997) Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J 16:211–220. https://doi.org/10.1093/emboj/16.2.211
Obermann WM, van der Ven PF, Steiner F, Weber K, Furst DO (1998) Mapping of a myosin-binding domain and a regulatory phosphorylation site in M-protein, a structural protein of the sarcomeric M band. Mol Biol Cell 9:829–840. https://doi.org/10.1091/mbc.9.4.829
Muhle-Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H (2001) Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin S1-a possible role in the frank-starling mechanism of the heart. J Mol Biol 313:431–447. https://doi.org/10.1006/jmbi.2001.5017
Wang S-M, Jeng C-J, Sun M-C (1992) Studies on the interaction between titin and myosin. Histology and histopathology.
Kontrogianni-Konstantopoulos A, Huang S-C, Benz EJ Jr (2000) A nonerythroid isoform of protein 4.1 R interacts with components of the contractile apparatus in skeletal myofibers. Mol Biol Cell 11:3805–3817. https://doi.org/10.1091/mbc.11.11.3805
Fielitz J, Kim M-S, Shelton JM, Latif S, Spencer JA, Glass DJ, Richardson JA, Bassel-Duby R, Olson EN (2007) Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Investig 117:2486–2495. https://doi.org/10.1172/JCI32827
Bujalowski PJ, Nicholls P, Oberhauser AF (2014) UNC-45B chaperone: the role of its domains in the interaction with the myosin motor domain. Biophys J 107:654–661. https://doi.org/10.1016/j.bpj.2014.05.045
Ashrafian H, Watkins H (2007) Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications: Cardiomyopathies: Therapeutics based on molecular phenotype. J Am Coll Cardiol 49:1251–1264. https://doi.org/10.1016/j.jacc.2006.10.073
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults Echocardiographic analysis of 4111 subjects in the CARDIAsStudy. coronary artery risk development in (Young) adults. Circulation 92:785–789. https://doi.org/10.1161/01.cir.92.4.785
Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO (1996) Sudden death in young competitive athletes: clinical, demographic, and pathological profiles. JAMA 276:199–204
Frey N, Luedde M, Katus HA (2012) Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol 9:91–100. https://doi.org/10.1038/nrcardio.2011.159
Rosenzweig A, Watkins H, Hwang D-S, Miri M, McKenna W, Traill TA, Seidman J, Seidman CE (1991) Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. N Engl J Med 325:1753–1760. https://doi.org/10.1056/NEJM199112193252501
Biddinger KJ, Jurgens SJ, Maamari D, Gaziano L, Choi SH, Morrill VN, Halford JL, Khera AV, Lubitz SA, Ellinor PT (2022) Rare and Common Genetic Variation Underlying the Risk of Hypertrophic Cardiomyopathy in a National Biobank. JAMA cardiology. https://doi.org/10.1001/jamacardio.2022.1061
Lafreniere-Roula M, Bolkier Y, Zahavich L, Mathew J, George K, Wilson J, Stephenson EA, Benson LN, Manlhiot C, Mital S (2019) Family screening for hypertrophic cardiomyopathy: is it time to change practice guidelines? Eur Heart J 40:3672–3681. https://doi.org/10.1093/eurheartj/ehz396
Lopes LR, Brito D, Belo A, Cardim N (2019) Genetic characterization and genotype-phenotype associations in a large cohort of patients with hypertrophic cardiomyopathy–An ancillary study of the Portuguese registry of hypertrophic cardiomyopathy. Int J Cardiol 278:173–179. https://doi.org/10.1016/j.ijcard.2018.12.012
Homburger JR, Green EM, Caleshu C, Sunitha MS, Taylor RE, Ruppel KM, Metpally RPR, Colan SD, Michels M, Day SM (2016) Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc Natl Acad Sci 113:6701–6706. https://doi.org/10.1073/pnas.1606950113
García-Giustiniani D, Arad M, Ortíz-Genga M, Barriales-Villa R, Fernández X, Rodríguez-García I, Mazzanti A, Veira E, Maneiro E, Rebolo P (2015) Phenotype and prognostic correlations of the converter region mutations affecting the β myosin heavy chain. Heart 101:1047–1053. https://doi.org/10.1136/heartjnl-2014-307205
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–423. https://doi.org/10.1038/gim.2015.30
Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E (2018) Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med 20:351–359. https://doi.org/10.1038/gim.2017.218
Walsh R, Rutland C, Thomas R, Loughna S (2010) Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 115:49–60. https://doi.org/10.1159/000252808
Fan L-L, Guo S, Jin J-Y, He Z-J, Zhao S-P, Xiang R, Zhao W (2019) Whole exome sequencing identified a 13 base pair MYH7 deletion-mutation in a patient with restrictive cardiomyopathy and left ventricle hypertrophy. Ann Clin Lab Sci 49:838–840
Ramensky VE, Ershova AI, Zaicenoka M, Kiseleva AV, Zharikova AA, Vyatkin YV, Sotnikova EA, Efimova IA, Divashuk MG, Kurilova OV (2021) Targeted sequencing of 242 clinically important genes in the Russian population from the Ivanovo region. Front Genet 12:709419. https://doi.org/10.3389/fgene.2021.709419
Watkins H, Thierfelder L, Anan R, Jarcho J, Matsumori A, McKenna W, Seidman J, Seidman C (1993) Independent origin of identical beta cardiac myosin heavy-chain mutations in hypertrophic cardiomyopathy. Am J Hum Genet 53:1180
Moolman-Smook JC, De Lange WJ, Bruwer EC, Brink PA, Corfield VA (1999) The origins of hypertrophic cardiomyopathy–causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am J Hum Genet 65:1308–1320. https://doi.org/10.1086/302623
Van der Linde I, Hiemstra Y, Bökenkamp R, van Mil A, Breuning M, Ruivenkamp C, Ten Broeke SW, Veldkamp R, van Waning J, van Slegtenhorst M (2017) A Dutch MYH7 founder mutation, p. (Asn1918Lys), is associated with early onset cardiomyopathy and congenital heart defects. Neth Hear J 25:675–681. https://doi.org/10.1007/s12471-017-1037-5
Moolman-Smook J, De Lange W, Corfield V, Brink P (2000) Expression of HCM causing mutations: lessons learnt from genotype-phenotype studies of the South African founder MYH7A797T mutation. J Med Genet 37:951–956. https://doi.org/10.1136/jmg.37.12.951
Ross SB, Bagnall RD, Ingles J, Van Tintelen JP, Semsarian C (2017) Burden of recurrent and ancestral mutations in families with hypertrophic cardiomyopathy. Circulation 10:e001671. https://doi.org/10.1161/CIRCGENETICS.116.001671
Kapplinger JD, Landstrom AP, Bos JM, Salisbury BA, Callis TE, Ackerman MJ (2014) Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J Cardiovasc Transl Res 7:347–361. https://doi.org/10.1007/s12265-014-9542-z
Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, Jenkins S, McKenna W, Plagnol V, Elliott PM (2013) Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet 50:228–239. https://doi.org/10.1136/jmedgenet-2012-101270
Gruner C, Care M, Siminovitch K, Moravsky G, Wigle ED, Woo A, Rakowski H (2011) Sarcomere protein gene mutations in patients with apical hypertrophic cardiomyopathy. Circulation 4:288–295. https://doi.org/10.1161/CIRCGENETICS.110.958835
Antoniutti G, Caimi-Martinez FG, Álvarez-Rubio J, Morlanes-Gracia P, Pons-Llinares J, Rodríguez-Picón B, Fortuny-Frau E, Torres-Juan L, Heine-Suner D, Ripoll-Vera T (2022) Genotype–phenotype correlation in hypertrophic cardiomyopathy: new variant p. Arg652Lys in MYH7. Genes 13:320. https://doi.org/10.3390/genes13020320
Marsiglia JDC, Credidio FL, De Oliveira TGM, Reis RF, de Oliveira AM, De Araujo AQ, Pedrosa RP, Barbosa-Ferreira JMB, Mady C, Krieger JE (2013) Screening of MYH7, MYBPC3, and TNNT2 genes in Brazilian patients with hypertrophic cardiomyopathy. Am Heart J 166:775–782. https://doi.org/10.1016/j.ahj.2013.07.029
Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, Alastalo T-P, Koskenvuo J, Hiippala A, Jokinen E (2018) Genetic basis of severe childhood-onset cardiomyopathies. J Am Coll Cardiol 72:2324–2338. https://doi.org/10.1016/j.jacc.2018.08.2171
Mathew J, Zahavich L, Lafreniere-Roula M, Wilson J, George K, Benson L, Bowdin S, Mital S (2018) Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin Genet 93:310–319. https://doi.org/10.1111/cge.13157
Zhang L, Cheng X, Chen J, Zhou M, Qian T, Zhang Z, Yin J, Zhang H, Dai G, Qin Y (2020) Left bundle pacing for left bundle branch block and intermittent third-degree atrioventricular block in a MYH7 mutation-related hypertrophic cardiomyopathy with restrictive phenotype in a child. Front Pediatr 8:312. https://doi.org/10.3389/fped.2020.00312
Bobkowski W, Sobieszczańska M, Turska-Kmieć A, Nowak A, Jagielski J, Gonerska M, Lebioda A, Siwińska A (2007) Mutation of theMYH7 gene in a child with hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome. J Appl Genet 48:185–188. https://doi.org/10.1007/BF03194677
Greenway SC, Wilson GJ, Wilson J, George K, Kantor PF (2012) Sudden death in an infant with angina, restrictive cardiomyopathy, and coronary artery bridging: an unusual phenotype for a β-myosin heavy chain (MYH7) sarcomeric protein mutation. Circulation 5:e92–e93. https://doi.org/10.1161/CIRCHEARTFAILURE.112.969303
Ritter A, Leonard J, Gray C, Izumi K, Levinson K, Nair DR, O’Connor M, Rossano J, Shankar V, Chowns J (2022) MYH7 variants cause complex congenital heart disease. Am J Med Genet A. https://doi.org/10.1002/ajmg.a.62766
Wang B, Wang J, Wang LF, Yang F, Xu L, Li WX, He Y, Zuo L, Yang QL, Shao H (2019) Genetic analysis of monoallelic double MYH7 mutations responsible for familial hypertrophic cardiomyopathy. Mol Med Rep 20:5229–5238. https://doi.org/10.3892/mmr.2019.10754
Zhang M, Sun X, Wu G, Wang D, Wang L, Zhang C, Zou Y, Wang J, Song L (2022) Effect of cis-compound variants in MYH7 on hypertrophic cardiomyopathy with a mild phenotype. Am J Cardiol 167:104–110. https://doi.org/10.1016/j.amjcard.2021.11.049
Wang B, Guo R-Q, Wang J, Yang F, Zuo L, Liu Y, Shao H, Ju Y, Sun C, Xu L (2017) The cumulative effects of the MYH7-V878A and CACNA1C-A1594V mutations in a Chinese family with hypertrophic cardiomyopathy. Cardiology 138:228–237. https://doi.org/10.1159/000478900
Wang L, Zuo L, Hu J, Shao H, Lei C, Qi W, Liu Y, Miao Y, Ma X, Huang CL-H (2016) Dual LQT1 and HCM phenotypes associated with tetrad heterozygous mutations in KCNQ1, MYH7, MYLK2, and TMEM70 genes in a three-generation Chinese family. Europace 18:602–609. https://doi.org/10.1093/europace/euv043
Rodríguez-López R, García-Planells J, Martínez-Matilla M, Pérez-García C, García Banacloy A, Guzmán Luján C, ZomeñoAlcalá O, Belchi Navarro J, Martínez-León J, Salguero-Bodes R (2022) Homozygous Pro1066Arg MYBPC3 pathogenic variant in a 26Mb region of homozygosity associated with severe hypertrophic cardiomyopathy in a patient of an apparent non-consanguineous family. Life 12:1035. https://doi.org/10.3390/life12071035
Hsieh J, Becklin KL, Givens S, Komosa ER, Lloréns JEA, Moriarity BS, Webber BR, Singh BN, Ogle BM (2022) Myosin heavy chain converter domain mutations drive early-stage changes in extracellular matrix dynamics in hypertrophic cardiomyopathy. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2022.894635
Richard P, Isnard R, Carrier L, Dubourg O, Donatien Y, Mathieu B, Bonne G, Gary F, Charron P, Hagege A (1999) Double heterozygosity for mutations in the β-myosin heavy chain and in the cardiac myosin binding protein C genes in a family with hypertrophic cardiomyopathy. J Med Genet 36:542–545
Suzuki T, Saito K, Yoshikawa T, Hirono K, Hata Y, Nishida N, Yasuda K, Nagashima M (2022) A double heterozygous variant in MYH6 and MYH7 associated with hypertrophic cardiomyopathy in a Japanese family. J Cardiol Cases 25:213–217. https://doi.org/10.1016/j.jccase.2021.09.011
Hougs L, Havndrup O, Bundgaard H, Køber L, Vuust J, Larsen LA, Christiansen M, Andersen PS (2005) One third of Danish hypertrophic cardiomyopathy patients have mutations in MYH7 rod region. Eur J Hum Genet 13:161–165. https://doi.org/10.1038/sj.ejhg.5201310
Selvi Rani D, Nallari P, Dhandapany PS, Rani J, Meraj K, Ganesan M, Narasimhan C, Thangaraj K (2015) Coexistence of digenic mutations in both thin (TPM1) and thick (MYH7) filaments of sarcomeric genes leads to severe hypertrophic cardiomyopathy in a South Indian FHCM. DNA Cell Biol 34:350–359. https://doi.org/10.1089/dna.2014.2650
Kolokotronis K, Kühnisch J, Klopocki E, Dartsch J, Rost S, Huculak C, Mearini G, Störk S, Carrier L, Klaassen S (2019) Biallelic mutation in MYH7 and MYBPC3 leads to severe cardiomyopathy with left ventricular noncompaction phenotype. Hum Mutat 40:1101–1114. https://doi.org/10.1002/humu.23757
Hershkovitz T, Kurolap A, Ruhrman-Shahar N, Monakier D, DeChene ET, Peretz-Amit G, Funke B, Zucker N, Hirsch R, Tan WH (2019) Clinical diversity of MYH7-related cardiomyopathies: Insights into genotype–phenotype correlations. Am J Med Genet A 179:365–372. https://doi.org/10.1002/ajmg.a.61017
Hirono K, Hata Y, Ibuki K, Yoshimura N (2014) Familial Ebstein’s anomaly, left ventricular noncompaction, and ventricular septal defect associated with an MYH7 mutation. J Thorac Cardiovasc Surg 148:e223–e226. https://doi.org/10.1016/j.jtcvs.2014.08.049
Neagoe O, Ciobanu A, Diaconu R, Mirea O, Donoiu I, Militaru C (2019) A rare case of familial restrictive cardiomyopathy, with mutations in MYH7 and ABCC9 genes. Discoveries. https://doi.org/10.15190/d.2019.12
Dorn GW, McNally EM (2014) Two strikes and you’re out: gene–gene mutation interactions in HCM. Circ Res 115(2):208–210
Blankenburg R, Hackert K, Wurster S, Deenen R, Seidman JG, Seidman CE, Lohse MJ, Schmitt JP (2014) β-Myosin heavy chain variant Val606Met causes very mild hypertrophic cardiomyopathy in mice, but exacerbates HCM phenotypes in mice carrying other HCM mutations. Circ Res 115:227–237. https://doi.org/10.1161/CIRCRESAHA.115.303178
Richard P, Charron P, Leclercq C, Ledeuil C, Carrier L, Dubourg O, Desnos M, Bouhour J-B, Schwartz K, Daubert JC (2000) Homozygotes for a R869G mutation in the β-myosin heavy chain gene have a severe form of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 32:1575–1583. https://doi.org/10.1006/jmcc.2000.1193
Tyska M, Hayes E, Giewat M, Seidman C, Seidman J, Warshaw D (2000) Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res 86:737–744. https://doi.org/10.1161/01.res.86.7.737
Palmer BM, Wang Y, Teekakirikul P, Hinson JT, Fatkin D, Strouse S, VanBuren P, Seidman CE, Seidman JG, Maughan DW (2008) Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am J Physiol Heart Circ Physiol 294:H1939–H1947. https://doi.org/10.1152/ajpheart.00644.2007
Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman J, Ingwall JS (1998) Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Investig 101:1775–1783. https://doi.org/10.1172/JCI1940
Georgakopoulos D, Christe ME, Giewat M, Seidman CM, Seidman J, Kass DA (1999) The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an α-cardiac myosin heavy chain missense mutation. Nat Med 5:327–330. https://doi.org/10.1038/6549
Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, Giewat M, Seidman CE, Seidman J (2000) An abnormal Ca2+ response in mutant sarcomere protein–mediated familial hypertrophic cardiomyopathy. J Clin Investig 106:1351–1359. https://doi.org/10.1172/JCI11093
Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM (2010) Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J Clin Investig 120:3520–3529. https://doi.org/10.1172/JCI42028
Kim JB, Porreca GJ, Song L, Greenway SC, Gorham JM, Church GM, Seidman CE, Seidman J (2007) Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science 316:1481–1484. https://doi.org/10.1126/science.1137325
Lowey S (2002) Functional consequences of mutations in the myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Trends Cardiovasc Med 12:348–354. https://doi.org/10.1016/s1050-1738(02)00181-0
Sata M, Ikebe M (1996) Functional analysis of the mutations in the human cardiac beta-myosin that are responsible for familial hypertrophic cardiomyopathy. Implication for the clinical outcome. J Clin Investig 98:2866–2873. https://doi.org/10.1172/JCI119115
Toepfer CN, Garfinkel AC, Venturini G, Wakimoto H, Repetti G, Alamo L, Sharma A, Agarwal R, Ewoldt JF, Cloonan P (2020) Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation 141:828–842. https://doi.org/10.1161/CIRCULATIONAHA.119.042339
Lee KH, Sulbarán G, Yang S, Mun JY, Alamo L, Pinto A, Sato O, Ikebe M, Liu X, Korn ED (2018) Interacting-heads motif has been conserved as a mechanism of myosin II inhibition since before the origin of animals. Proc Natl Acad Sci USA 115:E1991–E2000. https://doi.org/10.1073/pnas.1715247115
Hooijman P, Stewart MA, Cooke R (2011) A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J 100:1969–1976. https://doi.org/10.1016/j.bpj.2011.02.061
McNamara JW, Li A, Dos Remedios CG, Cooke R (2015) The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys Rev 7:5–14. https://doi.org/10.1007/s12551-014-0151-5
Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE, Padrón R (2017) Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 6:e24634. https://doi.org/10.7554/eLife.24634
Vander Roest AS, Liu C, Morck MM, Kooiker KB, Jung G, Song D, Dawood A, Jhingran A, Pardon G, Ranjbarvaziri S (2021) Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state. Proc Natl Acad Sci USA 118:e2025030118. https://doi.org/10.1073/pnas.2025030118
Riaz M, Park J, Sewanan LR, Ren Y, Schwan J, Das SK, Pomianowski PT, Huang Y, Ellis MW, Luo J (2022) Muscle LIM protein force-sensing mediates sarcomeric biomechanical signaling in human familial hypertrophic cardiomyopathy. Circulation 145:1238–1253. https://doi.org/10.1161/CIRCULATIONAHA.121.056265
Adhikari AS, Trivedi DV, Sarkar SS, Song D, Kooiker KB, Bernstein D, Spudich JA, Ruppel KM (2019) β-Cardiac myosin hypertrophic cardiomyopathy mutations release sequestered heads and increase enzymatic activity. Nat Commun 10:1–10. https://doi.org/10.1038/s41467-019-10555-9
Singh RR, McNamara JW, Sadayappan S (2021) Mutations in myosin S2 alter cardiac myosin-binding protein-C interaction in hypertrophic cardiomyopathy in a phosphorylation-dependent manner. J Biol Chem. https://doi.org/10.1016/j.jbc.2021.100836
Tripathi S, Schultz I, Becker E, Montag J, Borchert B, Francino A, Navarro-Lopez F, Perrot A, Özcelik C, Osterziel K-J (2011) Unequal allelic expression of wild-type and mutated β-myosin in familial hypertrophic cardiomyopathy. Basic Res Cardiol 106:1041–1055. https://doi.org/10.1007/s00395-011-0205-9
Dos Remedios CG, Becker E, Ernstberger P (2017) Intrinsic MYH7 expression regulation contributes to tissue level allelic imbalance in hypertrophic cardiomyopathy. J Muscle Res Cell Motil. https://doi.org/10.1007/s10974-017-9486-4
Montag J, Kowalski K, Makul M, Ernstberger P, Radocaj A, Beck J, Becker E, Tripathi S, Keyser B, Mühlfeld C (2018) Burst-like transcription of mutant and wildtype MYH7-alleles as possible origin of cell-to-cell contractile imbalance in hypertrophic cardiomyopathy. Front Physiol 9:359. https://doi.org/10.3389/fphys.2018.00359
Wang J, Li W, Han Y, Chen Y (2019) Different clinical presentation and tissue characterization in a monozygotic twin pair with MYH7 mutation-related hypertrophic cardiomyopathy. Int Heart J 60:477–481. https://doi.org/10.1536/ihj.18-167
Rose J, Kraft T, Brenner B, Montag J (2020) Hypertrophic cardiomyopathy MYH7 mutation R723G alters mRNA secondary structure. Physiol Genomics 52:15–19. https://doi.org/10.1152/physiolgenomics.00100.2019
Wang J, Wan K, Sun J, Li W, Liu H, Han Y, Chen Y (2018) Phenotypic diversity identified by cardiac magnetic resonance in a large hypertrophic cardiomyopathy family with a single MYH7 mutation. Sci Rep 8:1–7. https://doi.org/10.1038/s41598-018-19372-4
Scolari FL, Torres MAR, Simon L, Freitas VCd, Giugliani R, Matte Ú (2016) Prevalence and phenotypic expression of mutations in the MYH7, MYBPC3 and TNNT2 genes in families with hypertrophic cardiomyopathy in the south of Brazil: a cross-sectional study. Arq Bras Cardiol 107:257–265. https://doi.org/10.5935/abc.20160133
Garcıa-Castro M, Reguero JR, Batalla A, Dıaz-Molina B, Gonzalez P, Alvarez V, Cortina A, Cubero GI, Coto E (2003) Hypertrophic cardiomyopathy: low frequency of mutations in the β-myosin heavy chain (MYH7) and cardiac troponin t (TNNT2) genes among spanish patients. Clin Chem 49:1279–1285. https://doi.org/10.1373/49.8.1279
Waldmüller S, Sakthivel S, Saadi AV, Selignow C, Rakesh PG, Golubenko M, Joseph PK, Padmakumar R, Richard P, Schwartz K (2003) Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 35:623–636. https://doi.org/10.1016/s0022-2828(03)00050-6
Goel N, Huddleston CB, Fiore AC (2018) A novel mutation of the MYH7 gene in a patient with hypertrophic cardiomyopathy. Turk J Pediatr 60:315–318. https://doi.org/10.24953/turkjped.2018.03.013
Wang H, Zou Y-B, Song L, Wang J-Z, Sun K, Song X-D, Gao S, Zhang C-N, Hui R-T (2009) The genotype-phenotype correlation of the MYH7 gene c. 1273G> a mutation in familial hypertrophic cardiomyopathy. Hereditas 31:485–488. https://doi.org/10.3724/sp.j.1005.2009.00485
Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO (2009) Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006. Circulation 119:1085–1092. https://doi.org/10.1161/CIRCULATIONAHA.108.804617
Dimitrow PP, Czarnecka D, Kawecka-Jaszcz K, Dubiel JS (2004) Sex-based comparison of survival in referred patients with hypertrophic cardiomyopathy. Am J Med 117:65–66. https://doi.org/10.1016/j.amjmed.2004.02.038
Ahmad F, Seidman J, Seidman CE (2005) The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet 6:185–216. https://doi.org/10.1146/annurev.genom.6.080604.162132
Foglieni C, Lombardi M, Lazzeroni D, Zerboni R, Lazzarini E, Bertoli G, Pisano A, Girolami F, Andolfo A, Magagnotti C (2022) Myosins and MyomiR network in patients with obstructive hypertrophic cardiomyopathy. Biomedicines 10:2180. https://doi.org/10.3390/biomedicines10092180
Mouton J, Van der Merwe L, Goosen A, Revera M, Brink P, Moolman-Smook J, Kinnear C (2016) MYBPH acts as modifier of cardiac hypertrophy in hypertrophic cardiomyopathy (HCM) patients. Hum Genet 135:477–483. https://doi.org/10.1007/s00439-016-1649-7
Ortlepp J, Vosberg H, Reith S, Ohme F, Mahon N, Schröder D, Klues H, Hanrath P, McKenna W (2002) Genetic polymorphisms in the renin-angiotensin-aldosterone system associated with expression of left ventricular hypertrophy in hypertrophic cardiomyopathy: a study of five polymorphic genes in a family with a disease causing mutation in the myosin binding protein C gene. Heart 87:270–275. https://doi.org/10.1136/heart.87.3.270
Perkins MJ, Van Driest SL, Ellsworth EG, Will ML, Gersh BJ, Ommen SR, Ackerman MJ (2005) Gene-specific modifying effects of pro-LVH polymorphisms involving the renin–angiotensin–aldosterone system among 389 unrelated patients with hypertrophic cardiomyopathy. Eur Heart J 26:2457–2462. https://doi.org/10.1093/eurheartj/ehi438
Tanjore R, RangaRaju A, Vadapalli S, Remersu S, Narsimhan C, Nallari P (2010) Genetic variations of β-MYH7 in hypertrophic cardiomyopathy and dilated cardiomyopathy. Indian J Hum Genet 16:67. https://doi.org/10.4103/0971-6866.69348
Kadota C, Arimura T, Hayashi T, Naruse TK, Kawai S, Kimura A (2015) Screening of sarcomere gene mutations in young athletes with abnormal findings in electrocardiography: identification of a MYH7 mutation and MYBPC3 mutations. J Hum Genet 60:641–645. https://doi.org/10.1038/jhg.2015.81
Marziliano N, Medoro A, Mignogna D, Saccon G, Folzani S, Reverberi C, Russo C, Intrieri M (2021) Sudden cardiac death caused by a fatal association of hypertrophic cardiomyopathy (MYH7, p. Arg719Trp), heterozygous familial hypercholesterolemia (LDLR, p. Gly343Lys) and SARS-CoV-2 B.1.1.7 infection. Diagnostics 11:1229. https://doi.org/10.3390/diagnostics11071229
Singh A, Kukreti S (2018) A triple stranded G-quadruplex formation in the promoter region of human myosin β (Myh7) gene. J Biomol Struct Dyn 36:2773–2786. https://doi.org/10.1080/07391102.2017.1374211
Coto E, Reguero JR, Palacín M, Gómez J, Alonso B, Iglesias S, Martín M, Tavira B, Díaz-Molina B, Morales C (2012) Resequencing the whole MYH7 gene (including the intronic, promoter, and 3′ UTR sequences) in hypertrophic cardiomyopathy. J Mol Diagn 14:518–524. https://doi.org/10.1016/j.jmoldx.2012.04.001
Andersen P, Havndrup O, Bundgaard H, Larsen L, Vuust J, Kjeldsen K, Christiansen M (1999) Adult-onset familial hypertrophic cardiomyopathy caused by a novel mutation, R694C, in the MYH7 gene. Clin Genet 56:244–246. https://doi.org/10.1034/j.1399-0004.1999.560313.x
Maron BJ, Spirito P, Wesley Y, Arce J (1986) Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med 315:610–614. https://doi.org/10.1056/NEJM198609043151003
Semsarian C, Ingles J, Maron MS, Maron BJ (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 65:1249–1254. https://doi.org/10.1016/j.jacc.2015.01.019
Marshall E (1997) ‘Playing chicken’ over gene markers. Science 278(5346):2046–2048
Debela AM, Thorimbert S, Hasenknopf B, O’Sullivan C, Ortiz M (2016) Electrochemical primer extension for the detection of single nucleotide polymorphisms in the cardiomyopathy associated MYH7 gene. Chem Commun 52:757–759. https://doi.org/10.1039/c5cc07762a
Ortiz M, Jauset-Rubio M, Kodr D, Simonova A, Hocek M, O’Sullivan CK (2022) Solid-phase recombinase polymerase amplification using ferrocene-labelled dNTPs for electrochemical detection of single nucleotide polymorphisms. Biosensors Bioelectron 198:113825–26. https://doi.org/10.1016/j.bios.2021.113825
Mattivi CL, Bos JM, Bagnall RD, Nowak N, Giudicessi JR, Ommen SR, Semsarian C, Ackerman MJ (2020) Clinical utility of a phenotype-enhanced MYH7-specific variant classification framework in hypertrophic cardiomyopathy genetic testing. Circulation 13:453–459. https://doi.org/10.1161/CIRCGEN.120.003039
Richmond CM, James PA, Pantaleo S-J, Chong B, Lunke S, Tan TY, Macciocca I (2021) Clinical and laboratory reporting impact of ACMG-AMP and modified ClinGen variant classification frameworks in MYH7-related cardiomyopathy. Genet Med 23:1108–1115. https://doi.org/10.1038/s41436-021-01107-y
Park J, Packard EA, Levin MG, Judy RL, Center RG, Damrauer SM, Day SM, Ritchie MD, Rader DJ (2022) A genome-first approach to rare variants in hypertrophic cardiomyopathy genes MYBPC3 and MYH7 in a medical biobank. Hum Mol Genet 31:827–837. https://doi.org/10.1093/hmg/ddab249
Vu MTT, Nguyen TV, Van Huynh N, Thai HTN, Nguyen VP, Huynh TDH (2019) Presence of hypertrophic cardiomyopathy related gene mutations and clinical manifestations in Vietnamese patients with hypertrophic cardiomyopathy. Circulation. https://doi.org/10.1253/circj.CJ-19-0190
Höller V, Seebacher H, Zach D, Schwegel N, Ablasser K, Kolesnik E, Gollmer J, Waltl G, Rainer PP, Verheyen S (2021) Myocardial Deformation analysis in MYBPC3 and MYH7 related sarcomeric hypertrophic cardiomyopathy—the Graz hypertrophic cardiomyopathy registry. Genes 12:1469. https://doi.org/10.3390/genes12101469
Lee S-P, Ashley EA, Homburger J, Caleshu C, Green EM, Jacoby D, Colan SD, Arteaga-Fernandez E, Day SM, Girolami F (2018) Incident atrial fibrillation is associated with MYH7 sarcomeric gene variation in hypertrophic cardiomyopathy: results from the International Sarcomeric Human Cardiomyopathy Registry. Circulation 11:e005191. https://doi.org/10.1161/CIRCHEARTFAILURE.118.005191
Weissler-Snir A, Hindieh W, Gruner C, Fourey D, Appelbaum E, Rowin E, Care M, Lesser JR, Haas TS, Udelson JE (2017) Lack of phenotypic differences by cardiovascular magnetic resonance imaging in MYH7 (β-myosin heavy chain)-versus MYBPC3 (myosin-binding protein C)-related hypertrophic cardiomyopathy. Circulation 10:e005311. https://doi.org/10.1161/CIRCIMAGING.116.005311
Witjas-Paalberends ER, Güçlü A, Germans T, Knaapen P, Harms HJ, Vermeer AM, Christiaans I, Wilde AA, Dos Remedios C, Lammertsma AA (2014) Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res 103:248–257. https://doi.org/10.1093/cvr/cvu127
Wang J, Yang F, Liu W, Sun J, Han Y, Li D, Gkoutos GV, Zhu Y, Chen Y (2020) Radiomic analysis of native T1 mapping images discriminates between MYH7 and MYBPC3-related hypertrophic cardiomyopathy. J Magn Reson Imaging 52:1714–1721. https://doi.org/10.1002/jmri.27209
Baulina N, Pisklova M, Kiselev I, Chumakova O, Zateyshchikov D, Favorova O (2022) Circulating miR-499a-5p is a potential biomarker of MYH7—associated hypertrophic cardiomyopathy. Int J Mol Sci 23:3791. https://doi.org/10.3390/ijms23073791
Yoneda ZT, Anderson KC, Ye F, Quintana JA, O’Neill MJ, Sims RA, Sun L, Glazer AM, Davogustto G, El-Harasis M (2022) Mortality among patients with early-onset atrial fibrillation and rare variants in cardiomyopathy and arrhythmia genes. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2022.0810
Li X, Tang J, Li J, Lin S, Wang T, Zhou K, Li Y, Hua Y (2021) Genetic clues on implantable cardioverter-defibrillator placement in young-age hypertrophic cardiomyopathy: a case report of novel MYH7 mutation and literature review. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2021.810291
Zhao J, Wang J, Liu L, Zheng Y, Wang B, Li W, Yang F, Kang N, Zuo L (2020) The role of three-dimensional speckle tracking imaging derived parameters on predicting outcome of hypertrophic cardiomyopathy patients with MYH7 mutations. Zhonghua Xin Xue Guan Bing Za Zhi 48:287–293. https://doi.org/10.3760/cma.j.cn112148-20190802-00451
Shao H, Zhang Y, Liu L, Ma Z, Zuo L, Ye C, Wei X, Sun C, Tao L (2016) Relationship between electrocardiographic and genetic mutation (MYH7-H1717Q, MYLK2-K324E and KCNQ1-R190W) phenotype in patients with hypertrophic cardiomyopathy. Zhonghua Xin Xue Guan Bing Za Zhi 44:50–54. https://doi.org/10.3760/cma.j.issn.0253-3758.2016.01.011
Zhao X, Wang B, Zhu X, Yang Q, Liu Y, Shao H, Zuo L, Luo Y, Wang Y, Liu L (2022) Analysis of phenotype and MYH7 gene variant in a family of patients with hypertrophic cardiomyopathy. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 39:873–876. https://doi.org/10.3760/cma.j.cn511374-20210317-00239
Christiaans I, ditDeprez RHL, van Langen IM, Wilde AA (2009) Ventricular fibrillation in MYH7-related hypertrophic cardiomyopathy before onset of ventricular hypertrophy. Heart Rhythm 6:1366–1369. https://doi.org/10.1016/j.hrthm.2009.04.029
Zhang S, Wilson J, Madani M, Feld G, Greenberg B (2018) Atrial arrhythmias and extensive left atrial fibrosis as the initial presentation of MYH7 gene mutation. JACC 4:1488–1490. https://doi.org/10.1016/j.jacep.2018.07.016
Chang AC, Chang AC, Kirillova A, Sasagawa K, Su W, Weber G, Lin J, Termglinchan V, Karakikes I, Seeger T (2018) Telomere shortening is a hallmark of genetic cardiomyopathies. Proc Natl Acad Sci USA 115:9276–9281. https://doi.org/10.1073/pnas.1714538115
Chang AC, Blau HM (2018) Short telomeres—a hallmark of heritable cardiomyopathies. Differentiation 100:31–36. https://doi.org/10.1016/j.diff.2018.02.001
Marian AJ, Braunwald E (2017) Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 121:749–770. https://doi.org/10.1161/CIRCRESAHA.117.311059
Xu H, Wang Z, Chen M, Zhao W, Tao T, Ma L, Ni Y, Li W (2021) YTHDF2 alleviates cardiac hypertrophy via regulating Myh7 mRNA decoy. Cell Biosci 11:1–16. https://doi.org/10.1186/s13578-021-00649-7
Yue P, Xia S, Wu G, Liu L, Zhou K, Liao H, Li J, Zheng X, Guo Y, Hua Y (2021) Attenuation of cardiomyocyte hypertrophy via depletion MYH7 using CASAAV. Cardiovasc Toxicol 21:255–264. https://doi.org/10.1007/s12012-020-09617-y
Anderson BR, Jensen ML, Hagedorn PH, Little SC, Olson RE, Ammar R, Kienzle B, Thompson J, McDonald I, Mercer S (2020) Allele-selective knockdown of MYH7 using antisense oligonucleotides. Mol Therapy Nucleic Acids 19:1290–1298. https://doi.org/10.1016/j.omtn.2020.01.012
Dainis A, Zaleta-Rivera K, Ribeiro A, Chang ACH, Shang C, Lan F, Burridge PW, Liu WR, Wu JC, Chang ACY (2020) Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol Genomics 52:293–303. https://doi.org/10.1152/physiolgenomics.00021.2020
Singh A, Joshi S, Kukreti S (2020) Cationic porphyrins as destabilizer of a G-quadruplex located at the promoter of human MYH7 β gene. J Biomol Struct Dyn 38:4801–4816. https://doi.org/10.1080/07391102.2019.1689850
Hill MG, Sekhon MK, Reed KL, Anderson CF, Borjon ND, Tardiff JC, Barber BJ (2015) Intrauterine treatment of a fetus with familial hypertrophic cardiomyopathy secondary to MYH7 mutation. Pediatr Cardiol 36:1774–1777. https://doi.org/10.1007/s00246-015-1250-1
Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman J (1996) A mouse model of familial hypertrophic cardiomyopathy. Science 272:731–734. https://doi.org/10.1126/science.272.5262.731
Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J (2008) Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an α-or β-myosin heavy chain backbone. J Biol Chem 283:20579–20589. https://doi.org/10.1074/jbc.M800554200
Nagueh SF, Chen S, Patel R, Tsybouleva N, Lutucuta S, Kopelen HA, Zoghbi WA, Quiñones MA, Roberts R, Marian A (2004) Evolution of expression of cardiac phenotypes over a 4-year period in the β-myosin heavy chain-Q403 transgenic rabbit model of human hypertrophic cardiomyopathy. J Mol Cell Cardiol 36:663–673. https://doi.org/10.1016/j.yjmcc.2004.02.010
Montag J, Petersen B, Flögel A, Becker E, Lucas-Hahn A, Cost G, Mühlfeld C, Kraft T, Niemann H, Brenner B (2018) Successful knock-in of Hypertrophic Cardiomyopathy-mutation R723G into the MYH7 gene mimics HCM pathology in pigs. Sci Rep 8:1–12. https://doi.org/10.1038/s41598-018-22936-z
Bu H, Ding Y, Li J, Zhu P, Shih Y-H, Wang M, Zhang Y, Lin X, Xu X (2021) Inhibition of mTOR or MAPK ameliorates vmhcl/myh7 cardiomyopathy in zebrafish. JCI insight. https://doi.org/10.1172/jci.insight.154215
Schipper T, Van Poucke M, Sonck L, Smets P, Ducatelle R, Broeckx BJ, Peelman LJ (2019) A feline orthologue of the human MYH7 c. 5647G>A (p. (Glu1883Lys)) variant causes hypertrophic cardiomyopathy in a Domestic Shorthair cat. Eur J Hum Genet 27:1724–1730. https://doi.org/10.1038/s41431-019-0431-4
Fontaine V, Duboscq-Bidot L, Jouve C, Hamlin M, Curjol A, Briand V, Janiak P, Hulot J-S, Pruniaux-Harnist M-P, Charron P (2021) Generation of iPSC line from MYH7 R403L mutation carrier with severe hypertrophic cardiomyopathy and isogenic CRISPR/Cas9 corrected control. Stem Cell Res 52:102245. https://doi.org/10.1016/j.scr.2021.102245
Li X, Fu W, Guo G, Liu M, Du W, Zhao J, Liu Y, Wang L, Dong J, Zhao X (2021) A heterozygous MYH7 (c. 2156G> A) mutant human induced pluripotent stem cell line (ZZUNEUi020-A) generated from a patient with hypertrophic cardiomyopathy. Stem Cell Res 51:102158. https://doi.org/10.1016/j.scr.2021.102158
Guo G, Fu W, Li X, Dong J, Zhao X, Zhang Y (2021) Generation of an iPSC line (ZZUNEUi016-A) derived from a hypertrophic cardiomyopathy patient with the heterozygote mutation in MYH7 gene. Stem Cell Res 53:102262–64. https://doi.org/10.1016/j.scr.2021.102262
Merkert S, Wunderlich S, Beier J, Franke A, Schwanke K, Göhring G, Kraft T, Francino A, Zweigerdt R, Martin U (2021) Generation of two iPSC clones (MHHi021-A and MHHi021-B) from a patient with hypertrophic cardiomyopathy with p. Arg723Gly mutation in the MYH7 gene. Stem Cell Res 52:102208. https://doi.org/10.1016/j.scr.2021.102208
Li X, Liu Y, Liu F, Wang X, Liu M, Du W, Zhao J, Wang M, Hu L, Wang C (2020) Generation of a hiPSC line ZZUNEUi007-A from a patient with hypertrophic cardiomyopathy caused by mutation in MYH7. Stem Cell Res 43:101699. https://doi.org/10.1016/j.scr.2020.101699
Dementyeva E, Kovalenko V, Zhiven M, Ustyantseva E, Kretov E, Vyatkin YV, Zakian S (2020) Generation of two clonal iPSC lines, ICGi019-A and ICGi019-B, by reprogramming peripheral blood mononuclear cells of a patient suffering from hypertrophic cardiomyopathy and carrying a heterozygous p. M659I mutation in MYH7. Stem Cell Res 46:101840. https://doi.org/10.1016/j.scr.2020.101840
Ross SB, Fraser ST, Nowak N, Semsarian C (2017) Generation of induced pluripotent stem cells (iPSCs) from a hypertrophic cardiomyopathy patient with the pathogenic variant p. Val698Ala in beta-myosin heavy chain (MYH7) gene. Stem Cell Res 20:88–90. https://doi.org/10.1016/j.scr.2017.02.015
Cao X, Jahng JW, Lee C, Zha Y, Wheeler MT, Sallam K, Wu JC (2021) Generation of three induced pluripotent stem cell lines from hypertrophic cardiomyopathy patients carrying MYH7 mutations. Stem Cell Research 55:102455. https://doi.org/10.1016/j.scr.2021.102455
Marian A (2021) Molecular genetic basis of hypertrophic cardiomyopathy. Circ Res 128:1533–1553. https://doi.org/10.1161/CIRCRESAHA.121.318346
Wu H, Yang H, Rhee J-W, Zhang JZ, Lam CK, Sallam K, Chang AC, Ma N, Lee J, Zhang H (2019) Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur Heart J 40:3685–3695. https://doi.org/10.1093/eurheartj/ehz326
Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, Mich-Basso J, Lis A, Hassan N, London B (2014) Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res 104:258–269. https://doi.org/10.1093/cvr/cvu205
Guo T, Jiang Y, Song Y, Ma S, Chang Y, Zhang S, Wang H, Dong T, Jiang H, Lu W (2021) Generation of a homozygous MYH7 gene knockout human embryonic stem cell line (WAe009-A-69) using an episomal vector-based CRISPR/Cas9 system. Stem Cell Res 57:102566. https://doi.org/10.1016/j.scr.2021.102566
Mosqueira D, Mannhardt I, Bhagwan JR, Lis-Slimak K, Katili P, Scott E, Hassan M, Prondzynski M, Harmer SC, Tinker A (2018) CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J 39:3879–3892. https://doi.org/10.1093/eurheartj/ehy249
Karakikes I, Termglinchan V, Cepeda DA, Lee J, Diecke S, Hendel A, Itzhaki I, Ameen M, Shrestha R, Wu H (2017) A comprehensive TALEN-based knockout library for generating human-induced pluripotent stem cell-based models for cardiovascular diseases. Circ Res 120:1561–1571. https://doi.org/10.1161/CIRCRESAHA.116.309948
Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547. https://doi.org/10.1038/nrcardio.2013.105
Mestroni L, Maisch B, McKenna W, Schwartz K, Charron P, Rocco C, Tesson F, Richter R, Wilke A, Komajda M (1999) Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J 20:93–102. https://doi.org/10.1053/euhj.1998.1145
Mestroni L, Maisch B, McKenna W, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M (1999) Collaborative research group of the European human and capital mobility project on familial dilated cardiomyopathy. Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J 20:93–102
McNally EM, Golbus JR, Puckelwartz MJ (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Investig 123:19–26. https://doi.org/10.1172/JCI62862
Codd M, Sugrue D, Gersh B, Melton L 3rd (1989) Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 80:564–572. https://doi.org/10.1161/01.cir.80.3.564
Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, Duboc D, Gimeno J, De Groote P, Imazio M (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37:1850–1858. https://doi.org/10.1093/eurheartj/ehv727
Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED (2000) Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 343:1688–1696. https://doi.org/10.1056/NEJM200012073432304
Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJ (2020) Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 141:387–398. https://doi.org/10.1161/CIRCULATIONAHA.119.037661
Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J (2021) Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 144:7–19. https://doi.org/10.1161/CIRCULATIONAHA.120.053033
Lanfear DE, Reza N (2022) Myosin-related dilated cardiomyopathy: another elephant emerges from darkness. J Am Coll Cardiol 80(15):1462–1464
Bansch D, Antz M, Boczor S, Volkmer M, Tebbenjohanns J, Seidl K, Block M, Gietzen F, Berger J, Kuck KH (2002) Primary prevention of sudden cardiac death in idiopathic dilated cardiomyopathy: the Cardiomyopathy Trial (CAT). Circulation 105:1453–1458. https://doi.org/10.1161/01.cir.0000012350.99718.ad
Goldberger JJ (2014) Sudden cardiac death risk stratification in dilated cardiomyopathy: climbing the pyramid of knowledge. Circ Arrhythm Electrophysiol 7(6):1006–1008
Kadish A, Dyer A, Daubert JP, Quigg R, Estes NM, Anderson KP, Calkins H, Hoch D, Goldberger J, Shalaby A (2004) Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med 350:2151–2158. https://doi.org/10.1056/NEJMoa033088
Debold EP, Schmitt JP, Patlak J, Beck S, Moore J, Seidman JG, Seidman C, Warshaw DM (2007) Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol 293:H284–H291. https://doi.org/10.1152/ajpheart.00128.2007
Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, Mende U, Lohse MJ, Warshaw D, Seidman CE (2006) Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci USA 103:14525–14530. https://doi.org/10.1073/pnas.0606383103
Abdallah AM, Carlus SJ, Al-Mazroea AH, Alluqmani M, Almohammadi Y, Bhuiyan ZA, Al-Harbi KM (2019) Digenic inheritance of LAMA4 and MYH7 mutations in patient with infantile dilated cardiomyopathy. Medicina 55:17. https://doi.org/10.3390/medicina55010017
Kuo K, Speranza R, Hackmon R (2020) Fetal dilated cardiomyopathy associated with variants of uncertain significance in MYH7 and DSG2 genes: a case report and review of the literature. J Obstet Gynaecol Can 42:1147–1150. https://doi.org/10.1016/j.jogc.2019.11.002
Petropoulou E, Soltani M, Firoozabadi AD, Namayandeh SM, Crockford J, Maroofian R, Jamshidi Y (2017) Digenic inheritance of mutations in the cardiac troponin (TNNT2) and cardiac beta myosin heavy chain (MYH7) as the cause of severe dilated cardiomyopathy. Eur J Med Genet 60:485–488. https://doi.org/10.1016/j.ejmg.2017.06.008
Samsa LA, Yang B, Liu J (2013) Embryonic cardiac chamber maturation: Trabeculation, conduction, and cardiomyocyte proliferation. Am J Med Genet C Semin Med Genet 163C(3):157–168
Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V (2013) The MOGE (S) classification for a phenotype–genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 62:2046–2072. https://doi.org/10.1016/j.jacc.2013.08.1644
Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow C, Wilkinson JL (2003) The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 348:1639–1646. https://doi.org/10.1056/NEJMoa021737
Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE (2003) The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 348:1647–1655. https://doi.org/10.1056/NEJMoa021715
Muchtar E, Blauwet LA, Gertz MA (2017) Restrictive cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 121:819–837. https://doi.org/10.1161/CIRCRESAHA.117.310982
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ (2008) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29:270–276. https://doi.org/10.1093/eurheartj/ehm342
Gallego-Delgado M, Delgado JF, Brossa-Loidi V, Palomo J, Marzoa-Rivas R, Perez-Villa F, Salazar-Mendiguchía J, Ruiz-Cano MJ, Gonzalez-Lopez E, Padron-Barthe L (2016) Idiopathic restrictive cardiomyopathy is primarily a genetic disease. J Am Coll Cardiol 67:3021–3023. https://doi.org/10.1016/j.jacc.2016.04.024
Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, Thaman R, Mogensen J, Elliott PM, Doi Y (2007) Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol 49:2419–2426. https://doi.org/10.1016/j.jacc.2007.02.061
Oechslin EN, AttenhoferJost CH, Rojas JR, Kaufmann PA, Jenni R (2000) Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol 36:493–500. https://doi.org/10.1016/s0735-1097(00)00755-5
Jenni R, Rojas J, Oechslin E (1999) Isolated noncompaction of the myocardium. N Engl J Med 340:966–967. https://doi.org/10.1056/NEJM199903253401215
Maron B, Towbin J (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113:1807–1816. https://doi.org/10.1161/CIRCULATIONAHA.106.174287
Kayvanpour E, Sedaghat-Hamedani F, Gi W-T, Tugrul OF, Amr A, Haas J, Zhu F, Ehlermann P, Uhlmann L, Katus HA (2019) Clinical and genetic insights into non-compaction: a meta-analysis and systematic review on 7598 individuals. Clin Res Cardiol 108:1297–1308. https://doi.org/10.1007/s00392-019-01465-3
Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, Greutmann M, Hurlimann D, Yegitbasi M, Pons L (2008) Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 117:2893–2901. https://doi.org/10.1161/CIRCULATIONAHA.107.746164
Mazzarotto F, Hawley MH, Beltrami M, Beekman L, de Marvao A, McGurk KA, Statton B, Boschi B, Girolami F, Roberts AM (2021) Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet Med 23:856–864. https://doi.org/10.1038/s41436-020-01049-x
van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck-Zwarts KY, Baas AF, Boekholdt SM, van Melle JP, Teske AJ, Asselbergs FW, Backx AP (2018) Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol 71:711–722. https://doi.org/10.1016/j.jacc.2017.12.019
Gati S, Chandra N, Bennett RL, Reed M, Kervio G, Panoulas VF, Ghani S, Sheikh N, Zaidi A, Wilson M (2013) Increased left ventricular trabeculation in highly trained athletes: do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart 99:401–408. https://doi.org/10.1136/heartjnl-2012-303418
van Waning JI, Moesker J, Heijsman D, Boersma E, Majoor-Krakauer D (2019) Systematic review of genotype–phenotype correlations in noncompaction cardiomyopathy. J Am Heart Assoc 8:e012993. https://doi.org/10.1161/JAHA.119.012993
Hesaraki M, Bora U, Pahlavan S, Salehi N, Mousavi SA, Barekat M, Rasouli SJ, Baharvand H, Ozhan G, Totonchi M (2022) A novel missense variant in actin binding domain of MYH7 is associated with left ventricular noncompaction. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2022.839862
Huang X, Zhao K, Jiang M, Qiu D, Zhou J, Yang Z (2022) The G4 resolvase RHAU regulates ventricular trabeculation and compaction through transcriptional and post-transcriptional mechanisms. J Biol Chem. https://doi.org/10.1016/j.jbc.2021.101449
Wang C, Hata Y, Hirono K, Takasaki A, Ozawa SW, Nakaoka H, Saito K, Miyao N, Okabe M, Ibuki K (2017) A wide and specific spectrum of genetic variants and genotype–phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc 6:e006210. https://doi.org/10.1161/JAHA.117.006210
AttenhoferJost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK (2007) Ebstein’s anomaly. Circulation 115:277–285. https://doi.org/10.1161/CIRCULATIONAHA.106.619338
Correa-Villaseñor A, Ferencz C, Neill CA, David Wilson P, Boughman JA (1994) Ebstein’s malformation of the tricuspid valve: genetic and environmental factors. Teratology 50:137–147. https://doi.org/10.1002/tera.1420500208
Mann R, Lie J (1979) The life story of Wilhelm Ebstein (1836–1912) and his almost overlooked description of a congenital heart disease. Mayo Clinic Proc 54(3):197–204
Postma AV, Van Engelen K, Van De Meerakker J, Rahman T, Probst S, Baars MJ, Bauer U, Pickardt T, Sperling SR, Berger F (2011) Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circulation 4:43–50. https://doi.org/10.1161/CIRCGENETICS.110.957985
Wang J, Zhang X, Wang X, Wang C, Wang F, Wang B (2017) MYH7 rare variant in a family with double-chambered left ventricle. Circulation 10:e001729. https://doi.org/10.1007/s10072-017-3192-2
Vermeer AM, Van Engelen K, Postma AV, Baars MJ, Christiaans I, De Haij S, Klaassen S, Mulder BJ, Keavney B (2013) Ebstein anomaly associated with left ventricular noncompaction: an autosomal dominant condition that can be caused by mutations in MYH7. Am J Med Genet C Semin Med Genet 163C(3):178–184
Bettinelli AL, Mulder TJ, Funke BH, Lafferty KA, Longo SA, Niyazov DM (2013) Familial ebstein anomaly, left ventricular hypertrabeculation, and ventricular septal defect associated with a MYH7 mutation. Am J Med Genet A 161:3187–3190. https://doi.org/10.1002/ajmg.a.36182
Basu R, Hazra S, Shanks M, Paterson DI, Oudit GY (2014) Novel mutation in exon 14 of the sarcomere gene MYH7 in familial left ventricular noncompaction with bicuspid aortic valve. Circulation 7:1059–1062. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001666
Tu P, Sun H, Zhang X, Ran Q, He Y, Ran S (2022) Diverse cardiac phenotypes among different carriers of the same MYH7 splicing variant allele (c. 732+1G>A) from a family. BMC Med Genomics 15:1–6. https://doi.org/10.1186/s12920-022-01186-z
Ge J, Hu T, Liu Y, Wang Q, Fan G, Liu C, Zhang J, Chen S, Maduray K, Zhang Y (2022) Case report: Double-chambered right ventricle diagnosed in a middle-aged female with hypertrophic cardiomyopathy and atrial flutter: a rare case. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2022.937758
Yu B-l, Xiang R, Hu D, Peng D-q (2015) A novel MYH7 mutation in a family with cardiomyopathy presenting with restrictive physiology and varying degrees of left ventricle hypertrophy. Eur Heart J 36:178–178. https://doi.org/10.1093/eurheartj/ehu435
van Engelen K, Postma A, Van De Meerakker J, Roos-Hesselink J, Helderman-Van Den Enden A, Vliegen H, Rahman T, Baars M, Sels J-W, Bauer U (2013) Ebstein’s anomaly may be caused by mutations in the sarcomere protein gene MYH7. Neth Hear J 21:113–117. https://doi.org/10.1007/s12471-011-0141-1
Piekutowska-Abramczuk D, Paszkowska A, Ciara E, Frączak K, Mirecka-Rola A, Wicher D, Pollak A, Rutkowska K, Sarnecki J, Ziółkowska L (2022) Genetic profile of left ventricular noncompaction cardiomyopathy in children—a single reference center experience. Genes 13:1334. https://doi.org/10.3390/genes13081334
Khan RS, Pahl E, Dellefave-Castillo L, Rychlik K, Ing A, Yap KL, Brew C, Johnston JR, McNally EM, Webster G (2022) Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J Am Heart Assoc 11:e022854. https://doi.org/10.1161/JAHA.121.022854
Hoedemaekers YM, Cohen-Overbeek TE, Frohn-Mulder I, Dooijes D, Majoor-Krakauer D (2013) Prenatal ultrasound diagnosis of MYH7 non-compaction cardiomyopathy. Ultrasound Obstet Gynecol 41:336–339. https://doi.org/10.1002/uog.12279
Nomura Y, Momoi N, Hirono K, Hata Y, Takasaki A, Nishida N, Ichida F (2015) A novel MYH7 gene mutation in a fetus with left ventricular noncompaction. Can J Cardiol 31:e101--103. https://doi.org/10.1016/j.cjca.2014.11.012
Yang K-C, Breitbart A, De Lange WJ, Hofsteen P, Futakuchi-Tsuchida A, Xu J, Schopf C, Razumova MV, Jiao A, Boucek R (2018) Novel adult-onset systolic cardiomyopathy due to MYH7 E848G mutation in patient-derived induced pluripotent stem cells. JACC Basic Transl Sci 3:728–740. https://doi.org/10.1016/j.jacbts.2018.08.008
Shih Y-H, Zhang Y, Ding Y, Ross CA, Li H, Olson TM, Xu X (2015) Cardiac transcriptome and dilated cardiomyopathy genes in zebrafish. Circulation 8:261–269. https://doi.org/10.1161/CIRCGENETICS.114.000702
Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, Duff RM, Beckman K, de Visser M, van der Graaff MM (2004) Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). The American Journal of Human Genetics 75:703–708. https://doi.org/10.1086/424760
Lamont PJ, Udd B, Mastaglia FL, de Visser M, Hedera P, Voit T, Bridges LR, Fabian V, Rozemuller A, Laing NG (2006) Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry 77:208–215. https://doi.org/10.1136/jnnp.2005.073825
Laing N, Laing B, Meredith C, Wilton S, Robbins P, Honeyman K, Dorosz S, Kozman H, Mastaglia F, Kakulas B (1995) Autosomal dominant distal myopathy: linkage to chromosome 14. Am J Hum Genet 56:422
Voit T, Kutz P, Leube B, Neuen-Jacob E, Schröder J, Cavallotti D, Vaccario M, Schaper J, Broich P, Cohn R (2001) Autosomal dominant distal myopathy: further evidence of a chromosome 14 locus. Neuromuscul Disord 11:11–19. https://doi.org/10.1016/s0960-8966(00)00158-9
Mastaglia F, Phillips B, Cala L, Meredith C, Egli S, Akkari P, Laing N (2002) Early onset chromosome 14-linked distal myopathy (Laing). Neuromuscul Disord 12:350–357. https://doi.org/10.1016/s0960-8966(01)00287-5
Alessi CE, Wu Q, Whitaker CH, Felice KJ (2020) Laing myopathy: report of 4 new families with novel MYH7 mutations, double mutations, and severe phenotype. J Clin Neuromuscul Dis 22:22–34. https://doi.org/10.1097/CND.0000000000000297
Oda T, Xiong H, Kobayashi K, Wang S, Satake W, Jiao H, Yang Y, Cha P-C, Hayashi YK, Nishino I (2015) A de novo mutation of the MYH7 gene in a large Chinese family with autosomal dominant myopathy. Hum Genome Variation 2:1–7. https://doi.org/10.1038/hgv.2015.22
Lamont PJ, Wallefeld W, Hilton-Jones D, Udd B, Argov Z, Barboi AC, Bonneman C, Boycott KM, Bushby K, Connolly AM (2014) Novel mutations widen the phenotypic spectrum of slow skeletal/β-cardiac myosin (MYH 7) distal myopathy. Hum Mutat 35:868–879. https://doi.org/10.1002/humu.22553
Muelas N, Hackman P, Luque H, Garcés-Sánchez M, Azorín I, Suominen T, Sevilla T, Mayordomo F, Gómez L, Martí P (2010) MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology 75:732–741. https://doi.org/10.1212/WNL.0b013e3181eee4d5
Liu X-Y, Zhang Y-S, Sun A-P, Zhong Y-F, Zheng D-F, Fan D-S (2019) A novel MYH7 mutation resulting in Laing distal myopathy in a Chinese family. Chin Med J 132:856–859. https://doi.org/10.1097/CM9.0000000000000148
Clarke NF, Amburgey K, Teener J, Camelo-Piragua S, Kesari A, Punetha J, Waddell LB, Davis M, Laing NG, Monnier N (2013) A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies. Neuromuscul Disord 23:432–436. https://doi.org/10.1016/j.nmd.2013.02.009
Darin N, Tajsharghi H, Östman-Smith I, Gilljam T, Oldfors A (2007) New skeletal myopathy and cardiomyopathy associated with a missense mutation in MYH7. Neurology 68:2041–2042. https://doi.org/10.1212/01.wnl.0000264430.55233.72
Homayoun H, Khavandgar S, Hoover JM, Mohsen A-W, Vockley J, Lacomis D, Clemens PR (2011) Novel mutation in MYH7 gene associated with distal myopathy and cardiomyopathy. Neuromuscul Disord 21:219–222. https://doi.org/10.1016/j.nmd.2010.12.005
Overeem S, Schelhaas H, Blijham P, Grootscholten M, Ter Laak H, Timmermans J, van den Wijngaard A, Zwarts M (2007) Symptomatic distal myopathy with cardiomyopathy due to a MYH7 mutation. Neuromuscul Disord 17:490–493. https://doi.org/10.1016/j.nmd.2007.02.007
Dubourg O, Maisonobe T, Behin A, Suominen T, Raheem O, Penttilä S, Parton M, Eymard B, Dahl A, Udd B (2011) A novel MYH7 mutation occurring independently in French and Norwegian Laing distal myopathy families and de novo in one Finnish patient. J Neurol 258:1157–1163. https://doi.org/10.1007/s00415-011-5900-9
Negrão L, Machado R, Lourenço M, Fernandez-Marmiesse A, Rebelo O (2020) Laing early-onset distal myopathy with subsarcolemmal hyaline bodies caused by a novel variant in the MYH7 gene. Acta Myol 39:24. https://doi.org/10.36185/2532-1900-004
Lefter S, Hardiman O, McLaughlin RL, Murphy SM, Farrell M, Ryan AM (2015) A novel MYH7 Leu1453pro mutation resulting in Laing distal myopathy in an Irish family. Neuromuscul Disord 25:155–160. https://doi.org/10.1016/j.nmd.2014.09.007
Park J-M, Kim YJ, Yoo JH, Hong YB, Park JH, Koo H, Chung KW, Choi B-O (2013) A novel MYH7 mutation with prominent paraspinal and proximal muscle involvement. Neuromuscul Disord 23:580–586. https://doi.org/10.1016/j.nmd.2013.04.003
Carbonell-Corvillo P, Tristán-Clavijo E, Cabrera-Serrano M, Servián-Morilla E, García-Martín G, Villarreal-Pérez L, Rivas-Infante E, Area-Gómez E, Chamorro-Muñoz M, Gil-Gálvez A (2018) A novel MYH7 founder mutation causing Laing distal myopathy in Southern Spain. Neuromuscul Disord 28:828–836. https://doi.org/10.1016/j.nmd.2018.07.006
Muelas N, Hackman P, Luque H, Suominen T, Espinós C, Garcés-Sánchez M, Sevilla T, Azorin I, Millán J, Udd B (2012) Spanish MYH7 founder mutation of Italian ancestry causing a large cluster of Laing myopathy patients. Clin Genet 81:491–494. https://doi.org/10.1111/j.1399-0004.2011.01667.x
Dahl-Halvarsson M, Olive M, Pokrzywa M, Ejeskär K, Palmer RH, Uv AE, Tajsharghi H (2018) Drosophila model of myosin myopathy rescued by overexpression of a TRIM-protein family member. Proc Natl Acad Sci USA 115:E6566–E6575. https://doi.org/10.1073/pnas.1800727115
Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 54:494–500. https://doi.org/10.1002/ana.10693
Laing N, Ceuterick-de Groote C, Dye D, Liyanage K, Duff R, Dubois B, Robberecht W, Sciot R, Martin J, Goebel H (2005) Myosin storage myopathy: slow skeletal myosin (MYH7) mutation in two isolated cases. Neurology 64:527–529. https://doi.org/10.1212/01.WNL.0000150581.37514.30
Bohlega S, Abu-Amero S, Wakil S, Carroll P, Al-Amr R, Lach B, Al-Sayed Y, Cupler E, Meyer B (2004) Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology 62:1518–1521. https://doi.org/10.1212/01.wnl.0000123255.92062.37
Dye DE, Azzarelli B, Goebel HH, Laing NG (2006) Novel slow-skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred. Neuromuscul Disord 16:357–360. https://doi.org/10.1016/j.nmd.2006.03.011
Ortolano S, Tarrío R, Blanco-Arias P, Teijeira S, Rodríguez-Trelles F, García-Murias M, Delague V, Lévy N, Fernández JM, Quintáns B (2011) A novel MYH7 mutation links congenital fiber type disproportion and myosin storage myopathy. Neuromuscul Disord 21:254–262. https://doi.org/10.1016/j.nmd.2010.12.011
Tajsharghi H, Oldfors A, Macleod DP, Swash M (2007) Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology 68:962–962. https://doi.org/10.1212/01.wnl.0000257131.13438.2c
Stalpers X, Verrips A, Braakhekke J, Lammens M, van den Wijngaard A, Mostert A (2011) Scoliosis surgery in a patient with “de novo” myosin storage myopathy. Neuromuscul Disord 21:812–815. https://doi.org/10.1016/j.nmd.2011.05.005
Beecroft SJ, van de Locht M, de Winter JM, Ottenheijm CA, Sewry CA, Mohammed S, Ryan MM, Woodcock IR, Sanders L, Gooding R (2019) Recessive MYH7-related myopathy in two families. Neuromuscul Disord 29:456–467. https://doi.org/10.1016/j.nmd.2019.04.002
Bohlega S, Lach B, Meyer B, Al Said Y, Kambouris M, Al Homsi M, Cupler E (2003) Autosomal dominant hyaline body myopathy: clinical variability and pathologic findings. Neurology 61:1519–1523. https://doi.org/10.1212/01.wnl.0000096022.09887.9d
Cancilla P, Kalyanaraman K, Verity M, Munsat T, Pearson C (1971) Familial myopathy with probable lysis of myofibrils in type I fibers. Neurology 21:579–579. https://doi.org/10.1212/wnl.21.6.579
Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R, Cenacchi G, Angelini C (2007) MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul Disord 17:321–329. https://doi.org/10.1016/j.nmd.2007.01.010
Masuzugawa S, Kuzuhara S, Narita Y, Naito Y, Taniguchi A, Ibi T (1997) Autosomal dominant hyaline body myopathy presenting as scapuloperoneal syndrome: clinical features and muscle pathology. Neurology 48:253–257. https://doi.org/10.1212/wnl.48.1.253
Uro-Coste E, Arné-Bes M-C, Pellissier J-F, Richard P, Levade T, Heitz F, Figarella-Branger D, Delisle M-B (2009) Striking phenotypic variability in two familial cases of myosin storage myopathy with a MYH7 Leu1793pro mutation. Neuromuscul Disord 19:163–166. https://doi.org/10.1016/j.nmd.2008.11.012
Armel TZ, Leinwand LA (2009) Mutations in the β-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci 106:6291–6296. https://doi.org/10.1073/pnas.0900107106
Parker F, Batchelor M, Wolny M, Hughes R, Knight PJ, Peckham M (2018) A1603P and K1617del, Mutations in β-cardiac myosin heavy chain that cause laing early-onset distal myopathy, affect secondary structure and filament formation in vitro and in vivo. J Mol Biol 430:1459–1478. https://doi.org/10.1016/j.jmb.2018.04.006
Postma A, Van Engelen K, Van De Meerakker J, Rahman T, Probst S, Baars M, Bauer U, Pickardt T, Sperling S, Berger F (2011) Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ Cardiovasc Genet 4:43–50. This study is the first to report mutations in MYH7 in individuals with Ebstein anomaly of the tricuspid valve
Clarke NF, North KN (2003) Congenital fiber type disproportion—30 years on. J Neuropathol Exp Neurol 62:977–989. https://doi.org/10.1093/jnen/62.10.977
Pajusalu S, Talvik I, Noormets K, Talvik T, Põder H, Joost K, Puusepp S, Piirsoo A, Stenzel W, Goebel HH (2016) De novo exonic mutation in MYH7 gene leading to exon skipping in a patient with early onset muscular weakness and fiber-type disproportion. Neuromuscul Disord 26:236–239. https://doi.org/10.1016/j.nmd.2015.11.011
Bánfai Z, Hadzsiev K, Pál E, Komlósi K, Melegh M, Balikó L, Melegh B (2017) Novel phenotypic variant in the MYH7 spectrum due to a stop-loss mutation in the C-terminal region: a case report. BMC Med Genet 18:1–7. https://doi.org/10.1186/s12881-017-0463-y
Bader I, Freilinger M, Landauer F, Waldmüller S, Mueller-Felber W, Rauscher C, Sperl W, Bittner R, Schmidt W, Mayr J (2022) A recurrent single-amino acid deletion (p. Glu500del) in the head domain of ß-cardiac myosin in two unrelated boys presenting with polyhydramnios, congenital axial stiffness and skeletal myopathy. Orphanet J Rare Dis 17:1–10. https://doi.org/10.1186/s13023-022-02421-7
Surikova Y, Filatova A, Polyak M, Skoblov M, Zaklyazminskaya E (2019) Common pathogenic mechanism in patients with dropped head syndrome caused by different mutations in the MYH7 gene. Gene 697:159–164. https://doi.org/10.1016/j.gene.2019.02.011
Li N, Zhao Z, Shen H, Bing Q, Guo X, Hu J (2018) MYH7 mutation associated with two phenotypes of myopathy. Neurol Sci 39:333–339
Romero NB, Xie T, Malfatti E, Schaeffer U, Böhm J, Wu B, Xu F, Boucebci S, Mathis S, Neau J-P (2014) Autosomal dominant eccentric core disease caused by a heterozygous mutation in the MYH7 gene. J Neurol Neurosurg Psychiatry 85:1149–1152. https://doi.org/10.1136/jnnp-2013-306754
Gil-Gálvez A, Carbonell-Corvillo P, Paradas C, Miranda-Vizuete A (2020) Cautionary note on the use of Caenorhabditis elegans to study muscle phenotypes caused by mutations in the human MYH7 gene. Biotechniques 68:296–299. https://doi.org/10.2144/btn-2020-0012
Murgiano L, Tammen I, Harlizius B, Drögemüller C (2012) A de novo germline mutation in MYH7 causes a progressive dominant myopathy in pigs. BMC Genet 13:1–7. https://doi.org/10.1186/1471-2156-13-99
Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L (2016) MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis 11:1–14. https://doi.org/10.1186/s13023-016-0476-1
Atemin S, Todorov T, Maver A, Chamova T, Georgieva B, Tincheva S, Pacheva I, Ivanov I, Taneva A, Zlatareva D (2021) MYH7-related disorders in two Bulgarian families: novel variants in the same region associated with different clinical manifestation and disease penetrance. Neuromuscul Disord 31:633–641. https://doi.org/10.1016/j.nmd.2021.04.004
Naddaf E, Waclawik AJ (2015) Two families with MYH7 distal myopathy associated with cardiomyopathy and core formations. J Clin Neuromuscul Dis 16:164–169. https://doi.org/10.1097/CND.0000000000000069
Ruggiero L, Fiorillo C, Gibertini S, De Stefano F, Manganelli F, Iodice R, Vitale F, Zanotti S, Galderisi M, Mora M (2015) A rare mutation in MYH7 gene occurs with overlapping phenotype. Biochem Biophys Res Commun 457:262–266. https://doi.org/10.1016/j.bbrc.2014.12.098
Finsterer J, Stöllberger C, Brandau O, Laccone F, Bichler K, Laing NG (2014) Novel MYH7 mutation associated with mild myopathy but life-threatening ventricular arrhythmias and noncompaction. Int J Cardiol 173:532–535. https://doi.org/10.1016/j.ijcard.2014.03.025
Yüceyar N, Ayhan Ö, Karasoy H, Tolun A (2015) Homozygous MYH7 R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy. Neuromuscul Disord 25:340–344. https://doi.org/10.1016/j.nmd.2015.01.007
Díaz-Manera J, Alejaldre A, Llauger J, Mirabet S, Rojas-García R, Ramos-Fransi A, Gallardo E, Illa I (2014) Cranial, axial and proximal myopathy and hypertrophic cardiomyopathy caused by a mutation in the globular head region of the MYH7 gene. Eur J Neurol 21:e51–e52. https://doi.org/10.1111/ene.12416
Brand P, Dyck PJB, Liu J, Berini S, Selcen D, Milone M (2016) Distal myopathy with coexisting heterozygous TIA1 and MYH7 Variants. Neuromuscul Disord 26:511–515. https://doi.org/10.1016/j.nmd.2016.05.012
Cullup T, Lamont P, Cirak S, Damian M, Wallefeld W, Gooding R, Tan S, Sheehan J, Muntoni F, Abbs S (2012) Mutations in MYH7 cause Multi-minicore Disease (MmD) with variable cardiac involvement. Neuromuscul Disord 22:1096–1104. https://doi.org/10.1016/j.nmd.2012.06.007
Zhang Y, Wang Q, Zhu T, Chen H (2022) Identification of cigarette smoking-related novel biomarkers in lung adenocarcinoma. BioMed Res Int. https://doi.org/10.1155/2022/9170722
Sun J, Li S, Wang F, Fan C, Wang J (2019) Identification of key pathways and genes in PTEN mutation prostate cancer by bioinformatics analysis. BMC Med Genet 20:1–9. https://doi.org/10.1186/s12881-019-0923-7
Mahapatra S, Bhuyan R, Das J, Swarnkar T (2021) Integrated multiplex network based approach for hub gene identification in oral cancer. Heliyon 7:e07418. https://doi.org/10.1016/j.heliyon.2021.e07418
Huang Y-H, Zhang CZ, Huang Q-S, Yeong J, Wang F, Yang X, He Y-F, Zhang X-L, Zhang H, Chen S-L (2021) Clinicopathologic features, tumor immune microenvironment and genomic landscape of Epstein-Barr virus-associated intrahepatic cholangiocarcinoma. J Hepatol 74:838–849. https://doi.org/10.1016/j.jhep.2020.10.037
Funding
This work was supported by Shandong key research and development plan (Grant No. 2019GSF108186 and 2014GSF118066) and Science Foundation of Qilu Hospital of Shandong University (Grant No. 2017QLQN33).
Author information
Authors and Affiliations
Contributions
CZ conceptualized the manuscript. YG and LP wrote the manuscript, and designed and prepared the figures and tables. CZ edited the final version of the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gao, Y., Peng, L. & Zhao, C. MYH7 in cardiomyopathy and skeletal muscle myopathy. Mol Cell Biochem 479, 393–417 (2024). https://doi.org/10.1007/s11010-023-04735-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-023-04735-x