
Abstract
Accurate characterization of exposure–response relationship of clinical endpoints is important in drug development to identify optimal dose regimens. Endpoints with ≥ 10 ordered categories are typically analyzed as continuous. This manuscript aims to show circumstances where it is advantageous to analyze such data as ordered categorical. The results of continuous and categorical analyses are compared in a latent-variable based Indirect Response modeling framework for the longitudinal modeling of Mayo scores, ranging from 0 to 12, which is commonly used as a composite endpoint to measure the severity of ulcerative colitis (UC). Exposure response modeling of Mayo scores is complicated by the fact that studies typically include induction and maintenance phases with re-randomizations and other response-driven dose adjustments. The challenges are illustrated in this work by analyzing data collected from 3 phase II/III trials of golimumab in patients with moderate-to-severe UC. Visual predictive check was used for model evaluations. The ordered categorical approach is shown to be accurate and robust compared to the continuous approach. In addition, a disease progression model with an empirical bi-phasic rate of onset was found to be superior to the commonly used placebo model with one onset rate. An application of this modeling approach in guiding potential dose-adjustment was illustrated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exposure–response (E–R) modeling of clinical endpoints is important for the selection of optimal dose regimen [1]. Longitudinal E–R modeling is particularly important for the understanding of time course of treatment effect. A widely used class of longitudinal E–R models includes the Types I-IV indirect response (IDR) models [2]. IDR models have been used to characterize various types of treatment effects with mechanistic delays, and have been argued as appropriately parsimonious for clinical endpoints [3]. In situations where clinical endpoints are not physiological variables but instead composite measures of disease severity with varying levels of possible response, IDR models have been successfully applied to categorical variables via the latent variable approach [3,4,5,6,7,8].
In exposure–response modeling of endpoints with small (e.g., < 6) possible response categories, the endpoint is typically analyzed as ordered categorical variable. When the number of response categories is ≥ 10, the endpoint is typically analyzed as continuous variable. Apart from the fact that predictions from the ordered categorical approach will always fall in legitimate categories while the continuous approach may not, the two approaches differ in residual variability modeling. The continuous approach requires normality assumptions, perhaps with transformations. In contrast, the ordered categorical approach requires as many intercept parameters as the number of categories − 1. Recently, it has been suggested that the ordered categorical approach may have advantages because it is scale-independent and therefore robust, and has good performance with adequate sample sizes [9].
Ulcerative colitis (UC) is an inflammatory bowel disease (IBD) that affects the colon. The disease activity in UC is most often evaluated with the Mayo score [10], which is the sum of 4 subscores (i.e., stool frequency, rectal bleeding, endoscopic findings, and a physician’s global assessment). Each subscore ranges from 0 to 3, with higher scores indicating more severe disease, and the total Mayo score ranges from 0 to 12. Longitudinal E–R modeling of IBD data is challenging for the following two main reasons. IBD clinical trials often employ complex study designs with the aim of evaluating treatment effectiveness during induction and maintenance phases, and treatment received in the maintenance phase typically depends on earlier responses: responders may be re-randomized, and non-responders may receive higher doses or be discontinued. In UC trials, the long-term placebo effect is usually not directly observed for ethical reasons, however its accurate assessment is important for drug effect evaluation. The lack of long-term placebo data also hampers the accurate characterization of the drug effect in the longitudinal modeling. Nevertheless, satisfactory model performance in each of the respective treatment phases is still needed to enable dose selection/optimization. To our knowledge, no longitudinal E–R modeling of Mayo scores in UC patients has yet been published.
Several tumor necrosis factor alpha (TNFα) antagonists, such as infliximab, golimumab (Simponi; Janssen Biotech, Inc., Horsham, PA) and adalimumab, have been used in the treatment of patients with moderate-to-severe UC [11, 12]. Golimumab is a subcutaneously (SC) administered fully human anti-TNFα antibody that is approved for the treatment of rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis [13,14,15,16,17], and more recently UC [18]. This manuscript reports the results of longitudinal E–R modeling of Mayo Scores, using data from 3 integrated phase II/III clinical trials of golimumab in patients with UC through a total length of 60 weeks [10, 19, 20]. The results of the continuous and categorical analysis approaches are compared and possible reasons for observed discrepancies are also discussed.
Methods
Data and information used for E–R modeling
Model development and evaluation were performed using data from 3 integrated phase II/III clinical trials: PURSUIT-IV, PURSUIT-SC, and PURSUIT-M [10, 19, 20]. These were randomized, double-blind, placebo-controlled, parallel, multicenter trials of golimumab in patients who have moderate to severe UC, with baseline Mayo score ≥ 6, and endoscopic subscore ≥ 2. PURSUIT-IV and PURSUIT-SC were induction studies; after week 6, all patients continued into the PURSUIT-M maintenance study. In PURSUIT-IV, 291 patients were randomized in an approximately 1:1:1:1 ratio to a single IV infusion of placebo, 1-, 2- or 4-mg/kg golimumab. In PURSUIT-SC, 1,064 patients were randomized (1:1:1:1) to receive SC injections of placebo or 1 of 3 golimumab induction regimens at weeks 0 and 2; golimumab doses were 100/50 mg, 200/100 mg, or 400/200 mg, respectively. Clinical response was defined as a decrease from the baseline value in the Mayo score ≥ 30% and ≥ 3 points, with either a decrease in the rectal bleeding subscore of 1 or more or a rectal bleeding subscore of 0/1.
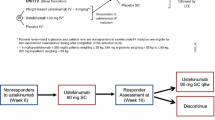
The PURSUIT-M maintenance study started at week 6 following the induction studies of PURSUIT-IV and PURSUIT-SC. The event times from the start of the induction treatment are given below. Patients who responded to golimumab induction therapy (n = 464) were randomized in a 1:1:1 ratio to receive SC placebo, golimumab 50 mg, or golimumab 100 mg every 4 weeks (q4w) through week 58. Placebo-induction responders (n = 129) received SC placebo q4w through week 58. Golimumab-induction (n = 405) or placebo-induction (n = 230) non-responders received golimumab 100 mg q4w from week 6 to week 18, and patients were discontinued from the study if disease activity was not improved based on investigator assessment at week 22. Induction-therapy responders who subsequently lost clinical response at any time during the study could have their treatment modified as follows: placebo-treated patients received golimumab 100 mg every 4 weeks, patients treated with golimumab 50 mg initially were rerandomized to receive golimumab 50 mg or 100 mg every 4 weeks, and patients treated with golimumab 100 mg initially were re-randomized to receive golimumab 100 mg or 200 mg every 4 weeks. After a protocol amendment, dose adjustment to 200 mg every 4 weeks was discontinued; patients initially randomized to 100 mg continued to receive 100 mg, and patients who already had their dose increased to golimumab 200 mg were decreased to golimumab 100 mg. A brief schema of the PURSUIT-M study design is given in Fig. 1.

Study design schema for PURSUIT-M
In PURSUIT-IV, serum golimumab concentrations were evaluated at weeks 0 (1 h post-infusion), 2, 4, 6. Mayo score was collected at weeks 0 and 6. In PURSUIT-SC, serum golimumab concentrations were evaluated at weeks 0 prior to study agent administration and weeks 2, 4, 6. Mayo score was collected at week 0 and 6. In PURSUIT-M, serum golimumab concentrations were evaluated at weeks 10, 14, 18, 26, 34, 36, 42, 50 and 60 from the start of the induction treatment. An additional random sample for the measurement of serum golimumab concentration was scheduled between weeks 22 and 30 from the start of induction treatment, and at least 24 h prior to or after a study agent injection. Mayo score was collected at weeks 36 and 60 from the start of the induction treatment.
In terms of E–R modeling, it may be helpful to view the three studies as one since there were no interruptions between the two induction studies and the subsequent maintenance study. It is noted that the maintenance (PURSUIT-M) study structure is complex, which essentially classified the patients into the following subgroups based on induction treatment groups and responder/non-responder status: induction placebo responders, induction placebo non-responders, induction golimumab non-responders, and induction golimumab responders who subsequently received placebo, golimumab 50 mg q4w, or golimumab 100 mg q4w. Understanding the performance of E–R modeling in these six subgroups, in addition to the induction phase, is important for the interpretation of clinical results.
Data available for E–R analysis were collected from a total of 1349 subjects, with a total of 4669 Mayo score evaluations across the three studies. Demographic characteristics of the subjects who were used in the current analysis can be found in previous work [21, 22].
Population PK modeling
Population PK modeling following SC administration of golimumab using a one-compartment model with first-order absorption and first-order elimination has previously been published [23]. The availability of the data from IV administration make a two-compartment population PK model possible, with body weight as the main covariate for both clearance and volume of distribution. The average clearance and absolute bioavailability were estimated as 0.54 (L/day) and 52%, respectively. The complete results will be reported elsewhere. Individual empirical Bayesian PK parameter estimates based on the population PK model were obtained and used for the subsequent E–R analysis.
Continuous E–R analysis model
In this approach, the Mayo score was modeled by adopting a semi-mechanistic approach applied in earlier E–R analyses [7] as
where Mayo(t) is the observed Mayo score at time t, b represents baseline Mayo score, fp(t) is placebo effect, and fd(t) is drug effect, and ε is residual error with a normal distribution [N(0, σ2)]. The placebo effect was modeled empirically as
where 0 ≤ Fp ≤ 1 is the fraction of maximum placebo effect and rp is the rate of onset. The drug effect was modeled as
where 0 ≤ Emax ≤ 1 represents maximum drug effect, with R(t) governed by:
where Cp is the model estimated individual drug concentration at time t, and kin (disease formation rate), IC50 (half-maximal inhibitory concentration), and kout (disease amelioration rate) are parameters in a Type I IDR model. It was further assumed that R = 1 at baseline, i.e., R(0) = 1, yielding kin = kout.
The standard IDR model form has the Emax term in Eq. (4) instead of in Eq. (3). In our experience, results are indistinguishable but estimation is faster in the current form. This IDR model representation corresponds to a change-from-baseline parameterization, where R(t) represents a latent variable of the disease process and kout may be interpreted as the rate of drug effect onset and offset. Theoretical characteristics of general change-from-baseline IDR models, which have 1 fewer parameter than their corresponding IDR models, have been derived [5, 24]. For more details on the theoretical characteristics of latent variable IDR models, see Hu [3].
Between-subject variability (BSV) on Fp and Emax was modeled assuming logit-normal distributions to restrict their values between [0, 1]. BSV on other parameters were modeled with lognormal distributions. Correlations between BSV were modeled on the normal scale.
Categorical E–R analysis model
In this approach, the Mayo score was analyzed as an ordered categorical variable, and the cumulative probability prob(Mayo score ≤ k), k = 0, 1,…,11, was modeled. The previously established latent variable IDR modeling framework was used, leading to a mixed-effect probit regression model, as follows:
where Φ is cumulative standard normal probability density function, αk are intercepts, fp(t) is placebo effect, fd(t) is drug effect, and η represents baseline BSV. The probit link was chosen because of the ease of calculating mean predictions of Eq. (5) [25], as well as potential future joint modeling with other endpoints [6, 26]. To stabilize parameter estimation, αk are reparameterized as (d0,…,d5, α6, d7,…, d11) with di > 0 such that αi = αi+1 − di for i = 5, 4, …, 0, and αi = αi-1 + di for i = 7, 8,…,11.
The placebo effect was modeled empirically:
where Pmax is the maximum placebo effect and rp is the rate of onset.
The drug effect was modeled using a latent variable R(t), governed by:
where Cp is drug concentration, and kin, IC50, and kout are parameters in a Type I IDR model. It was further assumed that at baseline R(0) = 1, yielding kin = kout. The reduction of R(t) was assumed to drive the drug effect through:
where DE is a parameter to be estimated that determines the magnitude of drug effect.
Theoretically, the representation of drug effect in Eqs. (5)–(8) has been shown to be equivalent to a change-from-baseline latent-variable IDR model [5], under which kout may be interpreted as the rate of drug effect onset and offset, and DE may be interpreted as the baseline of the latent variable prior to normalization [3]. Change-from-baseline latent IDR models are needed in the modeling of categorical endpoints because the latent variable is determined only up to a constant and therefore needs to be normalized [3, 4].
The categorical analysis model (Eqs. 5–8) has many more parameters than the continuous analysis model (Eqs. 1–4) due to the number of intercepts. While the placebo and drug effect components in the two approaches are similar, the parameters are not exactly comparable because they operate on different scales.
A 2-phase placebo effect model
A more flexible placebo effect modification was considered by allowing the rate of onset rp in Eqs. (2) and (6) to change over time, in the following form
where Tp is time of placebo effect onset rate change, rp,i is initial rate of onset when t < Tp, and 0 ≤ Pr ≤ 1 represents a fractional reduction of rate of onset when t > Tp,. Substituting Eqs. (2) or (6) by (9) results in a placebo effect model that increases in 2 phases, i.e., an initial rapid phase and a slow late phase.
Model estimation
A sequential approach described by Zhang et al. [27] was used for the E–R model estimation by first fixing individual PK parameters to their respective empirical Bayesian parameter estimates obtained from the population PK model. Parameter estimation for the E–R model was implemented in NONMEM using the Importance Sampling option with the aim to improve BSV estimation [28]. E–R model selection was based on the NONMEM objective function values (OFVs), which are approximately two times log likelihood. A change in OFV of 10.83 corresponding to a nominal p value of 0.001 was judged as significant evidence for including an additional parameter.
Model evaluation
Visual predictive checks (VPCs) [29] were used to evaluate model performance by simulating 500 replicates of the dataset and comparing simulated and model-predicted responses grouped by the planned observation times. For evaluations at the maintenance phase, ideally the model predictions should be conducted to match the appropriate responder population in the respective treatment subgroups [30]. A difficulty in VPC occurred because the rectal bleeding subscore used in the responder classification was not available for modeling. Therefore, only the Mayo score criteria (a decrease from the baseline value in the Mayo score ≥ 30% and ≥ 3) were used to classify subjects into the respective maintenance phase treatment groups in the VPCs. Another difficulty is that the induction non-responder study-discontinuation at week 16 due to lack of improvement was based on investigator assessment, not Mayo scores. To approximate this condition, the lack of improvement in Mayo score from baseline was used in the VPCs. The appropriateness of this approximation was further verified by comparing the results with actual responder/non-responder status.
To facilitate the comparisons between the continuous and the categorical modeling approaches, VPC of the categorical model was not generated on the probabilities of achieving the Mayo score levels as usually done with ordered categorical data modeling. Suitable VPC scales need to be chosen, because the continuous modeling approach works on the Mayo score whereas the categorical approach works on the probability of achieving Mayo scores. Therefore, a priori, the Mayo score scale may be expected to favor the continuous modeling approach and the probability scale may be expected to favor the categorical approach. For the purpose of demonstrating the benefit of the categorical approach, the continuous scale was chosen for the main comparison. That is, the prediction intervals (PIs) of the predicted Mayo scores were simulated for the continuous and the categorical models.
The normal residual error distribution of the continuous modeling approach allows the predicted Mayo score to be negative. It might seem reasonable to set negative simulated scores to 0. However, this would create a discrepancy between the simulation model and the fitted model, which would prevent accurate model evaluation using VPC, as will be shown in Results.
E–R model simulations
The current approved golimumab induction dose regimen is SC 200 mg at week 0 and SC 100 mg at week 2. The current approved golimumab maintenance doses for induction non-responders at week 6 however differs between the US and the EU. In the US, the approved dose regimen is 100 mg q4w SC. In the EU, the approved dose regimen depends on baseline body weight, i.e. 50 mg q4w SC for patients < 80 kg and 100 mg q4w SC for patients ≥ 80 kg. To compare these two different maintenance dose regimens, a population of 10,000 virtual patients with body weight < 80 kg and are non-responders at week 6 to the approved induction dose regimen (SC 200 mg at week 0 and SC 100 mg at week 2) was first simulated. The patients’ baseline body weight and PK parameters were bootstrapped (resampled with replacement) from data of 822 patients in the current study population. The simulated 10,000 patients were then divided evenly into two groups receiving 50 mg q4w SC or 100 mg q4w SC maintenance treatment starting from week 6. The average Mayo score for these two different maintenance dose regimens were compared at the study planned observation and responder-assessment times, i.e., week 0, 6, 14, 36, and 58.
Results
Continuous analysis—initial model
The standard continuous modeling approach using Eqs. (1)–(4), which was structurally similar to previous applications [30], was first considered. Initial exploration suggested that the baseline Mayo score distribution is better described by a normal distribution than a lognormal. Therefore, a normally distributed BSV on b was used. Attempting BSV on rp or IC50 resulted in difficulties with model convergence. Additional BSV effects were modeled for Fp, kout, and Emax, along with a correlation between b and Fp, in Eqs. (1)–(4). Parameter estimates are given in Table 1. The relative standard errors (RSE) varied and was largest for the placebo effect onset rate rp, as could be expected. VPC results for the induction phase by treatment groups were shown in Fig. 2, and the model reasonably described the data.

Visual predictive check of Mayo score in the induction phase for the initial continuous analysis model. The 5th, 50th and 95th percentiles of observed Mayo scores are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. The observed Mayo scores were included in the background in grey color. PBO placebo, SC subcutaneous, IV intravenous
VPCs results for the maintenance phase were shown in Fig. 3, separated by treatment groups and the induction responder/non-responder status. The accuracy of predicting the responder/non-responder status using only Mayo scores for the six treatment groups were 97.7%, 99.1%, 98.1%, 95.3%, 96.8%, and 98.7% respectively. Therefore, ignoring the rectal bleeding subscore criterion should not affect the quality of the VPC results. Some 5% PIs fell below 0, as the normal residual error distribution allowed. At first, this problem might seem removable by setting all negative simulated scores to 0. However this would misrepresent the characteristics of the model. For example, the 5% CIs would collapse to 0, which would not provide accurate understandings of model predicted variability at low Mayo scores. The under- and over-prediction of 5% and 95% percentiles, respectively, suggest that the model overpredicted data variability. For all subgroups, the model over-predicted observed Mayo scores in varying degrees, most notably for the induction placebo non-responders who received the 100 mg golimumab SC treatment. The phenomenon of the model adequately predicting the induction data but over-predicting the maintenance data was also observed previously with modeling data in Crohn’s disease [30], under similar type of complex study designs.

Visual predictive check of Mayo score in the maintenance phase for the initial continuous analysis model. The 5th, 50th and 95th percentiles of observed Mayo scores are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. The observed Mayo scores were included in the background in grey color. PBO placebo, ACT active (golimumab) treatment, PBO→PBO induction PBO responders receiving placebo in maintenance, PBO→100 induction PBO responders receiving 100 mg golimumab in maintenance, ACT→PBO induction active treatment responders receiving placebo in maintenance, NonResp→100 induction non-responders receiving 100 mg golimumab in maintenance, SC 50 mg, Induction active treatment responders receiving 50 mg golimumab in maintenance; SC 100 mg, Induction active treatment responders receiving 100 mg golimumab in maintenance
Continuous analysis—flexible-placebo-effect model
To improve model performance in the maintenance phase, the placebo effect onset rate was allowed to change in time in the initial continuous model by adding Eqs. (9) to (1)–(4) to fit with the data. This resulted in an OFV decrease of over 100 for the inclusion of two additional parameters, indicating significant improvement in the fit. Parameter estimates are given in Table 1. The onset rate was estimated to decrease substantially (1–0.169, or ~ 87%) after Day 47, which is right after the end of induction phase (week 6). The estimation of Tp was highly precise, with a low RSE of ~ 1.8%. Estimates of the fraction of maximum placebo effect and the initial effect placebo onset rate were similar compared with the initial model, but the RSE were notably reduced. RSEs for the remaining parameters also reduced substantially, further indicating model improvement.
VPC results for the induction phase were similar to Fig. 2 and therefore not shown. VPCs results for the maintenance phase were shown in Fig. 4. While the model still over-predicted the observed Mayo scores for all treatment groups, the magnitudes were much reduced for the two induction placebo treatment groups. It is noted that the distributions of the observed Mayo scores appeared to be skewed toward 0, more notably at week 60 (from induction). This may explain part of the difficulties of improving the model.

Visual predictive check of Mayo score in the maintenance phase for the continuous analysis model with flexible placebo effect model. The 5th, 50th and 95th percentiles of observed Mayo scores are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. The observed Mayo scores were included in the background in grey color. PBO placebo, ACT active (golimumab) treatment, PBO→PBO induction PBO responders receiving placebo in maintenance, PBO→100 Induction PBO responders receiving 100 mg golimumab in maintenance, ACT→PBO Induction active treatment responders receiving placebo in maintenance; NonResp→100 Induction non-responders receiving 100 mg golimumab in maintenance, SC 50 mg, Induction active treatment responders receiving 50 mg golimumab in maintenance; SC 100 mg, Induction active treatment responders receiving 100 mg golimumab in maintenance
Categorical analysis model
The ordered categorical modeling approach used similar structures of the fixed and random effects as in the continuous model with flexible placebo effect. That is, Eqs. (5)–(9) was fitted to the Mayo score data, with BSV effects on baseline, Pmax (maximum placebo effect), kout, and DE, along with a correlation between baseline (η) and Pmax. The parameter estimates are given in Table 2. It is difficult to exactly compare parameter estimations between the continuous and the categorical model due to the scale difference between the two approaches. On the other hand, RSE in the categorical model appeared to be much smaller, suggesting improved estimation stability. VPC results for the induction phase by treatment groups were shown in Fig. 5 At a first look, the model predictions may seem unusual as some PIs have 0 length. This is due to the fact that quantiles of categorical values, in this case integers between 0 and 12, usually may only be integers. With this in mind, the model appeared to reasonably describe the data.

Visual predictive check of Mayo score in the induction phase for the categorical analysis model. The 5th, 50th and 95th percentiles of observed Mayo scores are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. The observed Mayo scores were included in the background in grey color. PBO placebo, SC subcutaneous, IV intravenous
VPCs results for the maintenance phase are shown in Fig. 6. Compared with those results from the continuous analysis model with flexible placebo effect (Fig. 5), the model predictions were in much closer agreement with observed data. In Fig. 6, medians of observed data are generally covered by their corresponding PIs. In addition, the 5% PIs are all at or above 0, because the categorical model simulated Mayo scores are always in the legitimate range of 0 to 12.

Visual predictive check of Mayo score in the maintenance phase for the categorical analysis model. The 5th, 50th and 95th percentiles of observed Mayo scores are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. The observed Mayo scores were included in the background in grey color. PBO placebo, ACT active (golimumab) treatment, PBO→PBO induction PBO responders receiving placebo in maintenance, PBO→100 induction PBO responders receiving 100 mg golimumab in maintenance, ACT→PBO induction active treatment responders receiving placebo in maintenance, NonResp→100 induction non-responders receiving 100 mg golimumab in maintenance; SC 50 mg, Induction active treatment responders receiving 50 mg golimumab in maintenance; SC 100 mg, Induction active treatment responders receiving 100 mg golimumab in maintenance
Application simulation
Figure 7 shows the categorical model predicted average Mayo scores for the simulated patient population with body weight < 80 kg who receive the approved golimumab induction (SC 200 mg at week 0 and SC 100 mg at week 2) and are non-responders at week 6. Patients receiving 100 q4w SC as approved in the US posology were predicted to have lower Mayo scores than those receiving 50 q4w SC as approved in the EU posology. The model predicted clinical response rates for patients receiving 50 q4w SC at week 14, week 36, and week 58 were 38.5%, 54.1% and 57.0%, respectively. The corresponding predicted clinical response rates for patients receiving 100 q4w SC were higher, and were 42.7%, 62.2%, and 62.8%, respectively.

Average predicted average Mayo score over time under the US and EU approved golimumab dose regimens for Induction Week 6 non-responders to the approved induction dose regimen (SC 200 mg at week 0 and SC 100 mg at week 2) with body weight < 80 kg. 100 mg, receiving 100 mg q4w SC starting from week 6 in maintenance; 50 mg, receiving 50 mg q4w SC starting from week 6
Discussion
Clinical trial endpoints are often composite scores with varying possible number of categories that measure disease severity. Endpoints with ≥ 10 possible response categories are customarily analyzed as continuous data. Analyzing such data as categorical is attractive in that the model predictions are never outside the natural range of possible values. The categorical analysis approach does require many more parameters to model the intercepts, therefore may potentially lose analysis efficiency when the residual error distribution does not substantially deviate from normal. However, with sufficient number of observations (> 1000 at each time point) available, the loss of efficiency is limited. On the other hand, when skewness is present in the residual error distribution, the categorical analysis approach remains accurate but the continuous analysis approach is not. In this sense, the categorical analysis approach is robust [9].
From a related perspective, endpoints such as the Mayo scores have been classified as bounded outcome scores (BOS) which, by definition, report a discrete set of values on a finite range. Such endpoints often demonstrate non-normal distributions near the boundary, and analysis approaches involving various kind of transformations have been used. A direct application of beta-regression using a seemingly innocuous small correction factor to transform the endpoint to the open interval (0,1) may be intuitively appealing but ill-behaved [8]. Hutmacher et al. proposed a censoring approach that is essentially continuous but treats observations at the boundary as censored [31]. The coarsened grid approach [32] may be viewed as a categorical approach with default intercepts. A later extension [33] used parametric transformations to model the intercepts parsimoniously. Despite the conceptual difference, in our experience the censoring and the coarsened grid approach may perform similarly [8]. Ursino and Gasparini [34] applied beta-distribution on the latent variable scale. Most recently, a “bounded integer model” has been proposed [35], which motivates default intercepts differently than the coarsened grid approach and allows a broader class of models than used in [33]. The beta-distribution, the coarsened grid, and the bounded integer model approaches can be put under a general discrete data analysis framework [36]. Under this framework, the traditional categorical analysis, by estimating all intercepts, may be viewed as a “saturated” model in statistical terms, and models formally may be compared by using e.g., AIC or BIC. When sample sizes are large such as in phase 3 clinical trials, all intercept parameters may be estimated with reasonable accuracy, as Table 2 indicates. This may therefore lessen the need of searching for models with fewer parameters, e.g., through approximating the intercepts. This is consistent with our recent experience in psoriasis using PASI scores as a measure of disease severity, where a categorical model of PASI-score based criteria performed better than modeling the PASI scores, either as continuous data or some of the discrete data analysis approaches using fewer parameters [8]. When sample sizes are smaller, exploring more parsimonious approaches [36] should be beneficial.
The use of Mayo score scale for VPC requires some further discussion. It may seem that the continuous model predictions could be easily discretized to also allow a comparison of model performance on the probability scale, however the discretization may not be without controversy. For example, letting Y be the model predicted Mayo scores, the most intuitive method may be to first round off Y to the nearest integer [Y], and then calculate the proportion of ([Y] ≤ k). On the other hand, the rounding introduces noise, and to avoid this, the proportion ([Y] ≤ k) could be directly calculated without rounding. This illustrates in a way a fundamental difference in nature between continuous and categorical data, and therefore the lack of conceptually common scales for VPCs. On the other hand, a practically important modeling objective is to predict the responder/non-responder status. Additional VPCs on this scale were conducted for the final continuous model and the categorical model. The categorical model performed better than the continuous model in both Induction and Maintenance phases, and the results were included in Supplementary Material (Figs. S1–S4). The difficulty of accurately predicting derived criteria (e.g., responder/non-responder status) using models based on original clinical endpoints (e.g., Mayo score) has previously been noted [8].
VPC is practically effective but informal. One may therefore wonder whether any formal evaluations, e.g., AIC or BIC, can be used to compare the continuous and categorical approaches. Unfortunately this is not possible, because formal statistical comparisons require that the data stay the same. It is noted that although data values (or more accurately, notations) remained the same under the continuous and categorical approaches, the continuous approach presumes a much larger possible data space than the actual data values. It is noted that this restriction does not apply to the comparisons among the categorical type of models, e.g., the saturated and the parsimonious models [36], where the data space remain the same.
Although more complex placebo effect models have been used [37], the onset rate to maximum effect is typically modeled with one exponential term. The fact that data may not usually allow the identification of more complex models might discourage even the consideration of the more complex models. To our knowledge, more flexible models to describe placebo effect onset rate (such as Eq. 9) has not been used before. While the data were collected over a relatively long period (a total of 60 Weeks), but with only four scheduled Mayo score observation timepoints, one might normally expect Tp to be estimated anywhere between the two middle timepoints, and with low precision. The fact that Tp was estimated as shortly (< 6 days) after the induction period with high precision may therefore suggest a change in population characteristics after the induction period. Potential factors contributing to such change may include that induction non-responders may be less likely to continue onto the maintenance phase, patients’ perceptions may change after entering the maintenance phase, study conduct may change (as the name of the study changed), or any other confounding factors related to time. The existence or nonexistence of such factors cannot be directly observed by comparing the observed placebo outcomes between the induction and maintenance periods, because patients receiving placebo in maintenance must be placebo responders in induction and therefore a biased subgroup for assessing the overall maintenance placebo effect. Placebo effect plays an important role in longitudinal modeling of clinical trial data as it interacts with drug effect modeling. For example, in the continuous analysis model, IC50 estimation precisions were high in both the standard and the more flexible placebo model, but the magnitudes differed by 2-fold. This illustrates that high estimation precision of a parameter (e.g., IC50) may not guarantee that of a structural property (e.g., potency). Better structural models, including placebo effect models, allow more accurate practical interpretations and usages of the model structural parameters.
Longitudinal E–R modeling can provide unique insight at various stages to aid drug development and approval decisions [38]. Its conduct in IBD is however particularly challenging, in part due to the common use of complex study designs such as response-based rerandomizations [30]. Such complex designs create statistically dependent treatment subgroups with different sensitivities to placebo and drug treatments, which complicates data visualization and analysis interpretations. For example, the over-prediction of all maintenance treatment effect subgroups of the initial model in Fig. 2 might appear to suggest that placebo effect onset rate should be allowed to increase in the maintenance phase. To the contrary, Eq. (9), the more flexible placebo effect model, has the placebo effect decreased in the maintenance phase but better described the data. This illustrates the complexity of attempting model improvement under such complex study designs.
To our knowledge, this is a first attempt of longitudinal E–R modeling of Mayo scores in UC that involves complex clinical trial designs. Data in the induction phase were reasonably predicted. Certain degree of under-prediction of treatment effect appears to remain for the induction responder subgroups in maintenance. The reason for this is unclear, and could include (1) dosing adjustment of induction responders who subsequently lost response could occur any time during the study and thus difficult to be accounted for in VPC, (2) after the induction treatment, patients sensitivity to treatment might have changed, even with the use of the flexible bi-phasic onset-rate placebo effect model, or (3) informative dropout [39], e.g., patients with worse outcomes may be more inclined to leave the study. Further improving longitudinal modeling in the IBD area is the subject of future research.
References
Overgaard RV, Ingwersen SH, Tornoe CW (2015) Establishing good practices for exposure-response analysis of clinical endpoints in drug development. CPT 4(10):565–575. https://doi.org/10.1002/psp4.12015
Sharma A, Jusko WJ (1996) Characterization of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm 24(6):611–635
Hu C (2014) Exposure-response modeling of clinical end points using latent variable indirect response models. CPT 3:e117. https://doi.org/10.1038/psp.2014.15
Hutmacher MM, Krishnaswami S, Kowalski KG (2008) Exposure-response modeling using latent variables for the efficacy of a JAK3 inhibitor administered to rheumatoid arthritis patients. J Pharmacokinet Pharmacodyn 35:139–157
Hu C, Xu Z, Mendelsohn A, Zhou H (2013) Latent variable indirect response modeling of categorical endpoints representing change from baseline. J Pharmacokinet Pharmacodyn 40(1):81–91
Hu C, Szapary PO, Mendelsohn AM, Zhou H (2014) Latent variable indirect response joint modeling of a continuous and a categorical clinical endpoint. J Pharmacokinet Pharmacodyn 41(4):335–349. https://doi.org/10.1007/s10928-014-9366-0
Hu C, Zhou H (2016) Improvement in latent variable indirect response joint modeling of a continuous and a categorical clinical endpoint in rheumatoid arthritis. J Pharmacokinet Pharmacodyn 43(1):45–54
Hu C, Randazzo B, Sharma A, Zhou H (2017) Improvement in latent variable indirect response modeling of multiple categorical clinical endpoints: application to modeling of guselkumab treatment effects in psoriatic patients. J Pharmacokinet Pharmacodyn 44(5):437–448. https://doi.org/10.1007/s10928-017-9531-3
Liu Q, Shepherd BE, Li C, Harrell FE Jr (2017) Modeling continuous response variables using ordinal regression. Stat Med 36(27):4316–4335. https://doi.org/10.1002/sim.7433
Rutgeerts P, Feagan BG, Marano CW, Padgett L, Strauss R, Johanns J, Adedokun OJ, Guzzo C, Zhang H, Colombel JF, Reinisch W, Gibson PR, Sandborn WJ, group P-Is (2015) Randomised clinical trial: a placebo-controlled study of intravenous golimumab induction therapy for ulcerative colitis. Aliment Pharmacol Ther 42(5):504–514. https://doi.org/10.1111/apt.13291
Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJ, Present D, Sands BE, Colombel JF (2005) Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 353(23):2462–2476. https://doi.org/10.1056/NEJMoa050516
Sandborn WJ, van Assche G, Reinisch W, Colombel JF, D’Haens G, Wolf DC, Kron M, Tighe MB, Lazar A, Thakkar RB (2012) Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology 142(2):257–265. https://doi.org/10.1053/j.gastro.2011.10.032
Braun J, Deodhar A, Inman RD, van der Heijde D, Mack M, Xu S, Hsu B (2012) Golimumab administered subcutaneously every 4 weeks in ankylosing spondylitis: 104-week results of the GO-RAISE study. Ann Rheum Dis 71(5):661–667. https://doi.org/10.1136/ard.2011.154799
Kavanaugh A, van der Heijde D, McInnes IB, Mease P, Krueger GG, Gladman DD, Gomez-Reino J, Papp K, Baratelle A, Xu W, Mudivarthy S, Mack M, Rahman MU, Xu Z, Zrubek J, Beutler A (2012) Golimumab in psoriatic arthritis: one-year clinical efficacy, radiographic, and safety results from a phase III, randomized, placebo-controlled trial. Arthritis Rheum 64(8):2504–2517. https://doi.org/10.1002/art.34436
Kay J, Matteson EL, Dasgupta B, Nash P, Durez P, Hall S, Hsia EC, Han J, Wagner C, Xu Z, Visvanathan S, Rahman MU (2008) Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: a randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum 58(4):964–975. https://doi.org/10.1002/art.23383
Keystone E, Genovese MC, Klareskog L, Hsia EC, Hall S, Miranda PC, Pazdur J, Bae SC, Palmer W, Xu S, Rahman MU (2010) Golimumab in patients with active rheumatoid arthritis despite methotrexate therapy: 52-week results of the GO-FORWARD study. Ann Rheum Dis 69(6):1129–1135. https://doi.org/10.1136/ard.2009.116319
Smolen JS, Kay J, Doyle MK, Landewe R, Matteson EL, Wollenhaupt J, Gaylis N, Murphy FT, Neal JS, Zhou Y, Visvanathan S, Hsia EC, Rahman MU (2009) Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet 374(9685):210–221. https://doi.org/10.1016/S0140-6736(09)60506-7
Gibson PR, Feagan BG, Sandborn WJ, Marano C, Strauss R, Johanns J, Padgett L, Collins J, Tarabar D, Hebzda Z, Rutgeerts P, Reinisch W (2016) Maintenance of efficacy and continuing safety of golimumab for active ulcerative colitis: PURSUIT-SC maintenance study extension through 1 year. Clin Transl Gastroenterol 7:e168. https://doi.org/10.1038/ctg.2016.24
Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, Adedokun OJ, Guzzo C, Colombel JF, Reinisch W, Gibson PR, Collins J, Jarnerot G, Hibi T, Rutgeerts P, Group P-SS (2014) Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology 146(1):85–95. https://doi.org/10.1053/j.gastro.2013.05.048
Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, Adedokun OJ, Guzzo C, Colombel JF, Reinisch W, Gibson PR, Collins J, Jarnerot G, Rutgeerts P, Group PU-MS (2014) Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology 146(1):96–109. https://doi.org/10.1053/j.gastro.2013.06.010
Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, Blank MA, Johanns J, Gao LL, Miao Y, Adedokun OJ, Sands BE, Hanauer SB, Vermeire S, Targan S, Ghosh S, de Villiers WJ, Colombel JF, Tulassay Z, Seidler U, Salzberg BA, Desreumaux P, Lee SD, Loftus EV Jr, Dieleman LA, Katz S, Rutgeerts P, Group U-I-US (2016) Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med 375(20):1946–1960. https://doi.org/10.1056/nejmoa1602773
Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, Ghosh S, de Villiers WJ, Panaccione R, Greenberg G, Schreiber S, Lichtiger S, Feagan BG, Group CS (2012) Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med 367(16):1519–1528. https://doi.org/10.1056/nejmoa1203572
Xu Z, Vu T, Lee H, Hu C, Ling J, Yan H, Baker D, Beutler A, Pendley C, Wagner C, Davis HM, Zhou H (2009) Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-alpha human monoclonal antibody, in patients with psoriatic arthritis. J Clin Pharmacol 49(9):1056–1070. https://doi.org/10.1177/0091270009339192
Woo S, Pawaskar D, Jusko WJ (2009) Methods of utilizing baseline values for indirect response models. J Pharmacokinet Pharmacodyn 36:381–405
Hutmacher MM, French JL (2011) Extending the latent variable model for extra correlated longitudinal dichotomous responses. J Pharmacokinet Pharmacodyn 38:833–859
Hu C, Xu Y, Zhuang Y, Hsu B, Sharma A, Xu Z, Zhang L, Zhou H (2018) Joint longitudinal model development: application to exposure-response modeling of ACR and DAS scores in rheumatoid arthritis patients treated with sirukumab. J Pharmacokinet Pharmacodyn 45(5):679–691. https://doi.org/10.1007/s10928-018-9598-5
Zhang L, Beal SL, Sheiner LB (2003) Simultaneous vs sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn 30(6):387–404
Hutmacher MM (2016) Evaluation of estimation, prediction and inference for autocorrelated latent variable modeling of binary data-a simulation study. J Pharmacokinet Pharmacodyn 43(3):275–289. https://doi.org/10.1007/s10928-016-9471-3
Karlsson MO, Holford NHG (2008) A tutorial on visual predictive checks. www.page-meeting.org/?abstract=1434
Hu C, Adedokun OJ, Chen Y, Szapary PO, Gasink C, Sharma A, Zhou H (2017) Challenges in longitudinal exposure-response modeling of data from complex study designs: a case study of modeling CDAI score for ustekinumab in patients with Crohn’s disease. J Pharmacokinet Pharmacodyn 44(5):425–436. https://doi.org/10.1007/s10928-017-9529-x
Hutmacher MM, French JL, Krishnaswami S, Menon S (2011) Estimating transformations for repeated measures modeling of continuous bounded outcome data. Stat Med 30(9):935–949. https://doi.org/10.1002/sim.4155
Lesaffre E, Rizopoulos D, Tsonaka R (2007) The logistic transform for bounded outcome scores. Biostatistics 8(1):72–85
Hu C, Yeilding N, Davis HM, Zhou H (2011) Bounded outcome score modeling: application to treating psoriasis with ustekinumab. J Pharmacokinet Pharmacodyn 38(4):497–517
Ursino M, Gasparini M (2018) A new parsimonious model for ordinal longitudinal data with application to subjective evaluations of a gastrointestinal disease. Stat Methods Med Res 27(5):1376–1393. https://doi.org/10.1177/0962280216661370
Wellhagen GJK (2018) A bounded integer model for rating and composite scale data. PAGE 27 (2018) Abstr 8743 [www.page-meeting.org/?abstract=8743]
Iannario MP, Piccolo D (2016) A comprehensive framework of regression models for ordinal data. METRON 74(2):233–252. https://doi.org/10.1007/s40300-016-0091-x
Pilla Reddy V, Kozielska M, de Greef R, Vermeulen A, Proost JH (2013) Modelling and simulation of placebo effect: application to drug development in schizophrenia. J Pharmacokinet Pharmacodyn 40(3):377–388. https://doi.org/10.1007/s10928-012-9296-7
Hu C, Zhou H, Sharma A (2017) Landmark and longitudinal exposure-response analyses in drug development. J Pharmacokinet Pharmacodyn 44(5):503–507. https://doi.org/10.1007/s10928-017-9534-0
Hu C, Sale M (2003) A joint model for nonlinear longitudinal data with informative dropout. J Pharmacokinet Pharmacodyn 30(1):83–103
Funding
This research was funded by Janssen Research and Development, LLC.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
: Visual predictive check of responder/non-responder status calculated from only Mayo scores at the end of the induction phase for the initial continuous analysis model with flexible placebo effect model. The 5th, 50th and 95th percentiles of observed proportion of responders are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. PBO, placebo; SC, subcutaneous; IV, intravenous. Supplementary material 1 (EPS 6 kb)
Figure S2
: Visual predictive check of responder/non-responder status calculated from only Mayo scores in the maintenance phase for the continuous analysis model with flexible placebo effect model. The 5th, 50th and 95th percentiles of observed proportion of responders are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. PBO, placebo; ACT, active (golimumab) treatment; PBO→PBO, Induction PBO responders receiving placebo in maintenance; PBO→100, Induction PBO responders receiving 100 mg golimumab in maintenance; ACT→PBO, Induction active treatment responders receiving placebo in maintenance; NonResp→100; Induction non-responders receiving 100 mg golimumab in maintenance; SC 50 mg, Induction active treatment responders receiving 50 mg golimumab in maintenance; SC 100 mg, Induction active treatment responders receiving 100 mg golimumab in maintenance. Supplementary material 2 (EPS 15 kb)
Figure S3
: Visual predictive check of responder/non-responder status calculated from only Mayo scores at the end of the induction phase for the categorical analysis model. The 5th, 50th and 95th percentiles of observed proportion of responders are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. PBO, placebo; SC, subcutaneous; IV, intravenous. Supplementary material 3 (EPS 6 kb)
Figure S4
: Visual predictive check of Mayo score in the maintenance phase for the categorical analysis model. The 5th, 50th and 95th percentiles of observed proportion of responders are overlaid with the 90% prediction intervals (PI) of their model predictions at planned observation times by treatment. PBO, placebo; ACT, active (golimumab) treatment; PBO→PBO, Induction PBO responders receiving placebo in maintenance; PBO→100, Induction PBO responders receiving 100 mg golimumab in maintenance; ACT→PBO, Induction active treatment responders receiving placebo in maintenance; NonResp→100; Induction non-responders receiving 100 mg golimumab in maintenance; SC 50 mg, Induction active treatment responders receiving 50 mg golimumab in maintenance; SC 100 mg, Induction active treatment responders receiving 100 mg golimumab in maintenance.Supplementary material 4 (EPS 15 kb)
Rights and permissions
About this article

Cite this article
Hu, C., Adedokun, O.J., Zhang, L. et al. Modeling near-continuous clinical endpoint as categorical: application to longitudinal exposure–response modeling of Mayo scores for golimumab in patients with ulcerative colitis. J Pharmacokinet Pharmacodyn 45, 803–816 (2018). https://doi.org/10.1007/s10928-018-9610-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-018-9610-0