
Abstract
A method for the determination of 226Ra in drinking water samples by alpha spectrometry is described. The method uses 225Ra as yield tracer. Radium was purified using Ln Resin® and cation exchange resin, and electrodeposited onto a stainless steel disc from a solution of ammonium oxalate in nitric acid. This method produced high spectral resolutions and high recovery yields of 226Ra. Furthermore, the method allowed significant reduction in time required for the determination of 226Ra in drinking water samples, around 3 days, in comparison with other methods, which can reach 20 or 38 days to quantify this radionuclide.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Radium is an alkaline earth metal, and all isotopes are radioactive. Radium isotopes are present in the rocks and in groundwater; in geographical regions where there are naturally high concentrations of uranium and thorium in soil and rocks, the risk of their ingestion may be higher, as a result of consumption of water and agricultural products and livestock, which may lead to the accumulation of these nuclides, contributing to radiological dose. Radium isotopes can be easily incorporated into the bones due to its chemical and biological behavior similar to calcium, and the main recognized damage to the body is the production of bone tumors. 226Ra is one of the most important radioactive isotopes of the natural uranium series in terms of radiation dose, due to its relative long half-life (1600 years) and high radiotoxicity [1].
Many techniques can be employed for the determination of 226Ra; these include liquid scintillation counting [2–5], 222Rn emanation technique [6], γ-spectrometry [7–9], and α-particle spectrometry following radiochemical separation [10–12].
Routinely, the method for the determination of 226Ra, developed by Godoy [13], is performed by the radiochemical separation process through Ra coprecipitation with barium sulfate (BaSO4) followed by the filtration of precipitates. After filtration, it is necessary to wait for it to achieve the radioactive equilibrium condition with its daughter 222Rn, which takes about 38 days, corresponding to ten half-lives of daughter 222Rn. Expired this time, 222Rn is measured in ultra-low background gas-flow proportional counters.
Another method used, developed by Sill [14], is microprecipitation with Ba, Na2SO4 and acetic acid in a polypropylene filter, with determination of the chemical yield by quantification of 133Ba by γ-spectrometry, and determination of 226Ra by α-spectrometry.
Maxwell and Culligan [15] developed a method for the determination of 226Ra in environmental samples utilizing a rapid sodium hydroxide fusion method for solid samples, calcium carbonate precipitation to perform Ra preconcentration, and column separation steps to remove interferences. 225Ra was utilized as yield tracer. The separation process used cation exchange resin to remove calcium, Sr Resin® to remove barium and Ln Resin® to remove 225Ac and potential interferences. After the 226Ra purification, samples were prepared using barium sulfate microprecipitation in the presence of isopropanol for counting by α-spectrometry.
Crespo and Jiménez [16] developed a procedure for the purification of radium by cation exchange chromatography and source preparation by electrodeposition for α-spectrometry. Orlandini et al. [17] and Alvarado et al. [18] described on previous papers procedures for the determination of 226Ra in aqueous samples.
Crespo [19] reviewed some of the main electrodeposition methods for the preparation of alpha-radiation sources, and observed that there have been very few attempts to electrodeposit radium for routine determinations. Jia and Jia [20] reviewed the three most routinely used analytical techniques for radium isotopes determination in environmental samples, i.e., low-background gamma-spectrometry, liquid scintillation counting and alpha-spectrometry. Alpha-spectrometry, due to the high and stable yields, sensitivity, selectivity and traceability, as well as the high-energy resolution (FWHM: ~30 keV) of the alpha-spectra for environmental sample analyses has presented itself as a technique of wide applicability in the study fields of environmental science, health physics and geochemistry.
The main aim was to develop a rapid radiochemical method for radiological emergencies, which allows counting to begin as soon as possible after receipt of the sample.
The method described here allows that the radiochemical preparation and the electrodeposition of Ra to initiate the counting can be completed in 1 day (8 h). With the use of 225Ra as yield tracer, 226Ra is determined from a final count after deposition, and from the calculated 225Ra recovery determined after a suitable ingrowth period for 225Ac and daughters.
Experimental
Equipment and reagents
The radium sources were counted by α-spectrometry (Model Alpha Analyst with 12 Passivated Implanted Planar Silicon detectors and Genie 2000/Alpha Analyst spectroscopy systems, Canberra) with a counting efficiency of 18 %. The apparatus for electrodeposition was composed of an adjustable power supply (2 Adc) and acrylic cells of 22.3 mm internal diameter mounted on a cylindrical brass base; as cathode a stainless steel disc with 25 mm diameter, and as anode a platinum wire with 1 mm diameter and 80 mm length, with the spiral-shaped end of 7 mm diameter. The distance between cathode and anode was ~8 mm.
Composition of water samples of the Brazilian Intercomparison Program (PNI) were determined by inductively coupled plasma-optical emission spectrometer (ICP-OES) Model Liberty LR, Varian.
The platinum was added as a solution of dihydrogen hexachloroplatinate (H2PtCl6) taking amounts corresponding to ~400 µg of Pt. A standard solution of 226Ra, code 71L07 was supplied from Amersham and diluted to activity concentration of 24.1(12) × 10−2 Bq mL–1; a standard solution of 229Th(225Ra) code 16L97 was supplied from NBS and diluted to activity concentration of 75.8(11) × 10−2 Bq mL–1. Water samples of PNI used: sample 366, with activity concentration of 50(8) × 10−2 Bq L–1, sample 345, with activity concentration of 67(10) × 10−2 Bq L–1, sample 360, with activity concentration of 147(22) × 10−2 Bq L–1, sample 359, with activity concentration of 99(15) × 10−2 Bq L–1, and sample 282, with activity concentration of 128(19) × 10−2 Bq L–1. The following amounts of elements for these samples, determined by ICP-OES: Ba 31(3) × 10–6 g L−1; Ca 220(20) × 10–5 g L−1; Mg 147(17) × 10−6 g L−1.
Procedures
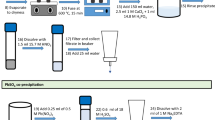
A description of the radiochemical and electrodeposition procedures is as follows:
-
1.
Prepare a Ln Resin® column (60 mL syringe attached to another 3 mL syringe; in this last syringe, 0.7 g of Eichrom Ln Resin® LN-B50-A, washed 3 times with deionized water, was packaged) by washing with 15 mL of 0.02 M HCl.
-
2.
Prepare a cation exchange column (Sigma-Aldrich DOWEX® 50WX8, 100–200 mesh, hydrogen form, 100 mm height, 8 mm i.d), by washing with 40 mL of 1 M HCl.
-
3.
Collect 50 mL of water and add 0.31 mL of standard solution of 229Th (225Ra). Evaporate to dryness and dissolve the residue in 10 mL of 0.02 M HCl.
-
4.
First purification step Pass the dissolved residue through a Ln Resin® column to retain actinium, and collect. Wash beaker with 5 mL of 0.02 M HCl, transfer to column and collect. Wash the column 2 times with 5 mL of 0.02 M HCl and collect. Time of separation Th and Ra from Ac is approximately 10 min. Join all the fractions and add 25 drops of 2 M HCl to reach pH ~1.
-
5.
Second purification step Pass the solution from step 4 through a cation exchange column to retain radium. Wash beaker with 5 mL of 0.02 M HCl and transfer to column. Wash the column with 50 mL of 2.5 M HCl and discard; time for this operation is approximately 2 h. Replace the beaker, elute Ra with 80 mL of 6 M HCl, collect and add 10 μL of H2PtCl6. Time of separation Th/Ra is approximately 2 h. The flow rate is 0.8–1.0 mL/min. Note this elution time.
-
6.
Evaporate the eluted radium to dryness and add 4 mL of HNO3, 2 drops of HClO4 and 2 mL of H2O2, to destroy any existing organic matter. Evaporate one more time.
-
7.
Add 4 mL of HNO3 and 2 mL of H2O2, and evaporate. Repeat this procedure 3 times.
-
8.
Dissolve the residue from step 7 in 10 mL of 0.17 M of ammonium oxalate adjusting the pH to 2.6 with drops of nitric acid, and transfer it to a deposition cell. Complete the transfer with the help of other 5 mL of 0.17 M of ammonium oxalate at pH 2.6. Perform the electrodeposition onto a highly polished stainless steel disc, at a constant current at 800 mA and 90 min electrodeposition time.
-
9.
Once the electrodeposition has finished, remove the disc and place it on a hot plate for fast drying at 250 °C for ~10 s. Then insert it into the alpha spectrometer for counting for a period of 150,000 s.
The methods developed by Hancock and Martin [21], Crespo [22] and Jia et al. [23] are based on the use of 225Ra as yield tracer and they all describe the calculation of recovery of 225Ra.
The activity of 226Ra in the sample is calculated using the net counts (background-correct), according to Eq. (1):
where C s is the net count of 226Ra in sample, ε is the efficiency of counter, T is the counting time, Q is the sample quantity (volume or mass), and Y is the chemical yield.
Results and discussion
The method for determination of 226Ra, developed by Godoy [13], although widely used, has some disadvantages: (a) a long time for analysis, which is inappropriate when you need quick answers to take protective measures; (b) the counting of 226Ra is performed indirectly through its daughter 222Rn, which can escape from the crystalline network formed in precipitation; (c) Ba is the chemical element used to monitor the chemical recovery process; this element is not the most appropriate: even though it is chemically similar, it has differences in solubility and that can lead to errors on the determination of chemical recovery of Ra.
There are several advantages in using α-spectrometry instead of other alternative methods, such as: higher sensitivity due to high-yield α decay process, low detector background and the elimination of interferences by a chemical separation process; the use of radioactive tracers to determine the chemical yield in each sample analyzed, the ability to determine activity concentrations of 226Ra without an ingrowth period of 222Rn and daughters, and the ability to determine activity concentrations of all the α-emitters Ra isotopes. However, problems in the chemical separation process of Ra from other Group II elements by alpha spectrometry may result in incorrect chemical yields, a spectrum with poor resolution, and the application of time-consuming analytical techniques. The method described here was developed to minimize these problems [21].
The experimental procedure of the method developed includes a column separation process using Ln Resin® as a first purification step to remove 225Ac and potential interferences from the sample, improving the separation step 225Ra/225Ac.
Since the 225Ac present in the sample was removed in the first purification step, the second purification step allowed the removal of calcium and barium, which were scarce in these samples. It was observed that the cation exchange column retains 229Th, once in the blank test (with the tracer alone) there was no vestige of the 229Th in the spectrum obtained in the energy of 4.84 MeV (58 %). Analysis of the blank suggests that there is no need to pass the sample by the anion exchange column for retention of thorium. According to Korkisch [24], thorium exhibits an extremely high affinity for strongly acidic cation exchange resins. Separations of thorium in an hydrochloric acid media of acidities ranging from 1 to 4 N allow this element to be separated from all mono- and divalent, and also from many higher-valent metal ions. The DOE method RP450 [25] describes the separation of 226Ra in aqueous samples through ion exchange, electrodeposition, and analysis by alpha spectrometry. In this procedure an AG 50 W-X8 cation-exchange resin in hydrochloric acid media of acidity 1.5 M, which retained thorium, was used.
For the blank test, 50 mL of deionized water was collected and 0.31 mL of a standard solution of 229Th was added. This solution was evaporated to dryness and dissolved the residue in 0.02 M HCl, and then all steps of the procedures were performed.
This procedure ensures the complete separation 229Th/225Ra. There are two purification steps: in the first, Ac is separated from Th and Ra and starts its growth. In the second, Th is separated from Ra. At this point the Ra is unsupported and starts to decay, however, if the time between the Ln Resin separation that removes Ac and the cation resin elution the correction is minimal (~1 % for approximately 4 h time difference). Therefore, the time to be recorded is only the time of the second purification step, although if both steps are made immediately one after the other, the difference in time is very short.
To check the validity of both the radiochemical procedure and the use of 225Ra as a tracer for the determination of 226Ra, three standard samples were tested. Each sample consisted of 1 mL of a standard solution of 226Ra and 0.31 mL of a standard solution of 229Th (225Ra). These samples were evaporated to dryness and the residue was dissolved in 10 mL of 0.02 M HCl. The radiochemical procedure was applied to these samples.
Table 1 presents the results obtained for the determination of 226Ra in three tested samples using standard solutions of 226Ra and 225Ra.
The activities of 226Ra found, as shown in Table 1, indicate a high recovery yield, confirming the high efficiency of electrodeposition of Ra and Ac.
The result obtained in sample 3 was slightly lower, probably due to some imprecision in the radiochemical procedure.
Figure 1 shows the spectrum obtained from a sample with a certified standard solution; the stainless steel disc was counted immediately after electrodeposition.

Spectrum obtained from sample no. 2, with resolution of 23 keV FWHM and recovery yield of 97 %
The spectra of 226Ra presented good resolutions, ranging from 23 keV to 28 keV FWHM. The counts of 226Ra were measured directly from peaks at 4.60 MeV and 4.78 MeV. The chemical recovery of 225Ra was determined from 217At peak (7.07 MeV).
Another test to check the validity of the radiochemical procedure for the determination of 226Ra was performed with PNI water samples; the complete radiochemical procedure was also applied to these samples.
The PNI water samples presented little volume, insufficient for performing at least three tests per sample; however, the results were within the limits determined by the certificates of these samples. The results obtained by the developed method were then compared with the methods currently used.
Table 2 compares the results obtained for the determination of the activity concentration of 226Ra in PNI water samples by the methods currently used, and by the method developed in this work.
Although the counts under the 217At peak were low, the calculated uncertainties were relatively small. The calculation of recovery of 225Ra was performed according to what was described in Jia et al. [23] and Hancock and Martin [21], the calculation of activity of 226Ra in the samples was according to Eq. (1).
The calculation of the combined standard uncertainty was defined from various sources of uncertainty, such as: uncertainty of activity of the certified tracer solution; uncertainty of volume, taken with certified pipettes, uncertainty of mass taken with certified scales, and estimated uncertainty of counting.
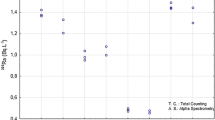
The methods employed for the determination of 226Ra in PNI water samples can be compared by applying a paired t test. This test shows with a statistical probability of approximately 96 % that both methods lead to the same results, once the Student’s t criterion 0.946 is considerably smaller than the critical value 2.13. The scatterplot of the data set concerning the activity concentration of 226Ra in PNI water samples, shown in Fig. 2, displays a linear regression with good correlation between the results obtained by the methods currently used, and by the method developed in this work.

Scatterplot of the data set of the activity concentration of PNI water samples, comparing the results obtained by the methods currently used, and by the method developed
Figure 3 shows the spectrum of PNI water sample; the stainless steel disc was counted immediately after electrodeposition. All spectra of activity concentration of 226Ra in PNI water samples showed high resolutions, ranging from 24 keV to 29 keV FWHM.

Spectrum of activity concentration of 226Ra in PNI 360 water sample, with resolution of 28 keV FWHM, and recovery yield of 92 %
The results indicated that the purification of radium was effective, with use of Ln Resin® for retention of actinium present in the sample, and use of cation exchange resin for retention and subsequent washing of interferences such as magnesium, calcium, and low content of barium in the sample, as can be observed by the high resolution of spectrum obtained in Fig. 3.
Few steps of radiochemical procedures were performed in this study, if compared to works of other authors, which carried out a much larger sequence of procedures for chemical separation of 226Ra, before radioactive source preparation for counting by alpha spectrometry.
The necessary time for execution of this method was around 3 days, which represents a significant reduction if compared to other methods.
Conclusions
The method described in this paper was developed to enable the preparation of high-resolution sources for the determination of 226Ra in drinking water samples by alpha spectrometry, and simultaneously to minimize the number of chemical operations and the operator time. The method allows the separation of 226Ra and effective removal of interferences, with high recovery yields and high spectral resolutions. The use of 225Ra as a tracer enables an accurate measure of chemical yield, via alpha counting of 217At. This method allows a significant reduction in the time required for determination of 226Ra in drinking water samples, around 3 days, in comparison with other methods, which can reach 20 or 38 days to quantify this radionuclide.
References
Analytical methodology for the determination of radium isotopes in environmental samples. International Atomic Energy Agency. http://www-pub.iaea.org/MTCD/Publications/PDF/IAEA-AQ-19_web.pdf. Accessed 20 Apr 2014
Escobar VG, Tomé FV, Lozano JC, Sanchez AM (1996) Determination of 222Rn and 226Ra in aqueous samples using a low-level liquid scintillation counter. Appl Radiat Isot 47:861–867
Przylibski TA, Gorecka J, Kula A, Fijalkowska-Lichwa L, Zagożdżon K, Zagożdżon P, Miśta W, Nowakowski R (2014) 222Rn and 226Ra activity concentrations in groundwaters of southern Poland: new data and selected genetic relations. J Radioanal Nucl Chem. doi:10.1007/s10967-014-3215-x
Fernandes PCP, Sousa WO, Julião LMQC, Dantas BM (2011) Development and validation of a technique for the determination of 226Ra and 228Ra by liquid scintillation in liquid samples. Radiat Prot Dosim. doi:10.1093/rpd/ncq434
Schettler G, Oberhänsli H, Hahne K (2015) Ra-226 and Rn-222 in saline water compartments of the Aral Sea region. Appl Geochem. doi:10.1016/j.apgeochem.2015.04.005
Wardaszko T, Grzybowska D, Nidecka J (1986) 222Rn and 226Ra in fresh waters: measurement method and results. Nucl Instrum Methods Phys Res B17:530–534
Tinker RA, Smith JD, Cooper MB (1995) An assessment of the selection criteria for an analytical method for radium-226 in environmental samples. J Radioanal Nucl Chem 193:329–336
Sartandel SJ, Jha SK, Bara SV, Tripathi RM (2014) Assessment of 226Ra and 228Ra activity concentration in west coast of India. J Radioanal Nucl Chem. doi:10.1007/s10967-014-3037-x
Küsters M, Schraven W (2009) Determination and differentiation of 226Ra and 222Rn by gamma-ray spectrometry in drinking water. J Radioanal Nucl Chem. doi:10.1007/s10967-009-7460-3
Erden PE, Dirican A, Seferinoğlu M, Yeltepe E, Şahin NK (2014) 238U, 234U and 226Ra concentrations in mineral waters and their contribution to the annual committed effective dose in Turkey. J Radioanal Nucl Chem. doi:10.1007/s10967-014-3105-x
Lozano JC, Rodríguez PB, Tomé FV, Leal-Cidoncha E (2012) Improvement of a method for the sequential determination of 210Pb, 226Ra, and uranium isotopes by LSC and alpha-particle spectrometry. Appl Radiat Isot. doi:10.1016/j.apradiso.2011.12.031
Jobbágy V, Dirican A, Wätjen U (2013) Radiochemical characterization of mineral waters for a European interlaboratory comparison. Microchem J. doi:10.1016/j.microc.2013.08.008
Godoy JM (1990) Methods for measuring radium isotopes: gross alpha and beta counting. In: Technical reports series 310—the environmental behaviour of radium. International Atomic Energy Agency. Chapter 3–5, pp 205–211
Sill CW (1987) Determination of radium-226 in ores, nuclear wastes and environmental samples by high-resolution alpha spectrometry. Nucl Chem Waste Manag 7:239–256
Maxwell SL, Culligan BK (2012) Rapid determination of 226Ra in environmental samples. J Radioanal Nucl Chem. doi:10.1007/s10967-012-1627-z
Crespo MT, Jiménez AS (1997) On the determination of radium by alpha-spectrometry. J Radioanal Nucl Chem 221:149–152
Orlandini KA, Gaffney JS, Marley NA (1991) An improved technique for the rapid assay of radium isotopes in water. Radiochim Acta 55:205–207
Alvarado JS, Orlandini KA, Erickson MD (1995) Rapid determination of radium isotopes by alpha spectrometry. J Radioanal Nucl Chem 194:163–172
Crespo MT (2012) A review of electrodeposition methods for the preparation of alpha-radiation sources. Appl Radiat Isot. doi:10.1016/j.apradiso.2011.09.010
Jia G, Jia J (2012) Determination of radium isotopes in environmental samples by gamma spectrometry, liquid scintillation counting and alpha spectrometry: a review of analytical methodology. J Environ Radioact. doi:10.1016/j.jenvrad.2011.12.003
Hancock GJ, Martin P (1991) Determination of Ra in environmental samples by α-particle spectrometry. Appl Radiat Isot 42:63–69
Crespo MT (2000) On the determination of 226Ra in environmental and geological samples by α-spectrometry using 225Ra as yield tracer. Appl Radiat Isot 53:109–114
Jia G, Torri G, Innocenzi P, Ocone R, Di Lullo A (2006) Determination of radium isotopes in mineral and environmental water samples by alpha-spectrometry. J Radioanal Nucl Chem 267:505–514
Korkish J (1969) Modern methods for the separation of rarer metal ions. Pergamon Press, London
RP-450—Determination of radium-226 in aqueous samples. United States Department of Energy. http://www.caslab.com/Test-Methods-Search/PDF/DOE-Method-RP450.pdf. Accessed 20 Apr 2014
Acknowledgments
This work was supported by the Poços de Caldas Laboratory of Brazilian Nuclear Energy Commission, and by the Federal University of Alfenas.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bergamini, G., Taddei, M.H.T., Rosa, M.M.L. et al. Determination of 226Ra in drinking water samples by alpha spectrometry. J Radioanal Nucl Chem 307, 829–834 (2016). https://doi.org/10.1007/s10967-015-4299-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-015-4299-7