
Abstract
A new routine method to measure 227Ac in water samples was developed. Actinium-227 was separated from potential radioactive and matrix interferences using a DGA resin (Eichrom) at optimal extraction and elution conditions (2 M and 15.7 M HNO3, respectively). This was possible because Ac was pre-concentrated and separated from Ca using titanium phosphate co-precipitation at pH 3.5. Finally, 227Ac was counted by alpha spectrometry for 48 h after a cerium fluoride micro-precipitation. A minimal detection limit of 0.03 ± 0.01 Bq l−1 was obtained. The method was validated using spiked water samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Actinium-227 is present at trace level in the environment as part of the 235U decay series. It has a radiological half-life (t1/2) of 21.772 ± 0.003 y [1], and decays mainly through beta emission to 227Th (98.58%) (t1/2 = 18.7 d) and slightly through alpha emission to 223Fr (1.42%) (t1/2 = 22.00 ± 0.07 min) [1]. Actinium-227 is considered a very toxic radionuclide because it deposits into the skeleton and liver following ingestion [2] and then decays to a series of short-lived alpha emitters [1]: 223Ra, 219Rn, 215Po, and 211Bi, which have radiological half-lives of 11.43 ± 0.03 d, 3.98 ± 0.03 s, 1.781 ± 0.004 ms, and 2.15 ± 0.02 min, respectively.
Actinium-227 monitoring could be needed for drinking, waste, and environmental waters in the vicinity of some uranium, rare-earth, and radiopharmaceutical facilities to ensure public health and to comply with national regulations. Actinium-227 is a waste product of the uranium and rare-earth mining and purification industries [3,4,5,6]. Also, 227Ac is of great importance in the medical field for the production of 223Ra, a medical isotope used for the treatment of some advanced prostate cancers [7,8,9]. The highest activity concentration guidance value issued by the World Health Organization (WHO) for safe consumption of drinking water for 227Ac is 0.1 Bq l−1 [10]. The exemption quantities for 227Ac in waste water in Canada [11] and the United Kingdom [12] are 10 and 0.1 Bq l−1, respectively. A method to monitor 227Ac activity in water with a minimum detectable activity (MDA) concentration of 0.1 Bq l−1 would be sufficiently sensitive for most routine monitoring applications and would safeguard public health.
Actinium-227 can be measured by alpha spectrometry [13], liquid scintillation counting (LSC) [14], and gas flow proportional counting [15]. Alpha spectrometry is the preferred method because it is more sensitive, there are fewer spectral interferences, and 225Ac can be used as a recovery tracer. To measure 227Ac by alpha spectrometry, 227Ac is pre-concentrated (e.g., with MnO2), purified, and deposited as a thin layer (e.g., electroplated) [13, 15]. Then, the 225Ac tracer is measured shortly after separation because it decays relatively quickly (t1/2 = 10.0 ± 0.1 d). The progenies of 227Ac are measured about 100 days after separation, when they reach their maximum activity, to obtain an optimal sensitivity. Most analytical methods to determine 227Ac in water have been developed to measure very low traces of 227Ac in sea water (~ 0.8–80 µBq l−1), mainly for oceanographic studies [13, 15,16,17,18]. These methods are too tedious and time-consuming for routine monitoring because a large volume of sea water (> 50 l) and a long time for the progenies to grow are needed [13]; thus, new routine monitoring methods more adapted to the routine determination of 227Ac in water samples are needed.
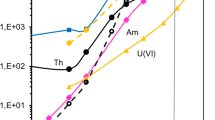
Precipitation [19], solvent extraction [6], ion exchange chromatography [16], and extraction chromatography (EXC) [20] can be used to separate Ac from potential matrix and radiological interferences. The commercially available EXC resin DGA [21] is commonly used to purify trivalent actinides and lanthanides. Dulaiova et al. [20] extracted Ac(III) in 4 M HCl (retention capacity (k′) of ~ 20) on the DGA resin (normal), as Ca(II) and Ra(II) are less retained (k′ < 1). Then Ac(III) was eluted with 2 M HCl (k′ = 0.5) while the other actinides remained on the resin (k′ > 20). The main disadvantage of this method is that the retention capacity for Ac(III) is very low in 4 M HCl (k′ = 20), which could lead to poor recoveries in some environmental water samples due to the presence of organic ligands. Actinium extraction on DGA resin differs significantly in nitric acid solutions from other actinides and lanthanides extraction: the Ac(III) retention capacity is optimal at 2 M HNO3 (k′ = 2000) and decreases at higher HNO3 molarities, whereas for other actinides and lanthanides the k′ increases with acid molarity to reach a maximum value at 10 M HNO3 [21]. The extraction of Ac(III) in 2 M HNO3 on the DGA resin would be ideal for its purification, but to our knowledge no such method has been developed because Ca(II) is also strongly extracted on the resin under the same conditions. Calcium interferes with Am(III) uptake on the DGA resin [21], and this is expected to be similar for Ac(III). Also, if the thin layer source of Ac for alpha spectrometry measurement is prepared using CeF3 micro-precipitation, Ca(II) will precipitate as CaF2 and directly decrease the alpha spectrometry resolution.
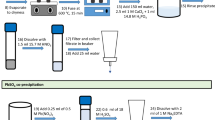
A novel and rapid method to purify and determine 227Ac in water samples using DGA resin was developed. Actinium was pre-concentrated with titanium phosphate at pH 3.5 from a 1 l water sample, which left Ca(II) in solution and prevented it from interfering with the purification and measurement steps (Fig. 1, steps 15–18). Then, the precipitate was dissolved in 2 M HNO3 and passed through a DGA resin. Actinium was eluted from the resin using 15.7 M HNO3, which left the other actinides and lanthanides strongly retained on the resin. Finally, Ac(III) was co-precipitated with CeF3 and immediately measured by alpha spectrometry for 48 h.

Schematic representation of the method
Experimental
Reagents and standards
All solutions used for this work were prepared using ultrapure water from a Millipore Direct-Q5 water purification system (Billerica, MA, USA). Trace metal grade acids (phosphoric acid (H3PO4), nitric acid (HNO3), hydrochloric acid (HCl), and hydrofluoric acid (HF)), hydrogen peroxide (H2O2), sodium hydroxide (NaOH), sodium chloride (NaCl), and cerium nitrate Ce(NO3)3 were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Titanium trichloride (TiCl3) and zirconyl chloride (ZrOCl2) in HCl were obtained from Sigma Aldrich (Oakville, ON, Canada). DGA® extraction chromatography resin (normal, 50–100 µm) containing N,N,N,N’-tetra-n-octyldiglycolamide and pre-packed in 2 ml cartridges was purchased from Eichrom Technologies (Lisle, IL, USA). Ethanol was obtained from Commercial Alcohols (Mississauga, ON, Canada). Certified solutions of 90Sr, 209Po, 147Pm, 226Ra, 227Ac, 231 Pa, 229Th, 237Np, 239Pu, and 244Cm were obtained from Eckert and Ziegler (Valencia, CA, USA). Solutions of natural Th and U were obtained from SCP Science (Baie d’Urfé, QC, Canada).
Instruments
All samples measurement were performed using an Octete Plus® alpha spectrometer with eight 450 mm2 ULTRA-AS ion-implanted silicon detectors (AMETEK/ORTEC, Oak Ridge, TN, USA). For method development, some beta emitters were measured using a Hidex 300 SL liquid scintillation counter (Hidex Oy, Turku, Finland).
Procedure
Sample preparation
A 1 l water sample was filtered using a 45 µm pore size filter to remove suspended particles and acidified with 10 ml of 15.7 M HNO3 (Fig. 1, steps 1 and 2). Actinium adsorbed on the suspended particles is not considered for this method. The filtered water sample and 20 mBq of 229Th tracer, in equilibrium with its daughter 225Ac, were weighed in a glass beaker (Fig. 1, steps 3 and 4). To the water sample, 0.6 ml of TiCl3 10% mv−1 in HCl and 0.6 ml of 14.8 M H3PO4 were added (Fig. 1, steps 5 and 6). The sample was mixed using a magnetic stirrer and a stirring plate until Ti(III) was fully oxidized to TiO2+ (absence of purple color), which took only a few minutes (Fig. 1, step 5). Note that it was preferable to add Ti(III) from a solution and oxidize it with HNO3 instead of adding Ti(IV) from a solution or salt, because Ti(III) solution is much easier to obtain commercially.
Actinium pre-concentration and calcium removal
The sample pH was adjusted to 3.50 with a 40% NaOH solution and mixed for 5 min to co-precipitate AcPO4 with titanium phosphate (Fig. 1, step 7). The pH was adjusted to 3.50 to optimize the precipitation of AcPO4 and minimize the precipitation of CaHPO4. The precipitate was isolated by centrifugation (3500 rpm for 3 min) using 500 ml centrifugation tubes (Fig. 1, step 8). The precipitate was rinsed and re-centrifuged three times with 100 ml of a 3% NaCl solution to remove as much Ca(II) as possible (Fig. 1, step 9). A NaCl solution was used instead of water to minimize the dispersion of the precipitate in solution during the centrifugation step, which increased the method’s recovery.
After centrifugation, the precipitate was dissolved with 1.25 ml of 15.7 M HNO3 and 0.25 ml of 30% vv−1 H2O2 (Fig. 1, step 10) (H2O2 is used to complex Ti(IV)). This solution was transferred to a 50 ml plastic centrifugation tube using a funnel, and the volume was completed to 10 ml with water to adjust the acid molarity to 2 M HNO3 (Fig. 1, steps 11). The transfer was necessary because the gradation on the 500 ml centrifugation tube was not precise enough to adjust the acid molarity. The 500 ml centrifugation tube was rinsed twice with about 5 to 10 ml of 2 M HNO3 and the rinsing solution transferred to the 50 ml centrifugation tube to minimize transfer losses (Fig. 1, step 12). The dissolved sample in the 50 ml centrifugation tube was centrifuged for 1 min at 3700 rpm and the supernatant filtered through a 0.1 µm pore size filter to remove remaining particles that could clog the resin (Fig. 1, steps 13 and 14).
Resin purification
Actinium was separated from potential interferences using a DGA resin in a 2 ml cartridge. The resin was put on top of a multi-hole vacuum box using the appropriate connectors and reservoirs. The resin was conditioned with 5 ml of 2 M HNO3 (Fig. 1, step 15) and the sample was passed through the resin at a flow rate of about 1 to 2 ml min−1 (Fig. 1, step 16). The resin was rinsed with 20 ml of 0.44 M H2O2 in 2 M HNO3 solution to remove TiO2+ and elements that were weakly retained on the resin (Fig. 1, step 17). Actinium was selectively eluted using 10 ml of 15.7 M HNO3 into a glass beaker (Fig. 1, step 18) and the elution time was recorded.
micro-precipitation
The eluate containing Ac was carefully evaporated to dryness on a hot plate to avoid losses by splattering (Fig. 1, step 19). The residue was re-dissolved with about 10 ml of 0.1 M HCl and transferred into a 50 ml plastic centrifuge tube (Fig. 1, steps 20 and 21). The nitric acid solution was evaporated to dryness because the yield of the CeF3 micro-precipitation is poor in highly acidic solutions. For each sample, 50 µg of Ce(III) and 0.5 ml of 29 M HF were added to co-precipitate AcF3 with a micro-precipitate of CeF3 (Fig. 1, steps 22 and 23). The solution was shaken, left aside for 5 min, and filtered through a 0.1 µm pore size filter (Fig. 1, steps 24 and 25). The filter was rinsed with 1 ml of ethanol and placed on a sticky metal planchet (Murphy Die & Machine, North Quincy, MA, USA) with the precipitate on top (Fig. 1, step 26). The filter was left to dry for a few minutes and then counted by alpha spectrometry for 48 h (Fig. 1, step 27).
Counting and activity calculation
The 227Ac activity concentration (AAc227) in Bq l−1 was calculated using Eq. 1:
where AAc225 is the activity of 225Ac tracer added (from 229Th) in Bq, CRAc227 the net count rate of 227Ac (count·s−1), b the fraction of 227Ac that decays through alpha emission (e.g., 0.0142) [1], and V the sample volume in liters. Note that the sample volume can be replaced by the sample mass. ACRAc225 is the corrected net count rate of 225Ac (count·s−1), calculated according to Eq. 2:
where CRAt217 is the net count rate of 217At in Bq, λAc225 is the decay constant of 225Ac (ln(2)/t1/2), t1 is the time elapsed between separation and counting in seconds, and t2 is the counting time in seconds. The count rate of 225Ac needs to be corrected because the isotope decays significantly while it is counted. The net count rate of 225Ac is obtained indirectly from 217At because the 217At peak is free from interferences. Astatine-217 is in transient equilibrium with 225Ac after about 30 min. The decay of 227Ac can usually be neglected if the sample is counted within a few days following the separation. The 227Ac activity measured should be decay-corrected to the sampling date.
Method development
Actinium elution profile from DGA Resin
To determine the elution profile of Ac(III) on the DGA resin, a 20 ml solution of 2 M HNO3 containing 100 mBq of 229Th (225Ac) was prepared. A DGA resin was conditioned with 5 ml of 2 M HNO3. The 299Th (225Ac) solution was passed through the resin and the resin was rinsed with 20 ml of 2 M HNO3. Actinium was eluted from the resin by fractions of 5 ml with 15.7 M HNO3. Each fraction was collected in an individual glass beaker. The load and rinse solutions were also collected to ensure that Ac(III) was completely retained by the resin under these conditions. The fractions eluted were evaporated to dryness and re-dissolved in 10 ml of 0.1 M HCl. The CeF3 micro-precipitation steps described in the procedure section (Fig. 1, steps 22–26) were performed and the test samples were measured by alpha spectrometry. The elution profile test was performed three times.
Purification and isolation of Ac
The method decontamination factor (DF) was determined by spiking water samples with a known activity or amount of an element (232Th, 238U, and 231 Pa (1 Bq); 209Po, 226Ra, 237Np, 239Pu, and 244Cm (0.1 Bq); 90Y (10 Bq); 147Pm (100 Bq); and Ca (13.5 g)) that could potentially interfere. The method was then applied. The DF was calculated by dividing the activity or amount added by the activity or amount measured. Each test was repeated twice. The isotopes were measured by alpha spectrometry after the micro-precipitation step (Fig. 1, step 27), except for 90Y, 147Pm, and Ca. The isotope 90Y was obtained by purifying a 90Sr solution using a Sr resin (2 ml cartridges, 50–100 µm, 4,4′(5′)-di-t-butylcyclohexano-18-crown-6 in 1-octanol) (Eichrom, Lisle, IL, USA) [21]. The isotopes 90Y and 147Pm were measured by dissolving the CeF3 precipitate on the disc used for alpha spectrometry measurement (Fig. 1, step 26) in 10 ml of 0.1 M HCl. The solution was transferred to an LSC vial, and 10 ml of Ultima Gold AB (PerkinElmer, Guelph, ON, Canada) liquid scintillation cocktail were added. The 90Y and 147Pm were then counted by LSC. Calcium was determined by measuring the mass of CaF2 on the final alpha disc (Fig. 1, step 26).
The optimal co-precipitation pH of Ac(III) with titanium phosphate (Fig. 1, step 7) was studied. Test samples were prepared with ultrapure water (1 l water and 30 mBq 229Th). The method procedure described above was applied, but the precipitation pH was varied between 0 and 7 at the titanium phosphate precipitation step (Fig. 1, step 7). The test was repeated twice.
Titanium phosphate
A rapid qualitative test was done to determine whether the salt obtained at the titanium phosphate co-precipitation step (Fig. 1, step 7) was a phosphate salt or a titanium oxide/hydroxide salt. In two separate 50 ml centrifuge tubes, 40 ml of water, 1 ml of 15.7 M HNO3, and 0.6 ml of a 10% TiCl3 solution were combined. In one of the tubes, 0.6 ml of 14.8 M H3PO4 was added. The pH was adjusted to 4. The precipitate obtained was centrifuged and rinsed three times with a 3% NaCl solution. The precipitate obtained was dissolved with 5 ml of 15.7 M HNO3 and 0.1 ml of 30% ZrOCl2 was added.
Figures of merit
The MDA was determined using 10 method blanks. The MDA was calculated using the Currie equation [22]. The MDA, relative bias (accuracy), and relative standard deviation (precision) were calculated as described in previous work [23]. The method was validated using river water and artificial sea water samples. The water samples were filtered and a known amount of 227Ac was added. The artificial sea water was prepared according to the method by Kester et al. [24] and the river water was collected at the Chalk River Laboratory site (Ottawa River, ON, Canada).
Results and discussion
Method development
Actinium extraction and elution on DGA resin
The elution profile of Ac(III) on the DGA resin using 15.7 M HNO3 is shown in Fig. 2. Most of the Ac(III) was eluted from the DGA resin using 10 ml of 15.7 M HNO3 (98 ± 3%). This volume was considered optimal to elute Ac(III) from the resin because it saved time during the elution and evaporation steps that followed (Fig. 1, steps 18 and 19), with only minor losses (~ 2%). About 95% (94 ± 5%) of the Ac(III) added was recovered and only 1 ± 1% of the Ac(III) passed through the resin (Fig. 2), which confirmed that Ac(III) was well retained by the resin in 2 M HNO3 and well eluted with 15.7 M HNO3.

Elution profile of Ac(III) on the DGA resin (extraction, 2 M HNO3; elution, 15.7 M HNO3)
Actinium purification
The DFs measured for potential radioactive and matrix interferences are presented in Tables 1 and 2, respectively. These DFs are high for all the radionuclides tested, including elements with oxidation state + 3, such as Am, Cm, Pm, and Y. This is in agreement with the retention capacity graphs published by Eichrom [21], which show that the retention capacity of elements of oxidation state + 3 increases when increasing the acid molarity, except for Ac(III). This new simple purification method allowed the efficient isolation of Ac(III) from potential interferences, and led to a very pure Ac fraction. The strategy could have further applications such as the separation of Ac(III) from lanthanides(III).
A high DF of 6000 ± 2000 was obtained for Ca(II) by co-precipitating Ac(III) at pH 3.5 with titanium phosphate (Table 2). The precipitation pH of CaHPO4 is about 4. Artificial sea water samples were prepared, and the method was first tested at pH 3.8 for the titanium phosphate co-precipitation step (Fig. 1, step 7). A small precipitate of CaF2 was randomly observed on some filters after the CeF3 micro-precipitation step (Fig. 1, step 26), which led to a poor alpha resolution (Fig. 3A). We chose to lower the precipitation pH of the titanium phosphate co-precipitation step to 3.5 to ensure a consistently good alpha resolution for all samples (Fig. 3B).

Example of alpha spectrum of 225Ac (~ 0.01 Bq) and.227Ac (~ 1 Bq) for an artificial sea water sample at A pH 3.8 (poor resolution) and B pH 3.5 (good resolution)
The Ac(III) recovery as a function of the pH at the titanium phosphate co-precipitation step (Fig. 1, step 7) is shown in Fig. 4. The chemical recovery was constant at pH ≥ 3 (54 ± 3%) and decreased significantly at lower pH. A pH of 3.5 was chosen for the titanium phosphate co-precipitation step because Ac was effectively co-precipitated without CaHPO4 and without affecting the chemical recovery.

Ac(III) recovery as a function of the pH at the titanium phosphate co-precipitation step
Titanium phosphate
Titanium was precipitated at pH 4 with and without H3PO4. Then, the salt obtained was rinsed and was dissolved with HNO3, and Zr(IV) was added. A precipitate was observed only in the tube in which H3PO4 was added. Zr3(PO4)4 is a very insoluble salt that precipitates in very acidic solutions. This test demonstrated that the precipitate obtained at the titanium phosphate co-precipitation step (Fig. 1, step 7) is in great part a phosphate salt, but this simple test does not exclude the possibility that some titanium oxide could also be present in the precipitate.
Figures of merit
Minimal detectable activity
An approximate MDA of 0.03 ± 0.01 Bq l−1 was obtained (Table 3). The MDA obtained is below the expected level of 0.1 Bq l−1, and is likely sufficient for assessing the safety of drinking and environmental waters.
River water and artificial sea water samples were spiked with a known amount of 227Ac and then measured using the developed method. The activity added as a function of the activity measured is shown in Fig. 5. In both cases, the activity measured corresponds to the activity added within the experimental uncertainty, which validates the method. The mean relative bias and the relative precision (all samples) were − 0.35% and 7.15%, respectively, and an average overall recovery of 67 ± 8% was obtained for these samples (Table 3). Note that the recovery could vary depending on the nature of the sample due to many factors such as particles in suspension (≤ 45 µm), salinity, and ligands present.

Spiked samples of A Ottawa River water and B artificial sea water (RL, reference level)
Method throughput
Four samples can be comfortably performed in one workday using this method (Table 3).
Conclusions
A new method was developed to measure 227Ac in water samples. A titanium phosphate co-precipitation was performed at pH 3.5 to pre-concentrate Ac(III) and separate it from Ca(II) interference. Then, Ac(III) was effectively separated from potential interferences, including lanthanides and actinides of oxidation state + 3, using a DGA resin. The method was validated using spiked river and artificial sea waters. The method is sufficiently sensitive to assess the safety of drinking water and to evaluate whether effluents can be released to the environment.
References
Tables of recommended data, Laboratoire National Henri-Becquerel. http://www.lnhb.fr/nuclear-data. Accessed Jan 2021
Storer J, Johnson O, Wellnitz J, Sanders P, Hale D (1952) Toxicology of actinium equilibrium mixture. United States Atomic Energy Commission, Oak Ridge
Petrow HG, Allen RJ (1963) Radiochemical determination of actinium in uranium mill effluents. Anal Chem 35(6):747–749
Rychkov VN, Semenishcev VS, Kirillov EV, Kirillov SV, Ryabukhina VG, Smyshlyaev DV, Bunkov GM, Botalov MS (2018) Radiochemical characterization and decontamination of rare-earth-element concentrate recovered from uranium leach liquors. J Radioanal Nucl Chem 317:203–213
Kosynkin VD, Moiseev SD, Vdovichev VS (1995) Cleaning rare earth elements from actinium. J Alloy Compd 225:320–323
Percival DR, Martin DB (1974) Sequential determination of radium-226, radium-228, actinium-227, and thorium isotopes in environmental and process waste samples. Anal Chem 46(12):1742–1749
Lewis B, Chalhoub E, Chalouhy C, Sartor O (2015) Radium-223 in bone-metastatic prostate cancer: current data and future prospects. Oncology 29(7):483–488
Abou DS, Pickett J, Mattson JE, Thorek DLJ (2017) A Radium-223 microgenerator from cyclotron-produced trace Actinium-227. Appl Radiat Isot 119:36–42
Soderquist CZ, McNamara BK, Fisher DR (2012) Production of high-purity radium-223 from legacy actinium beryllium neutron sources. Curr Radiopharm 5:244–252
Guidelines for drinking-water quality (2017) World Health Organization. 4th edition
Exemption quantities, schedule 1, Nuclear Substances and Radiation Devices Regulations, https://laws-lois.justice.gc.ca/eng/regulations/sor-2000-207/page-8.html#h-658291. Accessed Apr 2021
Radioactive substances act 1993, Schedule 1A, updated 2021 April 13. https://www.legislation.gov.uk/ukpga/1993/12/schedule/1A. Accessed Apr 2021
Geibert W, Vöge I (2009) Progress in the determination of 227Ac in sea water. Mar Chem 109(3–4):238–249
Eriksen DO, Ryningen B, Schoultz BW, Salberg G, Larsen RH (2012) Liquid scintillation spectroscopy of 227Ac and daughters. J Anal Sci Meth Instrum 2:33–36
Roy E, Sanial V, Lacana F, Beek P, Souhauta M, Charett MA, Handerson PB (2019) Insight into the measurement of dissolved 227Ac in seawater using radium delayed coincidence counter. Mar Chem 212:64–73
Geibert W, Rutgers van der Loeff MM, Hanfland C, Dauelsberg HJ (2002) Actinium-227 as a deep-sea tracer: sources, distribution and applications. Earth Planet Sci Lett 198:147–165
Shaw TJ, Moore WS (2002) Analysis of 227Ac in sea water by delayed coincidence counting. Mar Chem 78:197–203
Nozaki Y (1984) Excess 227Ac in deep ocean water. Nature 310:486–488
Rogers NE, Watrous RM (1955) Radiochemical separation of actinium and its daughters by means of lead sulfate. Anal Chem 27(12):2009–2012
Dulaiova H, Sims KWW, Charette MA, Prytulak J, Blusztajn JS (2013) A new method for the determination of low-level actinium-227 in geological samples. J Radioanal Nucl Chem 296:279–283
Eichrom technologies, Inc. https://eichrom.com/eichrom/products/dga-resins. Accessed Jan 2021
Currie LA (1968) Limits for qualitative detection and quantitative determination. Application to radiochemistry. Anal Chem 40:586–593
Guerin N, Dai X (2014) Determination of 210Po in drinking water and urine samples using copper sulfide microprecipitation. Anal Chem 86:6026–6031
Kester DR, Duedall IW, Connors DN, Pytkowicz RM (1967) Preparation of artificial sea water. Limnol Oceanogr 12(1):176–179
Funding
This work was supported by Atomic Energy of Canada Limited under the Canadian Federal Science and Technology program (project no. 51200.65.18.06).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial nor non-financial interests to disclose. The authors have no conflict of interest to disclose. The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guérin, N., Fabian, X., Leppinen, J. et al. Determination of 227Ac in water by alpha spectrometry after purification with titanium phosphate and DGA resin. J Radioanal Nucl Chem 331, 5781–5788 (2022). https://doi.org/10.1007/s10967-022-08619-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-022-08619-0