
Abstract
The super-relaxed (SRX) state of myosin was only recently reported in striated muscle. It is characterised by a sub-population of myosin heads with a highly inhibited rate of ATP turnover. Myosin heads in the SRX state are bound to each other along the thick filament core producing a highly ordered arrangement. Upon activation, these heads project into the interfilament space where they can bind to the actin filaments. Thus far, the population and lifetimes of myosin heads in the SRX state have been characterised in rabbit cardiac, and fast and slow skeletal muscle, as well as in the skeletal muscle of the tarantula. These studies suggest that the role of SRX in cardiac and skeletal muscle regulation is tailored to their specific functions. In skeletal muscle, the SRX modulates the resting metabolic rate. Cardiac SRX represents a “reserve” of inactive myosin heads that may protect the heart during times of stress, e.g. hypoxia and ischaemia. These heads may also be called up when there is a sustained demand for increased power. The SRX in cardiac muscle provides a potential target for novel therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Striated muscle contraction is driven by the interaction between the motor protein myosin and its major binding partner, actin (Huxley and Niedergerke 1954; Huxley and Hanson 1954). In the active state, the two globular heads of myosin undergo transient, non-processive interactions with actin resulting in a shortening of the sarcomeres. Relaxation has recently been shown to exist in a disordered relaxed state and an ordered relaxed or super-relaxed (SRX) state. In the disordered relaxed state, the myosin heads protrude into the interfilament space towards the actin filament (Wilson et al. 2014), but they are blocked or restricted from binding to actin by the thin filament regulatory proteins (the troponin–tropomyosin complex). The SRX state has a highly ordered arrangement of myosin heads, which interact with each other along the axis of the thick filament (Fig. 1a). These head-to-head interactions increase the lifetime of ATP turnover from less than 30 s per myosin head in the disordered relaxed state to over 100 s in the SRX state (Fig. 2) (Cooke 2011).

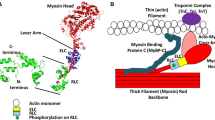
a The three states of myosin in striated muscle: active, disordered relaxed and super-relaxed. Active myosin produces force by pulling the actin filaments (red) towards the M-line of the sarcomere. Relaxed myosin in the disordered state protrudes into the interfilament space but is restricted from binding to actin by the thin filament regulatory proteins whilst myosin in the super-relaxed (SRX) state is bound on or close to the thick filament. This results in an ATP turnover lifetime that is at least 5-fold longer in SRX myosin than disordered myosin. b An atomic model of relaxed myosin heads bound to the thick filament core. In the SRX state the blocked (green) and free (red) myosin heads interact with each other forming the J-like structure of the interacting heads motif. These heads may also bind to the thick filament core (grey). (b) reproduced with permission from Craig and Woodhead (2006)

The three states of myosin in striated muscle. Active myosin has a rapid ATP turnover lifetime of under 1 s, while disordered relaxed myosin is generally less than 30 s. Activation of these myosin heads may occur through calcium binding to the thin filament regulatory proteins. The super-relaxed myosin has a highly inhibited ATP turnover time i.e. longer than 100 s. Phosphorylation of the thick filament proteins cMyBP-C and RLC may facilitate the transfer of myosin heads out of this state. Modified from Cooke (2011)
What is the super-relaxed state of myosin?
SRX was first discovered in rabbit skeletal muscle, and has since been identified in rabbit cardiac and tarantula skeletal muscle (Hooijman et al. 2011; Naber et al. 2011b; Stewart et al. 2010). These striated muscle types have distinct SRX populations and lifetimes, described below. A recent report suggests that the SRX state also exists in human cardiac muscle (McNamara et al. 2014).
The metabolic rate of ATP use by purified frog myosin in solution was five-fold greater than the rate observed in living frog muscle (Ferenczi et al. 1978; Kushmerick and Paul 1976). This effect was also observed in rabbit skeletal muscle (Gutierrez et al. 1989; Myburgh et al. 1995). The discovery of the SRX state of myosin accounted for this 30-year-old discrepancy.
The SRX state of myosin was discovered through a simple experiment. It took advantage of the known kinetics of the cross-bridge cycle and a fluorescent nucleotide, 2′–/3′-O-(N’-methylanthranioyl) adenosine triphosphate (MANT-ATP) that significantly changes its emission intensity when bound to myosin. MANT-ATP is an analogue of ATP that binds strongly to myosin but with a 2.5-fold increase in fluorescence emission. This increase in fluorescence is lost upon dissociation from myosin. These instantaneous changes in fluorescence enables the binding and dissociation of MANT-ATP to be measured, thus making it a useful probe of myosin ATPase kinetics (Cremo et al. 1990; Woodward et al. 1991). Confocal microscopy has also shown that MANT-ATP localises to the A-band of muscle fibres (Hooijman et al. 2011; Stewart et al. 2010).
The skeletal muscle experiment used rabbit psoas and soleus muscles that had been chemically skinned through glycerination. By disrupting the cell membrane, the skinning process permeabilises the fibre. This allows the experimenter to control exogenous Mg-ATP concentration while removing a portion of membrane bound ATPases such as channels and pumps (Wood et al. 1975). Single fibres were immobilised in a flow chamber, allowing the fast exchange of solutes in the fibre (Stewart et al. 2010).
Skeletal muscle fibres were initially incubated in a relaxing solution containing 250 μM MANT-ATP. This solution was then rapidly exchanged with a relaxing solution containing 4 mM unlabelled ATP resulting in a decrease in fluorescence intensity as the hydrolysed MANT-nucleotides were exchanged for ATP (Fig. 3, red symbols) (Stewart et al. 2010). The reverse experiment (Fig. 3, blue symbols) mirrors these results where the rate of intensity increases. This decay in fluorescence intensity, I, was fitted to a double exponential decay equation:

The change in fluorescence intensity as a function of time. The red trace shows the decay seen when 250 μM MANT-ATP is chased with 4 mM ATP. The blue trace shows the inverse experiment where the fibre was incubated with 4 mM ATP and chased with 250 μM MANT-ATP. The change in fluorescence intensity has two phases, a fast phase over the first 30 s and a slow phase over the next ~600 s. This second phase is credited to the slow release of nucleotides from the fraction of myosin heads in the super-relaxed state. Reproduced with permission from Cooke (2011)
P1 and P2 describe the proportion of fibre fluorescence attributed to each of the two exponential components. T1 and T2 represent their respective fluorescence lifetimes. These define the rate of ATP turnover per myosin head in each state, equivalent to 1/lifetime.
P1 and T1 describe the fast decay of fluorescence intensity within the first ~30 s, while P2 and T2 describe the slow decrease in fluorescence over ~600 s, as shown in Fig. 3. The rapid phase comprises multiple elements including:
-
(1)
The release of hydrolysed MANT-ATP by myosin in the disordered relaxed state. In rabbit skeletal myosin one ATP is hydrolysed every 6 s per myosin head (Myburgh et al. 1995);
-
(2)
The diffusion of unbound MANT-ATP out of the fibre, which takes about 10 s (Cooke and Pate 1985); and
-
(3)
The fast release of non-specifically bound MANT-nucleotides.
The slow phase, with a lifetime of ~230 s, was shown to be the slow release of nucleotides from the population of SRX myosin in the relaxed muscle fibre. Hence, the SRX state of myosin was found to have an ATP turnover lifetime more than 10-fold longer than purified myosin from skeletal muscle (Myburgh et al. 1995).
The slow release of nucleotides was the result of myosin heads in the SRX state. This was confirmed through a number of additional experiments, the first of which involved incubating the fibre with 250 μM MANT-ATP and a saturating concentration of 4 mM ATP that was chased with 4 mM ATP. At this saturating concentration, the majority of myosin heads bind ATP rather than MANT-ATP. This resulted in the elimination of the slow decay of fluorescence intensity, indicating that the SRX state arises from specific binding of nucleotides within the muscle fibres.
Additionally, the slow decay of fluorescence intensity associated with the SRX state was not observed when the fibre was chased with 4 mM ADP, indicating that the SRX is only observed in relaxed skeletal muscle fibres. This observation led to the hypothesis that myosin heads in the SRX state may be rapidly recruited into the disordered state via cooperative activation of the thick filament (Cooke 2011; Stewart et al. 2010). A simple, but unproven explanation for this mechanism is that the binding of a myosin head to actin disrupts adjacent myosin heads, thus bringing them out of the SRX (Moss and Fitzsimons 2010). Another mechanism for activation has been proposed from structural studies of tarantula thick filaments (Brito et al. 2011; Sulbaran et al. 2013). Each myosin molecule has a blocked head that is bound firmly to the core of the thick filament and has no interaction with actin, while the “swaying” head can extend away from the thick filament and interact with actin. It is possible that a similar structural configuration occurs in vertebrate skeletal muscle with strong actin bonds made by the swaying head cooperatively activating the whole thick filament (Brito et al. 2011; Sulbaran et al. 2013).
In the relaxed fibre, approximately 50 % of the myosin heads exist in the SRX state (Stewart et al. 2010). This proportion is adjusted for the level of non-specific binding determined by the co-incubation of 250 μM MANT-ATP with increasing ATP concentrations. ATP competes with MANTATP within the fibre where the fluorescence intensity falls rapidly within the first mM of ATP but begins to plateau thereafter. This enabled the proportion of fluorescence from MANT-ATP bound non-specifically to be calculated, which was found to be 41 % in rabbit skeletal muscle (Stewart et al. 2010).
Discovery of the SRX state in the heart
The SRX lifetime in cardiac muscle is considerably shorter than in skeletal muscle, with a single ATP hydrolysed approximately every 145 s per myosin head (Hooijman et al. 2011). This slow phase is present in fibres that were initially relaxed, but not in the rigor state (Hooijman et al. 2011). Cardiac muscle in rigor-ADP, measured by initial incubation with MANT-ADP and chased with ADP, reduces the slow decay to 9 % of the total fluorescence. This indicates a largely absent SRX state (Fig. 4b, open triangles). The SRX state was similarly eliminated when ADP was chased with MANT-ATP (Hooijman et al. 2011). Thus, myosin in the strongly bound state rapidly exchanges bound ADP for MANT-ATP, thereby inducing relaxation of the fibre.

Changes in the SRX state of cardiac and skeletal muscle fibres in ADP and Ca2+ chases. a The effect of ADP on the skeletal muscle SRX. The open circles show MANT-ATP chased by ATP, while the open triangles indicate MANT-ATP chased by ADP, where the slow decay of fluorescence intensity eliminated. The squares indicate the change in fluorescence intensity from an ATP incubation chased by MANT-ATP. b Effect of ADP on cardiac muscle SRX. Filled circles show the decay of fluorescence intensity in an ATP chase, whereas the open circles show the decay in response to ADP chase, where the SRX is diminished but still present. The open triangles show the change in fluorescence intensity when MANT-ADP is chased with ADP. c, d The effect of calcium activation on skeletal (c) and cardiac (d) muscle fibres. Red shows the decay in fluorescence intensity in ATP chase experiments, while the blue traces are the calcium activation chase (ATP plus calcium, pCa = 5.7). Skeletal SRX was lost upon activation while cardiac SRX was largely unchanged. (a) reproduced with permission from Stewart et al. (2010). (b–d) reproduced with permission from Hooijman et al. (2011)
Difference between cardiac and skeletal SRX
Intriguingly, unlike in skeletal fibres, the proportion of SRX was not fully abolished in cardiac fibres upon activation (Hooijman et al. 2011). When ADP was used to chase MANT-ATP in skeletal muscle, myosin binds strongly to actin, thereby eliminating the SRX state (Fig. 4a). However, when this experiment was performed in cardiac muscle, the slow phase was still observed, albeit in a slightly lower proportion than the ATP chase (Fig. 4b). This difference in myosin SRX between skeletal and cardiac muscle may be due to their different physiological functions. It is possible that cooperative activation of the myosin heads in skeletal muscle, as discussed above, is reduced in cardiac myosin.
Hooijman et al. (2011) found that cooperative activation of myosin heads from the SRX was largely absent in cardiac muscle. These fibres were incubated with MANT-ATP and then chased with a partial activating solution (4 mM ATP, pCa = 5.7). Cardiac muscle fibres under these conditions achieved 50 % activation in 4 s. Given the speed of this activation, it cannot be attributed to the SRX. The proportion of myosin in the SRX state was similar in cardiac muscle fibres regardless of whether it was fully relaxed or partially activated (Fig. 4d). However, its lifetime upon activation increased by more than 35 % (Hooijman et al. 2011). In comparison to relaxed skeletal muscle fibres, partial activation reduced the P2 by 18 % and T2 by ~80 % (Fig. 4c). Therefore, the cooperative interaction between adjacent myosin heads is present in skeletal but not cardiac thick filaments (Hooijman et al. 2011; Stewart et al. 2010).
Cardiac and skeletal muscles serve unique functions. Cardiac muscle must contract and relax consistently without fatigue, while skeletal muscle exists in a mostly relaxed state and generates force instantaneously. It therefore follows that skeletal muscle exhibits cooperative behaviour between myosin heads that facilitates rapid force generation. In contrast, cardiac muscle is not fully activated under physiological conditions, even during systole (Kobirumaki-Shimozawa et al. 2014). Thus, it would be energetically detrimental to recruit more myosin heads than required from the SRX state with each heartbeat. We postulate that the SRX state provides a means of modulating cardiac contractility in response to factors such as stress, changes to pre-load and peripheral pressures.
SRX and the Frank–Starling mechanism
Cardiac SRX state may play a role in the Frank–Starling mechanism by which end-diastolic volume determines cardiac output. The stroke volume increases as a result of myofilament stretch due to increased ventricular filling and venous return (Allen and Kentish 1985). In the stretched myofilament, thin filament calcium sensitivity is enhanced and interfilament lattice spacing is reduced to maintain this length–tension relationship (Kobirumaki-Shimozawa et al. 2014). This decreased lattice spacing would increase the probability of disordered myosin binding actin, while the increased calcium sensitivity would facilitate their transition into a strongly bound state. This strong binding may then cause a distortion of the thick filament resulting in the release of SRX myosin from the thick filament through the hypothetical mechanism suggested by Moss and Fitzsimons (2010).
It is believed that the giant myofibrillar protein titin decreases the interfilament lattice spacing at long sarcomere lengths and may also destabilise the thick filament generating more force producing myosins (Fukuda et al. 2001). At longer sarcomere lengths, where the ordering of the thick filament can be destabilised, myosin heads may shift from the SRX state into the disordered relaxed state. Here, they could bind more readily to actin due to both decreased interfilament spacing and increased Ca2+ sensitivity of the thin filament, resulting in an increase in force production. It would therefore be interesting to examine the length dependency of the SRX state in activated fibres at different sarcomere lengths.
SRX in the failing heart
SRX state may be involved in heart disease, particularly in hypertrophic cardiomyopathy (HCM). HCM is commonly associated with increased energy cost of tension generation through inefficient or excessive ATP usage (Ashrafian et al. 2003). This may come about by shifting myosin heads out of the energy-conserving SRX state into the disordered relaxed or active states, where ATP usage is greatly increased.
High incidences of HCM-related mutations occur in myosin. A number of these mutations appear close to the junction of myosin S2 rod and the “interacting heads” motif (discussed in detail below). One of these mutations occur in an area of the “free” myosin head termed the cardiomyopathy loop, while others occur in the region of the S2 rod with which this loop interacts (Alamo et al. 2008). Moore et al. (2012) suggest mutations near this interface may destabilise the ordered structure of the thick filaments thereby reducing the proportion of SRX and increasing the ATP utilisation leading to HCM.
Preserving the SRX state may be cardioprotective during stress, such as in early ischaemia or hypoxia (Hooijman et al. 2011), which enables the heart to reduce its energy usage and metabolic demands. This mechanism, known as myocardial hibernation, may result from an increased proportion of myosin in the SRX state. As discussed below, modulating the SRX state may also be useful in organ preservation for heart transplants (Hooijman et al. 2011).
Ultrastructure of the SRX state
Electron microscopy of tarantula thick filaments provided the first visualisation of intact myosin heads bound to the thick filament core (Crowther et al. 1985). The subsequent use of cryo-EM and improved image processing has shown that both myosin heads are bent “backwards” (Fig. 1) (Craig and Woodhead 2006; Woodhead et al. 2005). These heads anchor onto the thick filament surface via their interaction with the helical rod-like region of myosin (Woodhead et al. 2005). This binding of myosin heads to the core of the tarantula thick filament is consistent with the discovery of SRX myosin in the skeletal muscle of the tarantula (Naber et al. 2011b).
Multiple intra-molecular interactions were also found between the two heads of a single myosin molecule, forming a J-like structure termed the “interacting-heads motif” (Fig. 1b). These occur between the two heads and also between the essential and regulatory light chains (RLCs) (Alamo et al. 2008; Woodhead et al. 2005). Similar “interacting-heads motifs” have been observed in 3D reconstructions of vertebrate cardiac thick filaments from mice, fish and humans (AL-Khayat et al. 2013; Gonzalez-Sola et al. 2014; Zoghbi et al. 2008). In this motif, the converter domain of the “free” head interacts with the actin-binding domain of the “blocked” head switching both heads off. Not only does this prevent the “blocked” head from binding to actin, it also inhibits the ATPase activity of the “free” head (Fig. 1).
Intermolecular interactions between adjacent myosin dimers have been shown in tarantula thick filaments (see Fig. 1). This interaction occurs between the essential light chain (ELC) of the “blocked” head of one myosin dimer and the “free” head of the adjacent dimer (Woodhead et al. 2005). This structural framework supports the cooperative activation hypothesis of myosin in skeletal muscle. The “free” head of one myosin may directly induce the “blocked” head of the adjacent molecule to leave the surface of the thick filament (Brito et al. 2011; Sulbaran et al. 2013). The inhibition of ATP turnover seen in the SRX state is thought to result from the combination of these intra- and intermolecular interactions in addition to the binding of myosin heads to the thick filament (Cooke 2011).
Potential regulators of the SRX state
Regulatory light chain and SRX
The RLC of myosin is a potential regulator of the SRX state. RLC is a small protein that, together with the ELC stabilises the neck region of myosin (Rayment et al. 1993). Removal of these light chains decreases the sliding velocity of actin filaments in motility assays by more than 10-fold without affecting ATPase activity (Lowey et al. 1993). This indicates that the light chains are essential for converting chemical energy into mechanical work.
RLC in human cardiac muscle is phosphorylated at Ser-15 by the Ca2+/calmodulin-dependent myosin light chain kinase (MLCK) (Scruggs et al. 2010). The negative charge of the bound phosphate group repels the RLC causing the myosin head to move away from the thick filament core (Sweeney et al. 1994), disordering this highly organised structure (Levine et al. 1996). This repulsion is supported by electron micrographs and X-ray diffraction studies of tarantula thick filaments that show MLCK treatment disorders myosin heads in the relaxed state (Craig et al. 1987; Padron et al. 1991). Thus, RLC phosphorylation reduces the proportion of myosin heads in the SRX state.
RLC phosphorylation of vertebrate cardiac muscle by MLCK shows a greater density of cross-bridges near the thin filament, indicating a higher level of thick filament disorganisation (Colson et al. 2010). This increases resting and maximal force, Ca2+-sensitivity, and the rate of force development in cardiac trabeculae. Thus, RLC phosphorylation may shift myosin heads from the SRX into the disorganised relaxed state. Those relaxed myosin heads allow for the formation of weak-binding cross-bridges. Moreover, the presence of Ca2+ binds to and activates the thin filament regulatory proteins enabling the myosin heads to transition from the weak-to-strong actin binding state. Together, these mechanisms result in increased resting and active forces.
In permeabilised rat trabeculae exchanged with MLCK phosphorylated recombinant RLC enhances maximum power output and shortening velocity compared to dephosphorylated and native untreated trabeculae (Toepfer et al. 2013). The dephosphorylated trabeculae, and transgenic mice with non-phosphorylatable RLC, exhibited a 4-fold decrease in isometric force and reduced maximal power compared to RLC phosphorylated and native trabeculae (Scruggs et al. 2009; Toepfer et al. 2013). These transgenic mice also displayed systolic impairment with no change in end diastolic volume. Thus, RLC dephosphorylation may recruit myosin heads into the SRX state (Naber et al. 2011b; Stewart et al. 2010) that results in reduced force production.
In the tarantula, RLC phosphorylation disorders the helical structure of the thick filaments (Alamo et al. 2008; Craig et al. 1987) that thereby reduces the proportion of myosin in SRX (Naber et al. 2011b; Stewart et al. 2010). This is further supported by the highly organised orientation of spin-labelled nucleotide EPR probes seen in tarantula myosin, which is lost upon phosphorylation (Naber et al. 2011a).
The role of RLC phosphorylation in heart failure remains controversial. Toepfer et al. (2013) showed increased RLC phosphorylation in HCM hearts compared to donors. This is possibly due to the decrease of myosin heads in the SRX state that increases energy utilisation. This may contribute to the HCM phenotype described previously (Ashrafian et al. 2003). On the other hand, certain HCM-associated mutations in RLC inhibit its capacity to phosphorylate (Szczesna et al. 2001). Interestingly, RLC phosphorylation was also reduced in human dilated cardiomyopathy (DCM) compared to donors (van der Velden et al. 2003). This condition commonly presents with systolic dysfunction in patients that ultimately requires orthotopic heart transplantation (Jefferies and Towbin 2010). Presumably, drugs that reverse the RLC phosphorylation status by targeting the known phenotype of specific mutations would restore cardiac contractility by modulating the number of SRX and disordered myosins available for cross-bridge formation.
The role of myosin binding protein C
Cardiac myosin-binding protein C (cMyBP-C) is a thick filament-associated protein that modulates cross-bridge formation and kinetics (Koretz 1979; McClellan et al. 2001). It is localised in 7–9 transverse stripes across the A-bands (Craig and Offer 1976) and has multiple immunoglobulin-like (Ig) and fibronectin III-like domains (Gautel et al. 1995). The MyBP-C C-terminus binds to the thick filament, and its N-terminus has the ability to interact with both the myosin head and the actin filament (Moos et al. 1978; Shaffer et al. 2009). It is therefore a good candidate to regulate the SRX state.
cMyBP-C has four known serine residues that can be phosphorylated by a number of kinases (Barefield and Sadayappan 2010). Mice trabeculae treated with protein kinase A (PKA) which mimics beta-adrenergic activation has been demonstrated to enhance contractility. PKA treatment releases myosin from its binding site on the thick filament backbone, enabling them to move closer to the actin filaments (Colson et al. 2008, 2010). Moreover, while cMyBP-C mice had a larger radial displacement than wild-type mice, PKA phosphorylation of cMyBP-C knockout(KO) mice did not affect radial displacement of the myosin heads (Colson et al. 2007, 2008). This was supported by studies that used mouse models with constitutively dephosphorylated or phosphorylated mimetic cMyBP-C (Colson et al. 2012).
Electron micrographs of thick filaments from homozygous cMyBP-C KO mice display less order than wild-type mice, consistent with its role in thick filament stability (Kensler and Harris 2008). Notably, these thick filaments have a reduced proportion of myosin heads in the “interacting heads” motif (Zoghbi et al. 2008). This shift of myosin heads away from the thick filament supports the notion that cMyBP-C may regulate the SRX (Fig. 2).
Temperature
In skeletal muscle, temperature increases induce a higher proportion of myosin heads into the SRX state and elongates the lifetime of myosin ATP turnover (Stewart et al. 2010). While this has not been reported in cardiac muscle, it may provide a novel method for preserving hearts for transplantation. Donor hearts, usually chilled prior to and during its transportation, are limited to approximately 4 h due to factors such as ATP depletion leading to rigor mortis (Stringham et al. 1992). Thus, warmed explant hearts may induce higher portions of SRX myosin. This would conserve energy, and increase the time to rigor mortis, leading to a longer organ shelf life. Indeed, trials are underway that examine new strategies for warm transportation of donor hearts (Wagner 2011). However, the activity of other enzymes increase at higher temperatures adding to the metabolic rate. Thus, the development of a solution that sequestered myosin heads into the SRX state at a lower temperature could be the most beneficial strategy.
SRX as a therapeutic target
A misbalance between the intake and usage of calories is a known contributor of obesity. Thus, modulating the proportion of myosin heads in the disordered relaxed and SRX state in skeletal muscle may be a potential therapeutic pathway to increase the body’s energy consumption (for a detailed review, see Cooke 2011). Likewise, the increase in energy consumption by skeletal myosin could improve insulin resistance in skeletal muscle. This may facilitate an increase in insulin-mediated glucose uptake by the muscle, thus alleviating type two diabetes. Shifting 10 % of skeletal myosin in the SRX state into the relaxed state would result in a loss of 7 kg per year due to the global increase in skeletal myosin energy usage. One possibility is to develop pharmaceuticals that disrupt the “interacting-heads” motif. This decreases the proportion of heads in the SRX state and thus increases energy usage of the muscle. This would be particularly useful in the treatment of obesity and type two diabetes. This would be an ideal therapy for obese patients who may be unable to exercise; however, it may also be used in conjunction with exercise and healthy eating strategies for those who are more capable.
Presumably, modulating energetic output would also be of benefit to cardiovascular diseases. Coinciding with obesity, weight loss particularly around the gut region would reduce hypertensive strain on the heart (Poirier et al. 2006). SRX may also directly benefit the heart in cases such as DCM in which systolic function is impaired (Jefferies and Towbin 2010). Therapeutics with the ability to coax myosin out of SRX could invariably increase its contractility thereby maintaining cardiac output.
Numerous therapeutic small molecules have been reported; with at least two that target cardiac myosin, EMD 50733 and omecamtiv mecarbil (OM). EMD 50733 enhances contractility by activating myosin and may sensitise the thin filaments to calcium (Radke et al. 2014). This small molecule binds close to the converter region of myosin, and its presence decreased the lifetime of ATP turnover in purified β-cardiac myosin from approximately one ATP every 30 s to one ATP every 20 s per myosin head. It is therefore possible that EMD 50733 could destabilise the “interacting-heads” motif of SRX myosin in order to increase ATP utilisation. Despite its capacity to enhance contractility, EMD 50733 lacks specificity to myosin, and behaves as a calcium sensitiser in the myofilament. Increased calcium sensitivity in the heart has the potential to induce arrhythmias (Kass and Solaro 2006). Thus, compounds with more specificity than EMD 50733 are required.
Omecamtiv mecarbil (OM) is cardiac-specific myosin activator that has recently been undergoing clinical trials (Cleland et al. 2011). It was discovered in the search for small molecules that directly increased cardiac contractility without sensitising the regulatory proteins to calcium (Malik et al. 2011). This molecule also binds to the β-myosin heavy chain, close to the nucleotide binding site and the converter region. It speeds up the release of inorganic phosphate from the myosin heads, which is believed to increase the amount of myosin heads strongly bound to actin, thus increasing force production (Malik et al. 2011). In motility assays, OM causes a large decrease in actin sliding velocity, despite its dramatic increase in force production and modest increase in power (Wang et al. 2014). This decreased sliding velocity is the suggested cause of the lower resting heart rate seen in both human and canine trials of this drug (Cleland et al. 2011; Shen et al. 2010). Studies investigating the effect of OM on the SRX state of cardiac myosin may help to further understand the mechanical basis for this increased force production.
Summary
The existence of the SRX state of myosin provides new insights into the regulation of striated muscle contraction. Cardiac and skeletal SRX differ from each other (Hooijman et al. 2011; Stewart et al. 2010), suggesting that there is a different function for these two muscle types. Here, we propose that phosphorylation of thick filament proteins may alter the cardiac SRX leading to a modulation of the contractility of the heart. We also suggest that SRX may be altered in some forms of heart disease, particularly the cardiomyopathies. Drugs that alter the SRX may alter the contractility of the heart and thus may provide a novel treatment for heart failure in the future, while targeting the skeletal SRX may provide treatment for obesity. It is possible that cardiac SRX is cardioprotective during times of stress such as during ischaemia and could provide a new method for organ preservation during heart transplant.
References
Alamo L, Wriggers W, Pinto A, Bártoli F, Salazar L, Zhao F-Q, Craig R, Padrón R (2008) Three-dimensional reconstruction of tarantula myosin filaments suggests how phosphorylation may regulate myosin activity. J Mol Biol 384:780–797. doi:10.1016/j.jmb.2008.10.013
AL-Khayat HA, Kensler RW, Squire JM, Marston SB, Morris EP (2013) Atomic model of the human cardiac muscle myosin filament. Proc Natl Acad Sci U S A 110:318–323. doi:10.1073/pnas.1212708110
Allen DG, Kentish JC (1985) The cellular basis of the length-tension relation in cardiac muscle. J Mol Cell Cardiol 17:821–840. doi:10.1016/S0022-2828(85)80097-3
Ashrafian H, Redwood C, Blair E, Watkins H (2003) Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet 19:263–268. doi:10.1016/S0168-9525(03)00081-7
Barefield D, Sadayappan S (2010) Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol 48:866–875. doi:10.1016/j.yjmcc.2009.11.014
Brito R, Alamo L, Lundberg U, Guerrero JR, Pinto A, Sulbaran G, Gawinowicz MA, Craig R, Padron R (2011) A molecular model of phosphorylation-based activation and potentiation of tarantula muscle thick filaments. J Mol Biol 414:44–61. doi:10.1016/j.jmb.2011.09.017
Cleland JG, Teerlink JR, Senior R, Nifontov EM, Mc Murray JJ, Lang CC, Tsyrlin VA, Greenberg BH, Mayet J, Francis DP, Shaburishvili T, Monaghan M, et al (2011) The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet 378:676–683. doi:10.1016/s0140-6736(11)61126-4
Colson BA, Bekyarova T, Fitzsimons DP, Irving TC, Moss RL (2007) Radial displacement of myosin cross-bridges in mouse myocardium due to ablation of myosin binding protein-C. J Mol Biol 367:36–41. doi:10.1016/j.jmb.2006.12.063
Colson BA, Bekyarova T, Locher MR, Fitzsimons DP, Irving TC, Moss RL (2008) Protein kinase A–mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res 103:244–251. doi:10.1161/circresaha.108.178996
Colson BA, Locher MR, Bekyarova T, Patel JR, Fitzsimons DP, Irving TC, Moss RL (2010) Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J Physiol 588:981–993. doi:10.1113/jphysiol.2009.183897
Colson BA, Patel JR, Chen PP, Bekyarova T, Abdalla MI, Tong CW, Fitzsimons DP, Irving TC, Moss RL (2012) Myosin binding protein-C phosphorylation is the principal mediator of protein kinase a effects on thick filament structure in myocardium. J Mol Cell Cardiol 53:609–616. doi:10.1016/j.yjmcc.2012.07.012
Cooke R (2011) The role of the myosin ATPase activity in adaptive thermogenesis by skeletal muscle. Biophys Rev 3:33–45. doi:10.1007/s12551-011-0044-9
Cooke R, Pate E (1985) The effects of ADP and phosphate on the contraction of muscle fibers. Biophys J 48:789–798. doi:10.1016/S0006-3495(85)83837-6
Craig R, Offer G (1976) The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B 192:451–461
Craig R, Woodhead JL (2006) Structure and function of myosin filaments. Curr Opin Struct Biol 16:204–212. doi:10.1016/j.sbi.2006.03.006
Craig R, Padron R, Kendrick-Jones J (1987) Structural changes accompanying phosphorylation of tarantula muscle myosin filaments. J Cell Biol 105:1319–1327
Cremo CR, Neuron JM, Yount RG (1990) Interaction of myosin subfragment 1 with fluorescent ribose-modified nucleotides. a comparison of vanadate trapping and SH1-SH2 crosslinking. Biochemistry 29:3309–3319. doi:10.1021/bi00465a023
Crowther RA, Padron R, Craig R (1985) Arrangement of the heads of myosin in relaxed thick filaments from tarantula muscle. J Mol Biol 184:429–439
Ferenczi MA, Homsher E, Simmons RM, Trentham DR (1978) Reaction mechanism of the magnesium ion-dependent adenosine triphosphatase of frog muscle myosin and subfragment 1. Biochem J 171:165–175
Fukuda N, Sasaki D, Ishiwata S, Kurihara S (2001) Length dependence of tension generation in rat skinned cardiac muscle: role of titin in the Frank-Starling mechanism of the heart. Circulation 104:1639–1645
Gautel M, Zuffardi O, Freiburg A, Labeit S (1995) Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 14:1952–1960
Gonzalez-Sola M, Al-Khayat HA, Behra M, Kensler RW (2014) Zebrafish cardiac muscle thick filaments: isolation technique and three-dimensional structure. Biophys J 106:1671–1680. doi:10.1016/j.bpj.2014.01.050
Gutierrez G, Pohil RJ, Narayana P (1989) Skeletal muscle O2 consumption and energy metabolism during hypoxemia. J Appl Physiol (1985) 66:2117–2123
Hooijman P, Stewart MA, Cooke R (2011) A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J 100:1969–1976. doi:10.1016/j.bpj.2011.02.061
Huxley H, Hanson J (1954) Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature 173:973–976
Huxley AF, Niedergerke R (1954) Structural changes in muscle during contraction: interference microscopy of living muscle fibres. Nature 173:971–973
Jefferies JL, Towbin JA (2010) Dilated cardiomyopathy. Lancet 375:752–762. doi:10.1016/s0140-6736(09)62023-7
Kass DA, Solaro RJ (2006) Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation 113:305–315. doi:10.1161/circulationaha.105.542407
Kensler RW, Harris SP (2008) The structure of isolated cardiac Myosin thick filaments from cardiac Myosin binding protein-C knockout mice. Biophys J 94:1707–1718. doi:10.1529/biophysj.107.115899
Kobirumaki-Shimozawa F, Inoue T, Shintani SA, Oyama K, Terui T, Minamisawa S, Ishiwata S, Fukuda N (2014) Cardiac thin filament regulation and the Frank-Starling mechanism. J Physiol Sci 64:221–232. doi:10.1007/s12576-014-0314-y
Koretz JF (1979) Effects of C-protein on synthetic myosin filament structure. Biophys J 27:433–446. doi:10.1016/s0006-3495(79)85227-3
Kushmerick MJ, Paul RJ (1976) Aerobic recovery metabolism following a single isometric tetanus in frog sartorius muscle at 0 degrees C. J Physiol 254:693–709
Levine RJ, Kensler RW, Yang Z, Stull JT, Sweeney HL (1996) Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys J 71:898–907. doi:10.1016/s0006-3495(96)79293-7
Lowey S, Waller GS, Trybus KM (1993) Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature 365:454–456
Malik FI et al (2011) Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331:1439–1443. doi:10.1126/science.1200113
McClellan G, Kulikovskaya I, Winegrad S (2001) Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys J 81:1083–1092. doi:10.1016/S0006-3495(01)75765-7
McNamara J, Li A, MacDonald P, dos Remedios C, Cooke A (2014) Does the Super-relaxed State of Myosin Exist in the Human Heart? 18th International Biophysics Congress, Brisbane http://www.iupab2014.org/assets/IUPAB/NewFolder/iupababstracts.pdf
Moore JR, Leinwand L, Warshaw DM (2012) Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res 111:375–385. doi:10.1161/CIRCRESAHA.110.223842
Moos C, Mason CM, Besterman JM, Feng I, Nan M, Dubin JH (1978) The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J Mol Biol 124:571–586
Moss RL, Fitzsimons DP (2010) Regulation of contraction in mammalian striated muscles–the plot thick-ens. J Gen Physiol 136:21–27. doi:10.1085/jgp.201010471
Myburgh KH, Franks-Skiba K, Cooke R (1995) Nucleotide turnover rate measured in fully relaxed rabbit skeletal muscle myofibrils. J Gen Physiol 106:957–973
Naber N, Cooke R, Pate E (2011a) EPR spectroscopy shows oriented myosin heads in relaxed muscle fibers. Biophys J 100:130a doi:10.1016/j.bpj.2010.12.914
Naber N, Cooke R, Pate E (2011b) Slow myosin ATP turnover in the super-relaxed state in tarantula muscle. J Mol Biol 411:943–950. doi:10.1016/j.jmb.2011.06.051
Padron R, Pante N, Sosa H, Kendrick-Jones J (1991) X-ray diffraction study of the structural changes accompanying phosphorylation of tarantula muscle. J Muscle Res Cell Motil 12:235–241
Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH (2006) obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 american heart association scientific statement on obesity and heart disease from the obesity committee of the council on nutrition, physical activity, and metabolism. Circulation 113:898–918. doi:10.1161/circulationaha.106.171016
Radke MB, Taft MH, Stapel B, Hilfiker-Kleiner D, Preller M, Manstein DJ (2014) Small molecule-mediated refolding and activation of myosin motor function. eLife 3:e01603. doi:10.7554/eLife.01603
Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261:50–58
Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM, Solaro RJ (2009) Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem 284:5097–5106. doi:10.1074/jbc.M807414200
Scruggs SB, Reisdorph R, Armstrong ML, Warren CM, Reisdorph N, Solaro RJ, Buttrick PM (2010) A novel, in-solution separation of endogenous cardiac sarcomeric proteins and identification of distinct charged variants of regulatory light chain. Mol Cell Proteomics 9:1804–1818. doi:10.1074/mcp.M110.000075
Shaffer JF, Kensler RW, Harris SP (2009) The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem 284:12318–12327. doi:10.1074/jbc.M808850200
Shen Y-T, Malik FI, Zhao X, Depre C, Dhar SK, Abarzua P, Morgans DJ, Vatner SF (2010) Improvement of cardiac function by a cardiac myosin activator in conscious dogs with systolic heart failure. Circ Heart Fail 3:522–527
Stewart MA, Franks-Skiba K, Chen S, Cooke R (2010) Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci U S A 107:430–435. doi:10.1073/pnas.0909468107
Stringham JC, Southard JH, Hegge J, Triemstra L, Fields BL, Belzer FO (1992) Limitations of heart preservation by cold storage. Transplantation 53:287–294
Sulbaran G, Biasutto A, Alamo L, Riggs C, Pinto A, Mendez F, Craig R, Padron R (2013) Different head environments in tarantula thick filaments support a cooperative activation process. Biophys J 105:2114–2122. doi:10.1016/j.bpj.2013.09.001
Sweeney HL, Yang Z, Zhi G, Stull JT, Trybus KM (1994) Charge replacement near the phosphorylatable serine of the myosin regulatory light chain mimics aspects of phosphorylation. Proc Natl Acad Sci U S A 91:1490–1494
Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, Zhi G, Stull JT, Potter JD (2001) Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2 + binding, and phosphorylation. J Biol Chem 276:7086–7092. doi:10.1074/jbc.M009823200
Toepfer C, Caorsi V, Kampourakis T, Sikkel MB, West TG, Leung M, Al-Saud SA, MacLeod KT, Lyon AR, Marston SB, Sellers JR, Ferenczi MA (2013) Myosin regulatory light chain (RLC) phosphorylation change as a modulator of cardiac muscle contraction in disease. J Biol Chem 288:13446–13454. doi:10.1074/jbc.M113.455444
van der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PML, Hasenfuss G, Stienen GJM (2003) The effect of myosin light chain 2 dephosphorylation on Ca2 + −sensitivity of force is enhanced in failing human hearts. Cardiovasc Res 57:505–514. doi:10.1016/s0008-6363(02)00662-4
Wagner FM (2011) Donor heart preservation and perfusion. Appl Cardiopulm Pathophysiol 15:198–206
Wang Y, Ajtai K, Burghardt TP (2014) Analytical comparison of natural and pharmaceutical ventricular myosin activators. Biochemistry 53:5298–5306. doi:10.1021/bi500730t
Wilson C, Naber N, Pate E, Cooke R (2014) the myosin inhibitor blebbistatin stabilizes the super-relaxed state in skeletal muscle. Biophys J 107:1637–1646. doi:10.1016/j.bpj.2014.07.075
Wood DS, Zollman J, Reuben JP, Brandt PW (1975) Human skeletal muscle: properties of the “chemically skinned” fiber. Science 187:1075–1076. doi:10.1126/science.187.4181.1075
Woodhead JL, Zhao F-Q, Craig R, Egelman EH, Alamo L, Padron R (2005) Atomic model of a myosin filament in the relaxed state. Nature 436:1195–1199 http://www.nature.com/nature/journal/v436/n7054/suppinfo/nature03920_S1.html
Woodward SK, Eccleston JF, Geeves MA (1991) Kinetics of the interaction of 2′(3′)-O-(N-methylanthraniloyl)-ATP with myosin subfragment 1 and actomyosin subfragment 1: characterization of two acto-S1-ADP complexes. Biochemistry 30:422–430
Zoghbi ME, Woodhead JL, Moss RL, Craig R (2008) Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci U S A 105:2386–2390. doi:10.1073/pnas.0708912105
Conflict of interest
This article does not contain any studies performed by the any of the authors involving animal or human experimentation. J.M., A.L., C.d.R., R.C. each declare that they have no conflict of interest. Figure 3 (B)-(D) was reprinted from Biophysical Journal, 100(8), Hooijman P, Stewart MA, Cooke R, A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. pp. 1969–1976, 2011, with permission from Elsevier. Figure 1 (B) was reprinted from Current Opinion in Structural Biology, 16(2), Craig R, Woodhead JL, Structure and function of myosin filaments. pp. 204–2012, 2006, with permission from Elsevier.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: Biophysics of Human Heart Failure
Rights and permissions
About this article

Cite this article
McNamara, J.W., Li, A., dos Remedios, C. et al. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys Rev 7, 5–14 (2015). https://doi.org/10.1007/s12551-014-0151-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-014-0151-5