
Abstract
We developed a new method for the determination of 227Ac in geological samples. The method uses extraction chromatographic techniques and alpha-spectrometry and is applicable for a range of natural matrices. Here we report on the procedure and results of the analysis of water (fresh and seawater) and rock samples. Water samples were acidified and rock samples underwent total dissolution via acid leaching. A DGA (N,N,N′,N′-tetra-n-octyldiglycolamide) extraction chromatographic column was used for the separation of actinium. The actinium fraction was prepared for alpha spectrometric measurement via cerium fluoride micro-precipitation. Recoveries of actinium in water samples were 80 ± 8 % (number of analyses n = 14) and in rock samples 70 ± 12 % (n = 30). The minimum detectable activities (MDA) were 0.017–0.5 Bq kg−1 for both matrices. Rock sample 227Ac activities ranged from 0.17 to 8.3 Bq kg−1 and water sample activities ranged from below MDA values to 14 Bq kg−1of 227Ac. From the analysis of several standard rock and water samples with the method we found very good agreement between our results and certified values.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Actinium-227 is a member of the naturally occurring 235U decay series (Fig. 1). It is a beta-emitter with a half-life of T 1/2 = 21.77 years. The concentration of 227Ac in natural samples is extremely low, in seawater the reported activities range from 0.83 to 90 mBq m−3 [1], in some basalts, assuming equilibrium with 231Pa, the expected range can be as low as <0.1–5 Bq kg−1 [2]. Actinium-227 is used as an oceanographic tracer for studies of deep-ocean mixing and upwelling [1, 3], and it has the potential to be used as a tracer for geochemical characterization of rocks and other geological material. Some already existing techniques for 227Ac analysis include separation on ion exchange columns followed by measurement of its daughters 227Th and 223Ra by alpha-spectrometry [4, 5]. The disadvantage of the ion exchange columns is that chemical recoveries tend to be lower (~50–60 %) and may not be well suited for actinium separation from complex matrices. Another popular method uses a radium delayed coincidence counting (RaDeCC) system [6]. Actinium-227 in water samples is quantitatively sorbed on a MnO2-coated acrylic fiber and allowed to sit for 90 days for 223Ra ingrowth. The fiber is then measured by the RaDeCC system in which the 219Rn–215Po pair is counted. This technique works well for seawater analysis for example [6], but is impractical for rock samples.

Decay schemes of a 235U including 227Ac and its daughters 227Th–223Ra, and b the 229Th–225Ac pair used as a yield tracer. The vertical arrows indicate alpha decays and the tilted arrows represent transformations via beta decay. Half-lives are indicated below each isotope
We adapted a single-column extraction chromatographic technique (DGA column), which simplifies the separation step, reduces analysis time and the amount of reagents needed. The use of this technique assures clean separation of actinium from other elements and leads to close to quantitative chemical recoveries from diverse geological matrices. Here we report on its use for 227Ac separation from freshwater, seawater, and rock samples.
Experimental
Experimental design
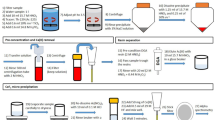
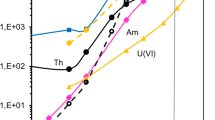
A commercially available extraction chromatographic resin containing N,N,N′,N′-tetra-n-octyldiglycolamide sorbed on 50–100 μm particle size AmberchromR CG-71 (DGA column manufactured by Eichrom Technologies, Inc [7]) was chosen for separation of actinium from other actinides and the sample matrix. This resin has high adsorption capacity for rare earth elements and actinides (0.086 mmol/mL of Eu) and various concentrations of acidic solutions allow a sequential elution of individual elements. The separation sequence for Ac was selected based on distribution coefficients of actinides and common ions on DGA derived by Horwitz et al. [7] using batch experiments for 1-h contact time of solutes with DGA at 22 °C. Figure 2 illustrates k′ for Ac, Th, Fe and alkaline earth cations in each separation and rinsing step on the column. K′ is defined as the volume distribution ratio of the element between the stationary extractant phase and the mobile aqueous phase times the ratio of the volumes of the stationary and mobile phases in a slurry packed column. We used 4 M HCl as load solution for which the k′ of Ac is 20, of Th, U and Am is >1,000, of Ba, Sr, Ca and Ra is 1–5, and of Fe is 1,000. Based on these k′ values, actinides and iron are well retained on the column with a 4 M HCl load solution while radium and other alkaline earth elements have no affinity and pass through the column without retention. We used 3 M HNO3 to rinse any leftover alkaline earth elements and iron from the column. In 3 M HNO3 the k′ of iron drops to <2, while all actinides including Ac have k′ >1,000. In the next step we eluted actinium with 2 M HCl, which is efficient for Ac but leaves Th and other actinides retained on the DGA column. If needed for further analysis, thorium (228Th for 228Ac or 227Th analysis) can be stripped from the column using dilute nitric acid [7]. We tested the procedure using an aliquot of NIST certified 229Th/225Ac standard solution (Fig. 1), which offered an ideal opportunity to demonstrate good separation between actinium and thorium on the DGA column. Consistent with the findings of Horwitz et al. [7] we observed that the presence of iron in the load solution positively affects the actinium uptake on DGA due to the salting out effect. Therefore, each load solution was spiked with 1 mg of iron as FeCl3.

Resin capacity factor k′ for Ac, Th, Fe and alkaline earth cations in each separation and rinsing step on the DGA column derived using batch experiments for 1-hour contact time of solutes with DGA at 22 °C [7]. The higher the k′ the higher the fraction of the element in the stationary extractant phase
Chemical procedure
Rock samples were crushed to <1 mm and then dissolved completely by a series of digestions using HF and HNO3, followed by HNO3 + H3BO3 and HClO4 to decompose fluorides [8]. In the final stage they were dissolved and stored in 4 M or higher HCl + sat. H3BO3 until analysis. Freshwater and seawater samples were acidified to pH 1. All samples were spiked using a NIST certified 229Th/225Ac tracer.
While it is possible for water samples to be evaporated, dissolved in 4 M HCl and loaded to the column directly, the high amount of salts in dissolved rock samples required a pre-concentration step. We used a PbSO4 co-precipitation technique after Martin et al. [4]. All sample volumes were reduced to 100 mL. We added 1 mL of 98 % H2SO4 to each sample after which 2 g of K2SO4 were added and dissolved. While stirring, 1 mL of 0.24 M Pb(NO3)2 solution was added to the sample drop-wise. The sample was then heated and the precipitation was allowed to settle. The supernatant was decanted and the precipitate was washed using 20 mL of 0.1 M K2SO4 + 0.2 M H2SO4. The precipitate was then easily dissolved in 20 mL of 4 M HCl. Some precipitates required heating and the addition of higher volumes of 4 M HCl; we tested the method with up to 50 mL of load solution for which we still achieved quantitative recoveries. A 1 mg Fe in solution was added to the solution prior to loading on the DGA column. The column was attached to a vacuum box (Eichrom Technologies, Inc.), which was used to achieve flow rates of 1 mL/min. The beaker was rinsed with 2 × 5 mL of 4 M HCl, which was loaded on the column. The DGA was then rinsed using 10 mL of 3 M HNO3 and the eluate, which contained iron and any leftover alkaline earth elements was discarded. Actinium was eluted using 20 mL 2 M HCl while Th and other actinides were retained on the DGA column.
The Ac fraction was saved and an alpha-source was prepared using CeF3 micro-precipitation [9]. 100 μg of Ce carrier in solution was added to the Ac fraction, stirred and 2 mL of concentrated HF were added while stirring. The solution rested for 30 min and then the precipitate was filtered using 0.1 μm polypropylene, 25 mm diameter 0.1 μm polypropylene Resolve™ filters (Eichrom Technologies, Inc.). The filters were dried and mounted on steel planchets.
Alpha-spectroscopy
The samples were counted using silicon surface barrier alpha detectors (Ortec, 450 mm) that were calibrated by a NIST certified 241Am solution prepared in the same geometry as the samples. The source detector distance was 10 mm and in this geometry the detector efficiencies ranged from 19.3 to 20.9 %. After source preparation the samples were stored for 15–20 min for the ingrowth of the 225Ac daughters 221Fr (T 1/2 = 4.8 min) and 217At (0.3 s). The sample was then counted immediately to obtain the highest count rate due to the 10-day half-life of 225Ac. From this measurement, counts from the region of 217At at 7.06 MeV (Fig. 3a) were used to calculate the chemical recovery of 225Ac and therefore also 227Ac. A second measurement was performed after 90 days of sample preparation, which allowed for the decay of 225Ac and the ingrowth of 227Th and 223Ra from 227Ac (Fig. 3b). Counts from the energy region of 5.38–6.10 MeV originating from 227Th and 223Ra (99.4 %) were used to calculate 227Ac. Any leftover 225Ac still present was subtracted from this region.

Alpha-spectrum of the actinium fraction of a seawater sample a counted immediately after source preparation showing peaks of 225Ac, 221Fr and 217At, and b the same source counted 90-days after preparation when 225Ac has mostly decayed and daughters 227Th and 223Ra are almost in equilibrium with 227Ac
The 227Ac activity was calculated using the count-rate in the 7.06 MeV peak of 217At for yield determination and in the 5.38–6.10 MeV originating from 227Th and 223Ra minus any decay corrected leftover counts from the 225Ac spike [4]. Actinium-227 at the time of separation \( A{}_{{{}^{227}{\text{Ac}}}} \)was then calculated using the branching ratios for 227Ac of 98.2 % by beta-decay to 227Th and 1.38 % by alpha-particle emission to 223Fr, while 100 % of 227Th and 99.4 % of 223Ra decay via alpha-particle emission:
where \( A_{{{}^{227}{\text{Th}} + {}^{223}{\text{Ra}}}} \) is the measured 227Th + 223Ra activity, and \( I_{{{}^{227}{\text{Th}}}} \) and \( I_{{{}^{223}{\text{Ra}}}} \) are the calculated ingrowth fractions of 227Th and 223Ra at the time of separation [4].
Minimum detectable activities (MDA) were calculated for a confidence limit of α = 0.05, 3-day counting times and one sigma standard deviation of the counts from the 227Th + 223Ra region of background spectra according to the method described by Currie [10].
Results and discussion
We tested the procedure with 229Th/225Ac spike in de-ionized water. As suggested by Horwitz et al. [7], in our test solutions the chemical recovery increased from 70 % (229Th/225Ac solution in de-ionized water) to 100 % with the addition of Fe (229Th/225Ac in de-ionized water with 50 mg Fe). Increasing Fe concentrations did not have further beneficial effects on the Ac sorption, the chemical recovery dropped to 70 % when we added up to 1 g of iron (Fig. 4). During these test runs we observed good separation of 229Th from the 225Ac fraction. One way to check the performance of our chemical separation was to evaluate the alpha-spectra for radium, polonium and thorium. Radium-226 (4.8 MeV), 222Rn (5.49 MeV) and 218Po (6.00 MeV) counts in the spectrum would be an indication of 226Ra impurity, 210Po has a peak at 5.30 MeV and finally 228Th and its daughters can be detected via the 212Po peak at 8.78 MeV. All our spectra including seawater and rock samples were free of these or only had minor interferences indicating that the extraction chromatography procedure reliably removes any interferences and impurities from the Ac fraction.

Actinium recoveries from a 229Th/225Ac spiked de-ionized water solution without and with the addition of variable amounts of iron
The analyzed water samples included de-ionized water, seawater and tap water spiked with 227Ac. The analysis of the de-ionized water sample was considered as blank and resulted in zero net counts in the 227Th + 223Ra region after background correction. Our analysis of a certified 227Ac standard (AEA Technology supplied by the IAEA [11]) resulted in (333 ± 16) Bq kg−1 and was in good agreement with the IAEA value of (329 ± 16) Bq kg−1 [11]. Actinium recoveries of water samples were 80 ± 8 % (number of analyses n = 14; where the standard deviation of the average of the 14 analyses is 8 %). The average minimum detectable activity for water samples for ~4 day counting time was 0.017 Bq kg−1. The sample activities ranged from below MDA values to 14 Bq kg−1of 227Ac.
The weights of analyzed rock samples were between 0.3 and 5.4 g and we analyzed various basalts and USGS and U-series community rock standards (BCR-2 [12], BHVO-1 [13], Hawaiian basalt (HK) [2], Samoan basalt (SAV) [14]). No certified value for 227Ac exists for these standards but assuming equilibrium through the decay chain (all basalts were over 100 years old) and a closed system we can compare 227Ac to 231Pa [2] (Table 1). There is a good agreement for 227Ac and 231Pa, for all samples the 227Ac/231Pa ratio is within 5 % of secular equilibrium. For the 24 rock analyses the chemical recoveries were 70 ± 12 % (n = 24). The lower recoveries were the results of (1) an incomplete PbSO4 precipitation because some rock samples could not be held dissolved unless kept in high acidity solution, (2) a high iron content (>1 g) remaining in the samples even after the PbSO4 precipitation negatively affects the Ac sorption on DGA (Figs. 3, 4) the presence of significant amount of lanthanides which negatively affects the co-precipitation of actinium by PbSO4 [4].
The minimum detectable activities for rock samples averaged 0.05 Bq kg−1 for 3-day counting and MDAs for individual sample measurements were in the range of 0.017–0.5 Bq kg−1. Sample activities ranged from 0.17 to 8.3 Bq kg−1.
Uncertainties were derived from counting statistics of the 217At (usually 3 % error) and 227Th + 223Ra (ranged from 3 to 10 %) region peaks and error propagation. Uncertainties of all analysis were <10 %. Overall the DGA separation method performed well, providing actinium fractions without interferences in alpha-spectrometry and resulting in high chemical recoveries. Our results agree well with certified values for water and 231Pa values for rock samples.
Conclusions
The advantages of extraction chromatographic techniques over ion exchange columns in general are a shorter analysis time and less acid waste produced. The method developed here has a high sensitivity due to the low backgrounds on the alpha-detectors and because of the use of the double-peak of 227Th and 223Ra, which doubles the counts and lowers the counting error [15]. Additional advantage of the DGA method is its possible extension to the analysis of thorium isotopes. Our typical 2 σ errors were 5 % which are much higher than those usually obtained for example by mass spectrometric analysis (ICP-MS, TIMS) of other isotopes, however due to the low abundance of 227Ac MS methods are currently not sensitive enough for its analysis. For comparison, in units of fg g−1 used for MS techniques, our method has a limit of detection of 0.02 fg g−1.
References
Geibert W, Rutgers van der Loeff MM, Hanfland C, Dauelsberg H-J (2002) Actinium-227 as a deep-sea tracer: sources, distribution and applications. Earth Planet Sci Lett 198(1–2):147–165
Sims KWW, Murrell MT, DePaolo DJ, Baldridge WS, Goldstein SJ, Clague D, Jull M (1999) Porosity of the melting zone and variations in the solid mantle upwelling rate beneath Hawaii: inferences from 238U–230Th–226Ra and 235U–231Pa disequilibria. Geochim Cosmochim Acta 63(23):4119–4138
Nozaki Y (1984) Excess 227Ac in deep ocean water. Nature 310:486–488
Martin P, Hancock GJ, Paulka S, Akber RA (1995) Determination of 227Ac by α-particle spectrometry. Appl Radiat Isot 46(10):1065–1070
Geibert W, Vöge I (2008) Progress in the determination of 227Ac in sea water. Mar Chem 109(3–4):238–249
Moore WS, Arnold R (1996) Measurement of 223Ra and 224Ra in coastal waters using a delayed coincidence counter. J Geophys Res (Oceans) 101(1):1321–1329
Horwitz EP, McAlister DR, Bond AH, Barrans RE (2005) Novel extraction chromatographic resins based on tetraalkyldiglycolamides: characterization and potential applications. Solvent Extr Ion Exch 23:219
Ball LA, Sims KWW, Schwieters J (2008) Measurement of 234U/238U and 230Th/232Th in volcanic rocks using the Neptune MC-ICP-MS. J Anal At Spectrom 23:173–180
Dulaiova H, Kim G, Burnett WC, Horwitz EP (2001) Separation and analysis of Am and Pu from large soil and sediment samples. Radioact Radiochem 12(3):4–15
Currie LA (1968) Limits for qualitative detection and quantitative determination. Application to radiochemistry. Anal Chem 40(3):586–593
Scholten JC, Pham MK, Blinova O, Charette MA, Dulaiova H, Eriksson M (2010) Preparation of Mn-fibers standards for the efficiency calibration of the delayed coincidence counting system (RaDeCC). Mar Chem 121(1–4):206–214
Prytulak J, Elliott T, Hoffmann DL, Coath CD (2008) Measurement of USGS BCR-2 as a reference material for siliscate rock U-Pa disequilibrium measurements. Geostand Geoanal Res 32(1):55–63
Pichat S, Sims KWW, Francois R, McManus JF, Brown Leger S, Albarede F (2004) Lower export production during glacial periods in the equatorial Pacific derived from (231Pa/230Th)xs,0 measurements in deep-sea sediments. Paleoceanography 19 PA4023. doi:10.1029/2003PA000994
Sims KWW, Hart SR, Reagan MK, Blusztajn J, Staudigel H, Sohn RA, Layne GD, Ball LA, Andrews J (2008) 238U–230Th–226Ra–210Pb–210Po, 232Th–228Ra, and 235U–231Pa constraints on the ages and petrogenesis of Vailulu’u and Malumalu lavas, Samoa. Geochem Geophys Geosyst 9:Q04003
Bojanowski R, Holm E, Whitehead NE (1987) Determination of 227Ac in environmental samples by ion-exchange and alpha-spectrometry. J Radioanal Nucl Chem 115(1):23–37
Acknowledgments
We would like to acknowledge Phil Horwitz and William Burnett for their advice on the extraction chromatographic techniques. Ken Sims’ lab at the Woods Hole Oceanographic Institution performed the rock dissolution procedures and Ken Buesseler (WHOI) provided the counting equipment for the alpha-spec analysis. Jan Scholten from the IAEA provided the actinium standard and spiked seawater samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dulaiova, H., Sims, K.W.W., Charette, M.A. et al. A new method for the determination of low-level actinium-227 in geological samples. J Radioanal Nucl Chem 296, 279–283 (2013). https://doi.org/10.1007/s10967-012-2140-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-012-2140-0