
Abstract
Accurate characterization of longitudinal exposure-response of clinical trial endpoints is important in optimizing dose and dosing regimens in drug development. Clinical endpoints are often categorical, for which much progress has been made recently in latent variable indirect response (IDR) modeling with single drugs. However, such applications have not yet been used for trials employing multiple drugs administered concurrently. This study aims to demonstrate that the latent variable IDR approach provides a convenient longitudinal exposure-response modeling framework to assess potential interaction effects of combination therapies. This is illustrated by an application to the exposure-response modeling of guselkumab, a monoclonal antibody in clinical development that blocks the interleukin-23p19 subunit, and golimumab, a monoclonal antibody that binds with high affinity to tumor necrosis factor-alpha. A Phase 2a study was conducted in 214 patients with moderate-to severe active ulcerative colitis for which longitudinal assessments of disease severity based on patient-reported measures of rectal bleeding, stool frequency, and symptomatic remission were evaluated as categorical endpoints, and fecal calprotectin as a continuous endpoint. The modeling results suggested independent pharmacodynamic guselkumab and golimumab effects on fecal calprotectin as a continuous endpoint, as well as interaction effects on the categorical endpoints that may be explained by an additional pathway of competitive interaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exposure-response (ER) modeling of clinical endpoints is important in drug development for facilitating informative dosing selection [1]. Longitudinal ER modeling is especially informative for dose-regimen decisions [2]. Clinical endpoints are often ordered categorical, for which the most common analysis approach may be logistic regression [3]. Probit regression is another often-used approach, and its choice vs. the logistic regression is usually the analyst’s preference [3]. A similarity between the two approaches has been suggested in a pharmacometric application [4]. A Markov transition approach has also been used and has the appeal of capturing individual response changes [5], although advantages of the mixed-effect logistic/probit regression have been argued [6].
For single drugs, a widely used class of longitudinal ER models is the indirect response (IDR) model [7] which can be applied to categorical clinical endpoints by the latent variable approach [6]. For multiple-drug combinations [8], longitudinal ER modeling has been evaluated in the context of fixed-dose drug combinations [9]. Various mechanism-based drug interaction models have been used for continuous data. In particular, the Agonist – Partial Agonist model for drugs A and B with effect-site concentrations CA and CB extends the commonly used Emax model shown below [10]:
where Emax,A, Emax,B, IC50,A, and IC50,B are the maximum effect and potency parameters for A and B. A general pharmacodynamic interaction model has also been proposed that allows the explicit modeling of the influence of one drug on the Emax or IC50 parameter of the other drug [11]. Conversely, longitudinal ER modeling of categorical endpoints for combination drug therapy has not yet been conducted.
Guselkumab (TREMFYA®) is a monoclonal antibody that specifically blocks the interleukin-23p19 subunit approved worldwide for the treatment of psoriasis and psoriatic arthritis and is currently under study in ulcerative colitis (UC) and Crohn’s disease [12]. Golimumab (SIMPONI®) is a monoclonal antibody that binds with high affinity to tumor necrosis factor alpha and is approved for the treatment of UC and other rheumatologic indications [13].
UC is a chronic immune-mediated gastrointestinal disorder of the colon characterized by symptoms of increased rectal bleeding, stool frequency, and abdominal pain. Disease activity and treatment response are frequently evaluated by patient-reported measures of rectal bleeding and stool frequency, with resolution of rectal bleeding and bowel habit normalization identified as important therapeutic targets for UC, in conjunction with endoscopic remission [14]. As endoscopy is an invasive and costly procedure, surrogate markers that reflect the severity of mucosal inflammation have been investigated. Fecal calprotectin, which can be readily measured from stool samples, is highly correlated with endoscopic and histologic activity and has been proposed as a reasonable surrogate for a treatment target [15, 16].
Dose/exposure-response modeling of drug interaction has a long history with multiple definitions of additivity or synergy, each having its advantages and disadvantages [8]. Defining additivity or synergy is not the purpose of this manuscript; instead, the aim was to demonstrate that the latent variable IDR framework can be readily used for mechanism-based assessment of drug-drug interaction with both categorical and continuous clinical endpoints. Data from a Phase 2a clinical trial in patients with moderate-to-severe active UC [17] that evaluated combination therapy with guselkumab (TREMFYA®) and golimumab (SIMPONI®) was used as an illustration.
Methods
Study design
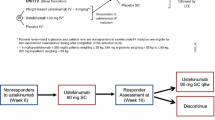
A Phase 2a, randomized, double-blind, active-controlled, parallel-group, proof-of-concept trial (ClinicalTrials.gov number, NCT03662542) was conducted in patients with moderate-to-severe active UC. The trial consisted of 2 distinct phases: a 12-week combination therapy comparison phase followed by a 26-week monotherapy phase (total treatment period of 38 weeks with last study treatment administered at Week 34). At Week 0, patients were randomly assigned, in a 1:1:1 ratio, to receive (1) golimumab monotherapy: golimumab 200 mg subcutaneous (SC) injection at Week 0, followed by golimumab 100 mg SC at Week 2 and then golimumab 100 mg SC every 4 weeks (q4w), (2) guselkumab monotherapy: guselkumab 200 mg intravenous (IV) infusion at Weeks 0, 4, and 8 followed by guselkumab SC 100 mg q8w, or (3) combination therapy→guselkumab monotherapy: guselkumab 200 mg IV and golimumab 200 mg SC at Week 0; golimumab 100 mg SC at Weeks 2, 6, and 10; guselkumab 200 mg IV at Weeks 4 and 8 followed by guselkumab 100 mg SC q8w. Patients randomized to combination therapy received guselkumab monotherapy after Week 12. The study design is shown in Fig. 1. More details have been described previously [12].

Study Design Schema
Pharmacokinetic and longitudinal clinical efficacy assessments
During Weeks 0–12, guselkumab and golimumab serum samples were collected every 2 weeks. At visits when patients received treatment, a pre-dose sample was also collected. Additionally, for IV infusion-related visits (i.e., Weeks 0, 4, and 8), another sample was taken pre-dose and approximately 1 hour post-dose. Afterwards, serum samples of guselkumab and golimumab were collected at Weeks 14, 16, 24, 32, 34, 38, at early termination when applicable, and at safety follow-up.
Patient-reported scores for rectal bleeding and stool frequency (both having possible values of 0, 1, 2, or 3, with higher scores indicating greater disease severity) were collected at Weeks 0, 2, 4, 6, 8, 10, 12, 14, 16, 18, 22, 24, 26, 30, 32, 34, 38, at early termination, and at safety follow-up. Rectal bleeding and stool frequency were modeled as ordered categorical endpoints. In addition, a binary endpoint of symptomatic remission (yes/no) was evaluated at the same time points and was defined as achieving a rectal bleeding score of 0, a stool frequency score of 0 or 1, where stool frequency had not increased from baseline. Fecal calprotectin concentrations were evaluated at Weeks 0, 2, 4, 8, 12, 24 and 38. The final dataset contained data from 214 randomized patients overall, with 1958 guselkumab pharmacokinetic (PK) measurements from 142 patients, 1686 golimumab PK measurements from 143 patients, 3553 symptomatic remission outcomes, 3441 rectal bleeding and stool frequency scores, and 1378 fecal calprotectin concentration measurements.
Population PK modeling
Population PK modeling following SC administration of guselkumab using a one-compartment model with first-order absorption and first-order elimination has been described previously [18]. The availability of the data from IV administration allowed for a two-compartment population PK model, with body weight as the main covariate for both clearance and volume of distribution [19]. The typical clearance and absolute bioavailability were estimated as 0.34 L/day and 74%, respectively. Population PK modeling following SC administration of golimumab using a one-compartment model with first-order absorption and first-order elimination has been described previously [20]. Following a confirmatory analysis approach [19], with body weight as the main covariate for both clearance and volume of distribution, the typical clearance was estimated as 0.84 L/day. Exploratory analyses did not suggest additional covariates. The detailed results will be reported in a separate publication. Individual empirical Bayesian PK parameter estimates based on the population PK model were obtained and used for the subsequent ER analysis.
Categorical endpoint model and latent variable motivation
For the categorical endpoints of symptomatic remission, rectal bleeding, and stool frequency, the following mixed-effect logistic regression model was used:
where Y is the response, k = 0 for symptomatic remission and k = 0, 1, or 2 for rectal bleeding and stool frequency scores, αk represents the intercept, t is time, and η is a normally distributed random variable representing between-subject variability (BSV). To stabilize parameter estimation, αk was re-parameterized as (α1, d0, d2) where d0, d2 > 0 are distances between intercepts such that α0 = α1 – d0, and α2 = α1 + d2. For symptomatic remission, there is only one intercept, namely α0, thus no re-parameterization is needed.
The logistic regression model has a latent variable motivation. Let L(t) be a latent variable indicating the underlying disease severity such that when it drops below thresholds {αk}, the outcome Y = k is observed. That is, Y ≤ k ⇔ L(t) < αk. Assume that L(t) is modeled as L(t) = M(t) + σε, where M(t) is the model predictor, ε is error with a logistic distribution with standard deviation σ. It can then be shown that σ is not separately identifiable and may be set to 1 [21], and prob[Y ≤ k] = prob[L(t) < αk] = F[αk - M(t)], where F is the inverse logit function. This is the same as Eq. 2, with -M(t) = f(t) + η. While standard in statistical theory [3], in the context of applying IDR models this derivation was first given by Hutmacher et al. [21]. The derivation allows the interpretation of f(t) in Eq. 2 as a physiological variable, and consequently the mechanistic consistency of applying IDR models. In this setting, Hu has proposed that f(t) be modeled in a reduction-from-baseline form [6].
The drug effect f(t) is given below separately for the scenarios of single and combination drugs.
Basic latent variable indirect response model
At the single drug level, the same structural drug effect model for both guselkumab and golimumab was used as follows:
where R(t) is a latent variable, C is drug concentration, and kin (disease formation rate), IC50 (half-maximal inhibitory concentration), and kout (disease amelioration rate) are parameters in a Type I IDR model that has previously been used for guselkumab in other disease areas and golimumab in UC [22, 23]. It was further assumed that at baseline R(0) = 1, yielding kin = kout. The reduction of R(t) was assumed to drive the drug effect through:
where DE is a parameter to be estimated that determines the magnitude of drug effect.
Theoretically, the representation of the drug effect in Eqs. 2–4 has been shown to be equivalent to a change-from-baseline latent-variable IDR model [24], for which kout may be empirically interpreted as the rate constant of drug effect onset and offset, and DE may be interpreted as the latent variable baseline prior to normalization. In this setting, the Imax/Emax parameter appearing in the general IDR model is usually not separately estimable and should be set to 1 as shown previously [6]. More details on the theoretical characteristics of latent variable IDR models are described elsewhere [6].
2-Pathway (independence) model
Initially, guselkumab and golimumab effects were modeled independently as.
where fgus(t) and fgol(t) follow the basic change-from-baseline latent-variable IDR form of Eqs. 3–4, namely
and
where kin, gus = kout, gus, and fgol(t) is defined similarly. Equation 5 may seem empirical, but guselkumab and golimumab may be interpreted as affecting the latent variable L(t) through two separate pathways, with maximal effects on each pathway determined by DEgus and DEgol, respectively [6].
Interaction model
It is not a priori clear whether guselkumab and golimumab would work independently in UC. To assess potential interaction effects, Eq. 5 was augmented with an additional component based on the Agonist – Partial Agonist model, which in this case appears more plausible than other interaction models given in [10]:
where
and
with kin, APA = kout, APA and IH taking the form of Eq. 1 with A = gus, B = gol for guselkumab and golimumab, respectively, and Emax,gus = Emax,gol = 0.5. Further refinement is possible by modeling the ratio of Emax,gus and Emax,gol as an additional parameter while keeping their sum at 1.
Under Eq. 8, guselkumab and golimumab may be interpreted as affecting endpoint Y through three separate pathways: one for guselkumab only, one for golimumab only, and one that is shared between both guselkumab and golimumab. The similarity of parameters, particularly IC50s between the Agonist – Partial Agonist component and the guselkumab and golimumab only components, were also assessed.
Continuous endpoint model
Fecal calprotectin, a non-invasive surrogate marker of colonic inflammation, was previously modeled in a single drug setting with a Type I IDR model [20]. Fecal calprotectin cut-off values, ranging from < 50 µg/g to < 150 µg/g, have been identified to differentiate active versus quiescent colonic disease endoscopically [12, 21, 22]. Similar cut-offs have also been associated with histologic remission [23, 24]. In this study, reduction of fecal calprotectin below 100 µg/g was used as a surrogate for endoscopic improvement. The Type I IDR model was extended to describe the combined guselkumab – golimumab effect on FC(t), the time course of fecal calprotectin, as
where
which may be viewed as a 1-pathway independence model. For ease of interpretation, kin,FC is re-parameterized as BFC ⋅ kout,FC, where BFC is baseline fecal calprotectin and will be estimated instead. BSV was modeled on BFC and explored on other parameters. A log-transform-both-sides approach was applied.
Model estimation and evaluation
A sequential pharmacokinetic/pharmacodynamic (PK/PD) modeling approach was used by first fixing the individual empirical Bayesian PK parameter estimates. NONMEM Version 7.4 with the LAPLACE estimation option was used for all modeling [25]. A decrease in the NONMEM minimum objective function value (OFV) of 10.83, corresponding to a nominal p-value of 0.001 for a χ2 distribution with 1 degree of freedom, was considered as the threshold criterion for including an additional model parameter. For categorical endpoints, residuals have been defined but are not practically useful [26]. Visual predictive check (VPC) with 500 replicates was used for model evaluation [27]. For categorical endpoints, the observed frequencies were overlayed with the VPC intervals (VPCI) constructed from the simulated median, 5%, and 95% percentiles of the model predicted frequencies [28]. Note that percentiles, e.g., 5% and 95%, are not meaningful for raw observed categorical endpoints.
Results
Demographics and baseline characteristics
Baseline body weight, the only influential PK covariate, ranged between 40 and 145 kg, with a mean (SD) of 71 (18) kg. Mean rectal bleeding and stool frequency scores and median fecal calprotectin concentrations were similar among treatment groups as shown in Table 1. More detailed demographics and baseline covariates were reported elsewhere [17].
Symptomatic remission
The 2-pathway independence model was fitted first. The parameter estimates and estimation precision were reasonable for the study size (data not shown). The VPC results are shown in Fig. 2 (left column). The model notably overpredicted the combination treatment response but underpredicted the golimumab treatment response. This suggests potential pharmacodynamic interactions.

Visual predictive check results of the symptomatic remission independence and interaction models by treatment group. Key: VPCI, visual predictive check interval; Independence: 2-way independence model; Interaction: 3-way interaction model
The Interaction model was fitted next. Compared with the 2-pathway independence model, the NONMEM OFV improved by more than 100, indicating significant improvement of the fit. An attempt of including the ratio of Emax,gus and Emax,gol as an additional parameter resulted in a point estimate of Emax,gus = 0.4, with an insignificant OFV change of 6 and convergence difficulties, thus it was not kept in the model. Subsequent exploration showed that neither individual component, fgus(t) or fgol(t), could be removed from the model, suggesting that all 3 pathways contributed to the combination effect of guselkumab and golimumab. Setting DE and kout parameters of different components to be equal mostly resulted in significantly worse fits. However, setting IC50,APA, gus = IC50,gus and IC50,APA, gol = IC50,gol resulted in a negligible NONMEM OFV change, indicating the lack of significant difference. This was considered the final model, and the parameter estimates are given in Table 2. Estimation precision was reasonable, with relative standard errors (RSEs) < 40% except for those of IC50 parameters where difficulties with parameter estimation may be expected with only single drug doses used. The condition number of the model, calculated as the ratio between the largest and smallest eigenvalues of the variance-covariance matrix, was ~ 100 which was reasonable. The VPC results improved notably from the independence model (Fig. 2, right column) and appeared to reasonably describe the observed data.
Rectal bleeding score
The 2-pathway independence model was also fitted first. The parameter estimates and estimation precision were reasonable for the study size (not shown). The VPC results are shown in Fig. 3 (left side). Also here, the model notably overpredicted the combination treatment response but underpredicted the single treatment responses. This suggests potential pharmacodynamic interactions.

Visual predictive check results of the rectal bleeding score independence and interaction models by treatment group. Key: VPCI, visual predictive check interval; Independence: 2-way independence model; Interaction: 3-way interaction model
The Interaction model was fitted next. Compared with the 2-pathway independence model, the NONMEM OFV improved more than 60, indicating significant improvement of the fit. Subsequent exploration showed that neither individual component, fgus(t) or fgol(t), could be removed from the model. However, setting IC50,APA, gus = IC50,gus and IC50,APA, gol = IC50,gol resulted in a NONMEM OFV increase of < 5, indicating the lack of significant difference. This was considered the final model, and the parameter estimates are given in Table 2. Estimation precision was reasonable, with relative standard errors (RSEs) < 40% for most parameters except DEgus and the independent pathway kout and IC50 parameters. The condition number of the model was ~ 200 which was reasonable. The VPC results, shown in Fig. 3 (right side), notably improved from the independence model and, with the overall variability, appeared to reasonably describe the observed data.
Fecal calprotectin
Equations 11–12 were fitted to the data. Estimates of Emax,gus and Emax,gol approached 1 and thus were fixed at 1. No additional BSV effects were supported by the data. The final model parameter estimates are given in Table 2. Estimation precision was reasonable, with RSE < 30% for all parameters. The condition number of the model was < 10 which was reasonable. No additional BSV terms were supported other than for the baseline. The VPC results for fecal calprotectin and the rate of achieving fecal calprotectin < 100 µg/g are shown in Figs. 4 and 5, respectively. The model reasonably described the observed data.

Visual predictive check results of the fecal calprotectin model by treatment group. Key: VPCI, visual predictive check interval

Visual predictive check results of the fecal calprotectin model on the proportion of subjects achieving < 100 mg/kg by treatment group. Key: VPCI, visual predictive check interval
Discussion
To our knowledge, longitudinal ER modeling of categorical clinical endpoints has not yet been conducted for combination drug therapy. We have shown that the latent variable IDR framework conveniently allows mechanism-based assessments. For the clinical symptoms endpoints (e.g., rectal bleeding score, stool frequency score and symptomatic remission), the 2-way independence models consistently overpredicted the combination treatment response but underpredicted the single treatment responses. This suggested potential pharmacodynamic interactions in the sense that, if the parameter estimations were to be changed to increase the single treatment predictions, the independence models would also push the combination treatment response prediction higher and thus even further away from the observed data. The findings that the combination treatment responses are less than the independence model-predicted single treatment responses are consistent with the Agonist – Partial Agonist mechanism. The 3-way interaction models notably improved the model fits as compared to the 2-way independence models. The estimated maximal effect and the rate of onset, i.e., DE and kout, under the Agonist – Partial Agonist pathway were much larger than those under the single pathways, indicating strong interaction effects with faster onset. The fact that the guselkumab and golimumab IC50 parameters could not be separately estimated between the different pathways may be attributed to the lack of data. The similarity of the final model structures also lends additional confidence in the appropriateness of the selected models. Other mechanism-based models, such as those given in [11], may be used if deemed appropriate, especially if the mechanism of drug interaction is known.
As symptomatic remission is defined in terms of rectal bleeding and stool frequency scores, the question arises as to why not use the developed rectal bleeding and stool frequency models to predict symptomatic remission. However, an attempt of this resulted in substantial biases (data not shown), despite the apparent adequacy of the rectal bleeding and stool frequency models in describing the observed data. An attempt of jointly modeling rectal bleeding and stool frequency using the shared random effect framework [29] did not help either. This finding may be explained because rectal bleeding and stool frequency models must predict not only mean outcomes, but also BSVs of all model parameters and their correlations. It recently has been shown that this task is difficult to achieve with only categorical data, and the joint modeling with an additional endpoint with continuous data granularity would be necessary to facilitate effective identification of the important random effects [30]. For this reason, a separate model was developed for symptomatic remission, a commonly used stringent clinical endpoint for signs and symptoms in UC.
For the continuous clinical endpoint fecal calprotectin, the 1-way independence model reasonably described the data. Including an additional Agonist – Partial Agonist interaction term into the model resulted in no improvement, which further confirmed the adequacy of the 1-way independence model. The ability of the model to describe the rate of achieving fecal calprotectin < 100 µg/g added further confirmation, as a model may describe the originally fitted endpoint but fail to describe a derived endpoint [31].
Longitudinal ER modeling of clinical endpoints facilitates effective decisions in clinical development [2]. For combination studies, using the full exposure and clinical endpoint time course information can be particularly important to assess the potential mechanism of drug interaction and to more effectively inform on possible clinical outcomes under alternative dosing regimens. Due to the limited number of dose groups and patients in this application, the findings, including the interaction mechanism, should be considered exploratory.
Conclusion
The latent variable IDR approach provides a convenient framework which allows mechanism-based interaction assessment of combination therapies using categorical clinical endpoints. The approach also extends readily to continuous clinical endpoints.
References
Overgaard RV, Ingwersen SH, Tornoe CW (2015) Establishing good practices for exposure-response analysis of clinical endpoints in drug development. CPT: pharmacometrics & systems pharmacology 4(10):565–575. https://doi.org/10.1002/psp4.12015
Hu C, Zhou H, Sharma A (2017) Landmark and longitudinal exposure-response analyses in drug development. J Pharmacokinet Pharmacodyn 44(5):503–507. https://doi.org/10.1007/s10928-017-9534-0
McCullagh P, Nelder JA (1989) Generalized linear models, 2nd edn. Chapman & Hall/CRC, New York
Hu C, Zhou H (2016) Improvement in latent variable indirect response joint modeling of a continuous and a categorical clinical endpoint in rheumatoid arthritis. J Pharmacokinet Pharmacodyn 43(1):45–54
Lacroix BD, Lovern MR, Stockis A, Sargentini-Maier ML, Karlsson MO, Friberg LE (2009) A pharmacodynamic Markov mixed-effects model for determining the effect of exposure to certolizumab pegol on the ACR20 score in patients with rheumatoid arthritis. Clin Pharmacol Ther 86(4):387–395. https://doi.org/10.1038/clpt.2009.136
Hu C (2014) Exposure-response modeling of clinical end points using latent variable indirect response models. CPT: pharmacometrics & systems pharmacology 3:e117. https://doi.org/10.1038/psp.2014.15
Dayneka NL, Garg V, Jusko WJ (1993) Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm 21(4):457–478. https://doi.org/10.1007/BF01061691
Geary N (2013) Understanding synergy. Am J Physiol Endocrinol Metab 304(3):E237–253. https://doi.org/10.1152/ajpendo.00308.2012
Nohr-Nielsen A, Lange T, Forman JL, Papathanasiou T, Foster DJR, Upton RN, Bjerrum OJ, Lund TM (2020) Demonstrating contribution of components of fixed-dose drug combinations through longitudinal exposure-response analysis. AAPS J 22(2):32. https://doi.org/10.1208/s12248-020-0414-y
Holford NH, Sheiner LB (1981) Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokinet 6(6):429–453. https://doi.org/10.2165/00003088-198106060-00002
Wicha SG, Chen C, Clewe O, Simonsson USH (2017) A general pharmacodynamic interaction model identifies perpetrators and victims in drug interactions. Nat Commun 8(1):2129. https://doi.org/10.1038/s41467-017-01929-y
Janssen Pharmaceutical Companies. Tremfya (guselkumab) [package insert].U.S. Food and Drug Administration
Janssen Pharmaceutical Companies. Simponi (golimumab) [package insert].U.S. Food and Drug Administration
Peyrin-Biroulet L, Sandborn W, Sands BE, Reinisch W, Bemelman W, Bryant RV, D’Haens G, Dotan I, Dubinsky M, Feagan B, Fiorino G, Gearry R, Krishnareddy S, Lakatos PL, Loftus EV Jr, Marteau P, Munkholm P, Murdoch TB, Ordas I, Panaccione R, Riddell RH, Ruel J, Rubin DT, Samaan M, Siegel CA, Silverberg MS, Stoker J, Schreiber S, Travis S, Van Assche G, Danese S, Panes J, Bouguen G, O’Donnell S, Pariente B, Winer S, Hanauer S, Colombel JF (2015) Selecting therapeutic targets in inflammatory bowel disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol 110(9):1324–1338. https://doi.org/10.1038/ajg.2015.233
D’Haens G, Ferrante M, Vermeire S, Baert F, Noman M, Moortgat L, Geens P, Iwens D, Aerden I, Van Assche G, Van Olmen G, Rutgeerts P (2012) Fecal calprotectin is a surrogate marker for endoscopic lesions in inflammatory bowel disease. Inflamm Bowel Dis 18(12):2218–2224. https://doi.org/10.1002/ibd.22917
Theede K, Holck S, Ibsen P, Ladelund S, Nordgaard-Lassen I, Nielsen AM (2015) Level of Fecal Calprotectin Correlates With Endoscopic and Histologic Inflammation and Identifies Patients With Mucosal Healing in Ulcerative Colitis. Clin Gastroenterol Hepatol 13(11):1929–1936. https://doi.org/10.1016/j.cgh.2015.05.038
Combination Therapy or Monotherapy with Golimumab and Guselkumab for Ulcerative Colitis.The VEGA Study Group. Submitted
Yao Z, Hu C, Zhu Y, Xu Z, Randazzo B, Wasfi Y, Chen Y, Sharma A, Zhou H (2018) Population Pharmacokinetic modeling of Guselkumab, a human IgG1lambda monoclonal antibody targeting IL-23, in patients with moderate to severe plaque psoriasis. J Clin Pharmacol 58(5):613–627. https://doi.org/10.1002/jcph.1063
Hu C, Zhang J, Zhou H (2011) Confirmatory analysis for phase III population pharmacokinetics. Pharm Stat 10(1):14–26. https://doi.org/10.1002/pst.403
Xu Z, Vu T, Lee H, Hu C, Ling J, Yan H, Baker D, Beutler A, Pendley C, Wagner C, Davis HM, Zhou H (2009) Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-alpha human monoclonal antibody, in patients with psoriatic arthritis. J Clin Pharmacol 49(9):1056–1070 0091270009339192 [pii]. https://doi.org/10.1177/0091270009339192
Hutmacher MM, Krishnaswami S, Kowalski KG (2008) Exposure-response modeling using latent variables for the efficacy of a JAK3 inhibitor administered to rheumatoid arthritis patients. J Pharmacokinet Pharmacodyn 35:139–157
Hu C, Yao Z, Chen Y, Randazzo B, Zhang L, Xu Z, Sharma A, Zhou H (2018) A comprehensive evaluation of exposure-response relationships in clinical trials: application to support guselkumab dose selection for patients with psoriasis. J Pharmacokinet Pharmacodyn 45(4):523–535. https://doi.org/10.1007/s10928-018-9581-1
Hu C, Adedokun OJ, Zhang L, Sharma A, Zhou H (2018) Modeling near-continuous clinical endpoint as categorical: application to longitudinal exposure-response modeling of Mayo scores for golimumab in patients with ulcerative colitis. J Pharmacokinet Pharmacodyn 45(6):803–816. https://doi.org/10.1007/s10928-018-9610-0
Hu C, Xu Z, Mendelsohn AM, Zhou H (2013) Latent variable indirect response modeling of categorical endpoints representing change from baseline. J Pharmacokinet Pharmacodyn 40(1):81–91. https://doi.org/10.1007/s10928-012-9288-7
Beal SL, Sheiner LB, Boeckmann A, Bauer RJ (2009) NONMEM User’s Guides (1989–2009). Icon Development Solutions, Ellicott City, MD, USA
Agresti A (1990) Categorical data analysis. John Wiley & Sons, Inc., New York.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear Mixed-Effects Models. AAPS J 13(2):143–151
Hu C (2022) Variability and uncertainty: interpretation and usage of pharmacometric simulations and intervals. J Pharmacokinet Pharmacodyn 49(5):487–491. https://doi.org/10.1007/s10928-022-09817-9
Hu C, Randazzo B, Sharma A, Zhou H (2017) Improvement in latent variable indirect response modeling of multiple categorical clinical endpoints: application to modeling of guselkumab treatment effects in psoriatic patients. J Pharmacokinet Pharmacodyn 44(5):437–448. https://doi.org/10.1007/s10928-017-9531-3
Hu C, Zhou H (2022) Improving categorical endpoint longitudinal exposure-response modeling through the joint modeling with a related endpoint. J Pharmacokinet Pharmacodyn 49(3):283–291. https://doi.org/10.1007/s10928-021-09796-3
Hu C, Zhou H, Sharma A (2020) Applying Beta distribution in analyzing bounded outcome score data. AAPS J 22(3):61. https://doi.org/10.1208/s12248-020-00441-4
Acknowledgements
The authors would like to thank James P Barrett, BS, a Professional Medical Writer employed by Janssen Scientific Affairs, LLC., for his editorial and submission support.
Author information
Authors and Affiliations
Contributions
CH wrote the manuscript text, analyzed the data, prepared the figures, and vouches for the integrity and veracity of the data. MV, AV, and DO reviewed and provided critical clinical input.
Corresponding author
Ethics declarations
Conflict of interest
C Hu, M Vetter, and D Ouellet are employees of Janssen Research & Development, LLC and own stock/stock options. A Vermeulen is an employee of Janssen Research & Development, a division of Janssen Pharmaceutica NV and owns stock/stock options.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, C., Vetter, M., Vermeulen, A. et al. Latent variable indirect response modeling of clinical efficacy endpoints with combination therapy: application to guselkumab and golimumab in patients with ulcerative colitis. J Pharmacokinet Pharmacodyn 50, 133–144 (2023). https://doi.org/10.1007/s10928-022-09841-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-022-09841-9