
Abstract
Silicon-based clusters have attracted particular attention because they are regard as building blocks for developing silicon-based nanomaterials. However, pure silicon clusters have low chemical stability owing to their dangling bonds. Doping with lanthanide atoms is a good way to form closed-shell of M@Sin clusters and alter their electronic and magnetic properties. Here, we systematically study the lanthanide element Pr doped neutral and anionic silicon clusters by using density functional theory. Extensive searches for ground-state structures of Sin+1λ and PrSinλ (n = 1–9, λ = 0, − 1) clusters were carried out based on the comparison between experimental photoelectron spectroscopy and simulated spectra. Furthermore, the calculated AEA values of our obtained structures show good agreement with the experimental values. Based on averaged binding energies, fragmentation energies and HOMO–LUMO gaps, their relative stabilities were analyzed. Furthermore, the patterns of HOMOs for the most stable isomers were investigated to gain insight into the nature of bonding. The results show that some σ-type and few π-type bonds are formed among Si and Pr atoms. To achieve a insight into localization of charge and charge-transfer information, the Mulliken population are analyzed and discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As is well known, silicon-based nanomaterials is the most important material for the modern microelectronics industry and the semiconductor devices [1,2,3,4,5]. Under this background, silicon-based clusters attracted particular attention due to that they are regard as building blocks for developing new and tunable silicon-based nanomaterials [6,7,8,9,10,11]. However, pure silicon cluster are unsuitable for the building block, because they have low chemical stability owing to the existence of dangling bonds [12]. After the addition of “impurity” atoms, especially the metal atoms, the M@Sin clusters tend to form closed-shell electronic structures that show extraordinary stabilities comparing with the pure species. Especially, doping with transition metal (TM) atoms possessing unfilled d-shells is a good way to alter the electronic and magnetic properties of silicon clusters. The unpaired d-electrons of transition metal atoms may retain their magnetic moments when TM atoms are embedded in a silicon cage.
Up to now, substantial experimental and theoretical works have been carried out on TM atoms doped silicon clusters, such as SinM (M = V, Mn, Ti, Cr, Mo, W and Cu) [13,14,15,16,17,18,19]. These previous studies showed that transition-metal (TM) atoms stabilize silicon cages by sitting endohedrally within them, TM@Sin. These Si-TM clusters might be served as basic materials for silicon-based semiconductor technology and the nascent field of spintronics. However, a considerable amount of evidence showed that the d-orbitals of endohedral transition metal atom interact strongly with the silicon’s sp-orbitals, thereby quenching the magnetic moment of cluster [20, 21].
To bypass this constraint, the endohedral doping of silicon clusters with lanthanide (Ln) atoms was proposed. As is well known, lanthanide (Ln) elements [22, 23] possess more localized f-electrons, which are, to a large extent, not responsible for bonding. Lanthanide atoms with lots of unpaired f-electrons may retain a significant portion of their atomic magnetic moments even in a silicon cage. Consequently, the magnetic properties [7] and potential applications of LnSin clusters attracted wide attentions in the past decade. For example, Ohara et al. [24] have studied the experimental photoelectron spectra and water reactivities of TbSin− (6 ≤ n ≤ 16). Kumar et al. [25] performed an theoretical study on the encapsulated fullerene like neutral and anionic M@Si20 (M = La, Ac, Sm, Gd, Tm, Ce, Pa, Pu, Th, Np, Pm) clusters. Their results showed that Pa@Si20, Sm@Si20, Pu@Si20, Tm@Si20, and Gd@Si20− retain rather significant magnetic moments in their ground state structures. Bowen’s group reported the photoelectron spectroscopic (PES) studies of EuSin− (3 ≤ n ≤ 7) [7] and LnSin− (Ln = Yb, Eu, Sm, Gd, Ho, Pr; 3 ≤ n ≤ 13) [26]. They found that the PES can be classified as three types according to their motifs. Several years ago, we have reported an extensive investigation on the small sized neutral and anionic SinSmλ (n = 1–9, λ = 0, − 1) clusters [27]. These previous studies provided the insights into the interaction between silicon and lanthanide atoms. However, the literature on lanthanide-containing silicon clusters is still limited.
In this paper, we reported an extensive investigation on the small sized neutral and anionic PrSinλ (n = 1–9, λ = 0, − 1) clusters based on density functional theory. The pure silicon clusters were also studied by using the identical method and basis sets for comparison. The main objective of this research is to investigate the nature of interaction between silicon and praseodymium atoms. The various ground-state structures for PrSinλ (n = 1–9, λ = 0, − 1) are also obtained, which can provide significant help for such kind of cluster assemble materials.
Computational Schemes
Geometrical optimizations and frequency analysis of PrSinλ (n = 1–9, λ = 0, − 1) clusters were carried out using density functional theory [28,29,30], as implemented in the Gaussian09 program package [31]. B3PW91 functional, which is Becke’s three-parameter functional (B3) [32] in conjunction with Perdew–Wang’s correlation [33], were first employed for studying this system. Afterwards, a carefully selected set of the low-energy optimized structures was tested by B3LYP [34] and CCSD (t) [35,36,37] methods. This can guarantee the accuracy of our calculations. The basis sets labeled GENECP are the combination of 6-311+G* [38] and MWB48 [39, 40] basis sets, which are employed for Si and Pr atoms, respectively. The ECP of MWB48 was developed by Dolg et al. for lanthanide metal atoms. A total of 48 electrons are included in the lanthanide core and the remaining 11 electrons are treated explicitly. Moreover, the adiabatic detachment energies (AEAs) were calculated using B3LYP, B3PW91 and CCSD (t) methods. The results comparing with experimental values [26] were presented in Table 1.
In this paper, the equilibrium geometries of pure silicon clusters were first studied based on the previous investigations. Our obtained structures of pure silicon clusters in the range of 2–10 atoms agree well with the previous results [41,42,43,44,45]. To search for the lowest energy structure of PrSinλ clusters, a large number of initial structures were obtained by placing the Pr atom on each possible site of the Sin0/− host clusters as well as by substituting one Si atom of Sin+10/− clusters using Pr. The previous studies [46, 47] on other lanthanide atoms doped silicon clusters were also employed as a guide. Due to the spin polarization, each initial structure was optimized at various possible spin multiplicities. Meanwhile, the vibrational frequency calculations were performed to make sure that the structures correspond to real local minima without imaginary frequency. As is well known, the well-resolved experimental PES can serve as electronic “fingerprints” of the underlying clusters. In order to prove the accuracy of our obtained structures, we have performed the simulated photoelectron spectroscopy spectra (PES) for the ground state isomers and compared them with Bowen’s experimental spectra [26].
Results and Discussion
Geometrical Structures
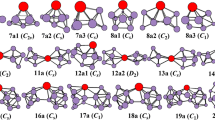
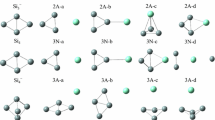
Based on the method that has been pointed out above, a large number of optimized isomers for PrSinλ (n = 1–9, λ = 0, − 1) clusters were obtained. We only select the four most likely candidate isomers for each size and list them in Figs. 1, 2 and 3. According to their energies from low to high, the neutral isomers are designated by nN-a, nN-b, nN-c and nN-d; the anions are designated by nA-a, nA-b, nA-c and nA-d. Where n represents the number of Si atoms in PrSin0/− clusters. In order to examine the effects of dopant Pr atom in silicon clusters, the lowest-energy structures of Sin0/− (n = 2–10) clusters, which are obtained by using the identical method and basis set, are also displayed for comparison. The corresponding relative energies, symmetries, electronic states, HOMO energies and LUMO energies, which are calculated based on B3PW91 functional, for the selected pure and doped clusters are summarized in Table 2. In addition, the electronic states, symmetries, HOMO energies, LUMO energies and HOMO–LUMO gaps of the lowest energy structures of PrSinλ (n = 2–9; λ = 0, − 1) clusters at CCSD(T) level are showed in Table 3. Most of the electronic states and symmetries based on the two levels are almost the same. Although the values of HOMO–LUMO gaps at CCSD(T) level are much larger than those at B3PW91 level, the general trend of the curve against the cluster size is similar roughly. Because B3PW91 is the first employed functional to study this system. Henceforth, the values of relative energies, HOMO and LUMO energies are given at B3PW91 level, unless mentioned otherwise.

The ground-state structures of Sin+1λ and PrSinλ (n = 2–4; λ = 0, − 1) clusters, and some low-lying isomers for doped clusters. The light blue and gray spheres represent the Pr and Si atoms, respectively (Color figure online)

The ground-state structures of Sin+1λ and PrSinλ (n = 5–7; λ = 0, − 1) clusters, and some low-lying isomers for doped clusters. The light blue and gray spheres represent the Pr and Si atoms, respectively (Color figure online)

The ground-state structures of Sin+1λ and PrSinλ (n = 8–9; λ = 0, − 1) clusters, and some low-lying isomers for doped clusters. The light blue and gray spheres represent the Pr and Si atoms, respectively (Color figure online)
As shown in Figs. 1, 2 and 3, our ground state structures of Sin and Sin− (n = 2–10) clusters are in good agreement with the results reported in previous studies [41, 48, 49]. Considering that the ground state structures of pure Sin and Sin− (n = 2–10) have been discussed everywhere, including our previous study [19]. In this section, we won’t describe pure silicon clusters in detail.
As for PrSinλ (n = 1–9, λ = 0, − 1) clusters, the first four low-lying structures, together with their corresponding symmetries, are presented in Figs. 1, 2 and 3, respectively. Our obtained ground state of PrSi is shown to be 6Σ, with 2.505 Å bond length at B3PW91 level. Upon an extra electron attachment, the ground state and bond length of PrSi− anion will change to be 5Σ and 2.611 Å. These results are in good agreement with the previous calculations [4]. All the possible initial structures of PrSi20/− clusters, i.e., linear structures (D∞h, C∞v), and triangle structures (acute angle or obtuse angle) are optimized with different spin multiplicities. The triangle structures (2N-a and 2A-a) with acute angle are found to be the lowest energy isomers for neutral and anionic clusters. Both of them can be obtained by replacing one Si atom by Pr in the corresponding ground-state Si30/− clusters. In addition, the ground state isomers of 3N-a, 3A-a, 4N-a and 4A-a can also be derived from the corresponding silicon clusters in the same way.
Starting at n = 4, the lowest energy structures of neutral and anionic PrSin clusters change from planar to three-dimensional geometries. When the number of silicon atoms is up to 5, no low-lying planar structure is obtained in our calculations. And almost all the neutral and anionic PrSin clusters have low symmetry. Moving on to the bigger clusters (n > 5), the ground state structures of neutral and anionic PrSin clusters begin to show different appearance. This indicates that attachment of an extra electron can exert a great influence on the structure. The praseodymium atom tends to occupy the low coordinated position, such as edge-capped or face-capped positions. Four Si atoms are allowed to bound atomically to Pr atom. This is in good agreement with the previous studies performed on the other Ln atoms doped silicon cluster, such as SmSin [27], EuSin [46] and so on, which indicates that Ln atoms don’t favor high coordinated position to form closed-shell structure. By comparing our results of the lowest energy structures of PrSin0/−1 (n = 3–9) clusters with the results reported in Ref. [4], it is found that our obtained lowest energy structures of PrSin0/−1 (n = 3–6) clusters are very similar to their obtained structures based on ABCluster global search technique. As for the lowest energy structures of PrSin0/−1 (n = 7–9) clusters, there are some differences between our results and Feng’s results because of the large number of atoms. However, the type of structure, in which Pr atoms tend to occupy the edge-capped or face-capped positions, is also similar. This further validates the reliability of our calculations. The reason why Pr atoms tends to occupy low coordinated position may be associated with its electronic configuration. It is well known that the outer shell electron distribution of lanthanide atoms is basically the same. With the increase of atomic number, only the inner orbitals (corresponding 4f orbital) are filled with electrons. In this case, the bonding characteristic between Ln and Si atoms, which is related with the outer shell electrons, is very similar. The few outer shell electrons allows few bonds with Si atoms, resulting in its low coordinated position in silicon frames.
AEAs and Simulated PES
The adiabatic detachment energies (AEAs) were calculated using B3LYP, B3PW91 and CCSD (t) methods and compared with the experimental values [26]. From Table 1, it can be seen that the theoretical values (particularly at B3LYP level) in good agreement with the experimental results with the deviations less than 4.0%, except for PrSi4 and PrSi9. This further validates the reliability of our calculations. With regard to the PrSi4 cluster, the value obtained at CCSD (t) level (1.697 eV) is more close to the experimental date (1.600 eV). Overall, the AEA values increase with the cluster size increasing, indicating the increase of the inherent electronic stabilization.
Bowen’s group [26] performed the PES experiment for PrSin− (n = 4–9) clusters using a magnetic-bottle time-of-flight photoelectron spectrometer equipped with a laser vaporization cluster source. As is well known, the experimental PES spectra could be used to compare with theoretical simulations and serve as electronic “fingerprints” of the underlying clusters [50]. In this case, we have performed the simulated spectra by adding the relative energies of the orbitals (∆En = E(HOMO-n) − EHOMO) to the vertical detachment energies (VDEs). The simulations were carried out using Multiwfn program package [51, 52]. The threshold energies correspond to the adiabatic electron affinities (AEAs) of the ground state clusters which are calculated based on B3LYP method. Then they are fitted with a unit-area Gaussian function of 0.26 eV full width at half maximum. The VDEs of each cluster anion corresponds to the first peak maximum of each spectrum in Fig. 4. Because the non-adiabatic interactions and anharmonic resonances were not included in our calculations, it is not possible to quantitatively compare calculated intensities with experimental ones. However, the positions and the general shape of the peaks can be compared with the experimental spectra. As can be seen in Fig. 4, the threshold energies of PrSi5−, PrSi6−, PrSi7 and PrSi8− clusters, which correspond to their AEAs at B3LYP level, in good agreement with the experimental ones. The amounts of distinct peaks of simulated PES for PrSi4−, PrSi5−, PrSi6− and PrSi7− clusters in the range of ≤ 4.5 eV general agree with the measured spectra. The agreement of locations and the amounts of distinct peaks increases the confidence in the reliability of the obtained ground-state structures.

Photoelectron spectra *of PrSin− (n = 4–9) measured at 266 nm. (The spectra are taken from Ref. [26]. Copyright 2009 American Chemical Society). Simulated photoelectron spectra for the lowest-energy structures of PrSin− (n = 2–9) clusters at the B3LYP level
Relative Stabilities
The relative stabilities of the ground state PrSinλ (n = 1–9, λ = 0, − 1) clusters were studied based on the calculation of averaged binding energies Eb(n) and fragmentation energies ΔE(n), which are defined by the following formulae:
where \(E(Si)\), \(E(\Pr )\), \(E(Si^{\lambda } )\), \(E(Si_{n} \Pr^{\lambda } )\) and \(E(Si_{{n{ - }1}} \Pr^{\lambda } )\) denote the energies of Si, Pr, Siλ, PrSinλ and PrSin−1λ, respectively.
Considering the influence of impurity atom on the small pure clusters, the calculations of Eb(n) and ΔE(n) were also performed for pure Sin+1λ (n = 1–9, λ = 0, − 1) clusters, which are defined as follows:
where \(E({\rm Si})\), \(E({\rm Si}^{\lambda } )\), \(E({\rm Si}_{n}^{\lambda } )\) and \(E({\rm Si}_{n + 1}^{\lambda } )\) denote the energies of Si, Siλ, Sinλ and Sin+1λ, respectively.
The averaged binding energies and fragmentation energies of the lowest energy Sin+1λ and PrSinλ (n = 1–9, λ = 0, − 1) clusters against the number of silicon atoms are shown in Fig. 5. From Fig. 5, the features of size evolution are best viewed and the peaks of curves correspond to those clusters with enhanced local stabilities. For pure Sin+1 and Sin+1− clusters, the averaged binding energies increase gradually with the cluster size increasing at n ≤ 5; and then, the curves begin to slow down. The curve of Sin+1− is higher than that of the corresponding sized Sin+1 clusters, reflecting that the stabilities of Sin+1 clusters are enhanced when they attach an extra electron. For both Eb(n) and ΔE(n) curves, the visible peak occurs at Si4 and Si10− hinting that they are more stable than their neighboring clusters.

Size dependence of the averaged binding energies Eb, fragmentation energies ΔE(n) and HOMO–LUMO energy gaps Egap for the ground-state structures of Sin+1λ and PrSinλ (n = 1–9; λ = 0, − 1) clusters
As for PrSinλ (n = 1–9, λ = 0, − 1) clusters, the averaged binding energies are almost degenerated with the corresponding sized pure Sinλ clusters, reflecting that the stability of silicon clusters can’t be enhanced with the dopant of Pr atom. The two Eb(n) curves of PrSin and PrSin− increase gradually with the cluster size at n ≤ 5; then, the curves flatten out. Fragmentation energies, which involve the energy that a Si atom separates from PrSin0/− clusters, are plotted in Fig. 5. The two curves of PrSin and PrSin− almost have the same tendency, which show an irregular odd–even oscillating behavior against the cluster size. The remarkable peaks appear at PrSi20/− and PrSi40/− clusters, indicating that these four clusters have slightly stronger relative stabilities than others, which is in accord with the analysis based on the averaged binding energies.
Orbital and Bonding Properties Analysis
HOMO–LUMO Gaps
The highest occupied-lowest unoccupied molecular orbital (HOMO–LUMO) energy gap, which represents the ability of a molecule to participate into chemical reaction in some degree, has been calculated. In a sense, it provides an important criterion to reflect the chemical stability of clusters. A small value of the HOMO–LUMO energy gap corresponds to a high chemical activity. In contrast, a large one is related to enhanced chemical stability. For the most stable Sinλ and PrSinλ (n = 1–9, λ = 0, − 1) clusters, HOMO and LUMO energies are shown in Table 2. Furthermore, the HOMO–LUMO energy gaps against the cluster size are also plotted in Fig. 5. As is shown in Fig. 5, the four curves show irregular behaviors. The HOMO–LUMO energy gaps of pure silicon clusters show the same tendency with our previous results [19]. For PrSin and PrSin− clusters, the remarkable peaks occur at PrSi2 and PrSi4− clusters. In other words, the HOMO–LUMO energy gaps of these two clusters are larger than those of others, indicating that they possess dramatically enhanced chemical stability. The result is in accord with the analysis of relative stabilities. For PrSi6 cluster, the gap is very small compared to the neighbor’s, indicating that its chemical activity is stronger than that of its neighboring clusters.
Orbital Analysis
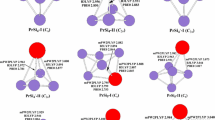
In order to gain insight into the nature of the bonding, the patterns of the highest occupied molecular orbital (HOMO) for the ground state PrSinλ (n = 1–9, λ = 0, − 1) clusters have been analyzed, and the results are displayed in Fig. 6. These MOs could provide insight into the special features of the natural bonding in PrSin0/− clusters. As for pure Si2 dimer, the σ-type bond is formed between two Si atoms, which may be originated from Si-p orbital. In its corresponding anions, the Si-p orbitals form bonding π-type bond. When Pr substitutes one Si atom, the bonding π-type bond is still observed but with mixed Pr-d character. The anti-bonding πp* bonds are found in 2N-a and 3A-a clusters. When the number of silicon atoms is up to 4, some σ-type and π-type bonds are formed among the Si and Pr atoms. Comparing with σ-type bonds, the π-type bonds between Si atoms are fewer, because the σ-type bonds are the dominant Si–Si bonding type. According to the contour maps of the HOMOs of the PrSin0/− clusters, one finds that the distribution of electron density around Si and Pr atoms is not very different, indicating that the hybridization between Si and Pr atoms is not obviously different from those between the Si and Si atoms.

Contour maps of the HOMOs of the ground-state PrSinλ (n = 1–9; λ = 0, − 1) clusters
Charge Distribution
In order to insightfully explore the charge-transfer information, the Mulliken charge population for neutral and anionic ground state PrSinλ (n = 1–9, λ = 0, − 1) clusters were calculated and listed in Tables 4 and 5, respectively. From Table 4, one can see that all the praseodymium atoms possess positive charge in neutral PrSin clusters, while most of the silicon atoms have negative charges, indicating that electrons transfer from Pr to Si atoms. That is to say, the Pr atoms act as electron donor in neutral clusters. This is consistent with their electronegativities that Si (1.90) is larger than Pr (1.13); therefore, the silicon has a stronger ability to attract electrons. The distribution of charges for PrSi dimer shows that 0.327e electrons transfer from Pr to Si, resulting in the weak bond between Si and Pr atoms. As regards Table 5, in PrSin− (n = 1–3) clusters, the charges of Pr atoms are negative which may be induced by the extra electron. However, the praseodymium atoms still possess positive charge in PrSin− (n = 4–9) clusters. Based on the comparison between neutral and anionic PrSin clusters, we found that the extra electron is mainly localized on silicon atoms.
Conclusions
The geometrical structures, stabilities, and electronic properties of PrSinλ (n = 1–9, λ = 0, − 1) have been investigated using density functional theory at B3PW91, B3LYP and CCSD (t) levels. All the results are summarized as follows.
Extensive searches for the ground-state structures of PrSinλ clusters were carried out based on the comparisons between the simulated spectra and experimental PES data. The results showed that the lowest energy structures tend to be planar structures for n = 1–3 and three-dimensional (3D) structures for n = 4–9. The praseodymium atom tends to occupy low coordinated position, such as edge-capped or face-capped position, and only four Si atoms are allowed to bound atomically to Pr atom. Based on averaged binding energies, fragmentation energies and HOMO–LUMO gaps, PrSi20/− and PrSi40/− clusters were found to have stronger relative stabilities. In order to gain insight into the nature of the bonding, the patterns of HOMOs for the ground state PrSinλ isomers were investigated. The results showed that π-type and σ-type bonds are always formed among the Si atoms, and the hybridization between Si and Pr atoms is not obviously different from those between the Si and Si atoms. The results of Mulliken population indicated that the electron transfer from Pr atom to the Sin frames in all the neutral clusters, namely, Pr atoms act as electron donor. In anionic PrSin− clusters, the extra electron is mainly localized on silicon atoms.
References
K. Tomioka, M. Yoshimura, and T. Fukui (2012). A III-V nanowire channel on silicon for high-performance vertical transistors. Nature. 488, 189–192.
B. Roche, R.-P. Riwar, B. Voisin, E. Dupont-Ferrier, R. Wacquez, M. Vinet, M. Sanquer, J. Splettstoesser, and X. Jehl (2013). A two-atom electron pump. Nat. Commun. 4, 1581.
Y. Chen, Y. Liu, S. Li, and J. Yang (2019). Theoretical study on the growth behavior and photoelectron spectroscopy of lanthanum-doped silicon clusters LaSin0/−(n = 6–20). J. Clust. Sci. 30, 789–796.
Y. Feng, J. Yang, and Y. Liu (2016). Study on the structures and properties of praseodymium-doped silicon clusters PrSin (n=3-9) and their anions with density functional schemes. Theor. Chem. Acc. 135, 258.
T. M. Fu, X. J. Duan, Z. Jiang, X. C. Dai, P. Xie, Z. G. Cheng, and C. M. Lieber (2014). Sub-10-nm intracellular bioelectronic probes from nanowire–nanotube heterostructures. Proc. Natl. Acad. Sci. USA 111, 1259–1264.
K. Koyasu, J. Atobe, S. Furuse, and A. Nakajima (2008). Anion photoelectron spectroscopy of transition metal- and lanthanide metal-silicon clusters: MSin− (n = 6–20). J. Chem. Phys. 129, 214301.
A. Grubisic, H. P. Wang, Y. J. Ko, and K. H. Bowen (2008). Photoelectron spectroscopy of europium-silicon clusters anions, EuSin− (3 ≤ n ≤ 17). J. Chem. Phys. 129, 054302.
C. G. Li, J. H. Gao, J. Zhang, W. T. Song, S. Q. Liu, S. Z. Gao, B. Z. Ren, and Y. F. Hu (2018). Structures, stabilities and electronic properties of boron-doped silicon clusters B3Sin (n=1–17) and their anions. Mol. Phys. 117, 1–13.
Y. Zhang, J. Yang, and L. Cheng (2018). Probing structure, thermochemistry, electron affinity and magnetic moment of erbium-doped silicon clusters ErSin (n = 3–10) and their anions with density functional theory. J. Clust. Sci. 29, 301–311.
T. D. Hang, H. M. Hung, and M. T. Nguyen (2016). Structural assignment, and electronic and magnetic properties of lanthanide metal doped silicon heptamers Si7M0/−with M = Pr, Gd and Ho. Phys. Chem. Chem. Phys. 18, 31054.
X. J. Li, Z. J. Yan, and S. N. Li (2016). The nature of structure and bonding between transition metal and mixed Si-Ge tetramers: a 20-electron superatom system. J. Comput. Chem. 37, 2316–2323.
I. Rata, A. A. Shvartsburg, M. Horoi, T. Frauenheim, K. W. M. Siu, and K. A. Jackson (2000). Single-parent evolution algorithm and the optimization of Si clusters. Phys. Rev. Lett. 85, 546–549.
V. T. Ngan, P. Gruene, P. Claes, E. Janssens, A. Fielicke, M. T. Nguyen, and P. Lievens (2010). Disparate effects of Cu and V on structures of exohedral transition metal-doped silicon clusters: a combined far-infrared spectroscopic and computational study. J. Am. Chem. Soc. 132, 15589–15602.
V. T. Ngan, E. Janssens, P. Claes, J. T. Lyon, A. Fielicke, M. T. Nguyen, and P. Lievens (2012). High magnetic moments in manganese-doped silicon clusters. Chem. Eur. J. 18, 15788–15793.
V. T. Ngan, K. Pierloot, and M. T. Nguyen (2013). Mn@Si14+: a singlet fullerene-like endohedrally doped silicon cluster. Phys. Chem. Chem. Phys. 15, 5493–5498.
P. Claes, V. T. Ngan, M. Haertelt, J. T. Lyon, A. Fielicke, M. T. Nguyen, P. Lievens, and E. Janssens (2013). The structures of neutral transition metal doped silicon clusters, SinX (n = 6–9; X = V, Mn). J. Chem. Phys. 138, 194301.
J. T. Lau, K. Hirsch, P. Klar, A. Langenberg, F. Lofink, R. Richter, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller, and B. V. Issendorff (2009). X-ray spectroscopy reveals high symmetry and electronic shell structure of transition-metal-doped silicon clusters. Phys. Rev. A. 79, 053201.
X. Y. Kong, H. G. Xu, and W. Zheng (2012). Structures and magnetic properties of CrSin− (n= 3–12) clusters: photoelectron spectroscopy and density functional calculations. J. Chem. Phys. 137, 4307.
P. Shao, X. Y. Kuang, L. P. Ding, M. M. Zhong, and Z. H. Wang (2012). Density-functional theory study of structures, stabilities, and electronic properties of the Cu2-doped silicon clusters: comparison with pure silicon clusters. Phys. B. 407, 4379–4386.
W. Zheng, J. M. Nilles, D. Radisic, and K. H. Bowen (2005). Photoelectron spectroscopy of chromium-doped silicon cluster anions. J. Chem. Phys. 122, 071101.
S. N. Khanna, B. K. Rao, and P. Jena (2002). Magic numbers in metallo-inorganic clusters: chromium encapsulated in silicon cages. Phys. Rev. Lett. 89, 016803.
P. Shao, L. P. Ding, D.-B. Luo, and C. Lu (2019). Probing the structures, electronic and bonding properties of multidecker lanthanides: Neutral and anionic Lnn(COT)m (Ln = Ce, Nd, Eu, Ho and Yb; n, m = 1, 2) complexes. J. Mol. Graph. Model. 90, 226–234.
W. G. Sun, X. Y. Kuang, H. D. J. Keen, C. Lu, and A. Hermann (2020). Second group of high-pressure high-temperature lanthanide polyhydride superconductors. Phys. Rev. B 102, 144524.
M. Ohara, K. Miyajima, A. Pramann, A. Nakajima, and K. Kaya (2007). Geometric and electronic structures of terbium-silicon mixed clusters (TbSin; 6 ≤ n ≤ 16). J. Phys. Chem. A. 111, 10884.
V. Kumar, A. K. Singh, and Y. Kawazoe (2006). Charged and magnetic fullerenes of silicon by metal encapsulation: predictions from ab initio calculations. Phys. Rev. B. 74, 125411.
A. Grubisic, Y. J. Ko, H. Wang, and K. H. Bowen (2009). Photoelectron spectroscopy of lanthanide-silicon cluster anions LnSin− (3 ≤ n ≤ 13; Ln = Ho, Gd, Pr, Sm, Eu, Yb): prospect for magnetic silicon-based clusters. J. Am. Chem. Soc. 131, 10783–10790.
C. G. Li, L. J. Pan, P. Shao, L. P. Ding, H. T. Feng, D. B. Luo, and B. Liu (2015). Structures, stabilities, and electronic properties of the neutral and anionic SinSmλ (n = 1–9, λ = 0, -1) clusters: comparison with pure silicon clusters. Theor. Chem. Acc. 134, 1–11.
C. Lu, W. G. Gong, Q. Li, and C. F. Chen (2020). Elucidating stress-strain relations of ZrB12 from first-principles studies. J. Phys. Chem. Lett. 11, 9165–9170.
B. L. Chen, L. J. Conway, W. G. Sun, X. Y. Kuang, C. Lu, and A. Hermann (2021). Phase stability and superconductivity of lead hydrides at high pressure. Phys. Rev. B. 103, 035131.
C. Lu and C. F. Chen (2021). Indentation strengths of zirconium diboride: intrinsic versus extrinsic mechanisms. J. Phys. Chem. Lett. 12, 2848–2853.
M.J. Frisch, et al. (2009). Gaussian 09 (Revision C.01), Gaussian, Inc., Wallingford
A. D. Becke (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652.
J. P. Perdew and Y. Wang (1992). Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B: Condens. Matter. 45, 13244–13249.
C. Lee, W. Yang, and R. G. Parr (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37, 785–789.
G. D. Purvis and R. J. Bartlett (1982). A full coupled-cluster singles and doubles model: the inclusion of disconnected triples. J. Chem. Phys. 76, 1910–1918.
G. E. Scuseria, C. L. Janssen, and H. F. Schaefer (1988). An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 89, 7382–7387.
G. E. Scuseria and H. F. Schaefer (1989). An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 90, 3700–3703.
R. Krishnan, J. S. Binkley, R. Seeger, and J. A. Pople (1980). Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654.
M. Dolg, H. Stoll, A. Savin, and H. Preuss (1989). Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta. 75, 173–194.
M. Dolg, H. Stoll, and H. Preuss (1989). Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 90, 1730–1734.
J. C. Yang, W. G. Xu, and W. S. Xiao (2005). The small silicon clusters Sin (n = 2–10) and their anions: structures, themochemistry, and electron affinities. J. Mol. Struct. Theochem. 719, 89–102.
C. Pouchan, D. Bégué, and D. Y. Zhang (2004). Between geometry, stability, and polarizability: density functional theory studies of silicon clusters Sin (n = 3–10). J. Chem. Phys. 121, 4628–4634.
K. Jackson, M. R. Pederson, D. Porezag, Z. Hajnal, and T. Frauenheim (1997). Density- functional-based predictions of Raman and IR spectra for small Si clusters. Phys. Rev. B. 55, 2549–2555.
N. Binggeli and J. R. Chelikowsky (1995). Photoemission spectra and structures of Si clusters at finite temperature. Phys. Rev. Lett. 75, 493–496.
O. Kostko, S. R. Leone, M. A. Duncan, and M. Ahmed (2010). Determination of ionization energies of small silicon clusters with vacuum ultraviolet radiation. J. Phys. Chem. A. 114, 3176–3181.
G. F. Zhao, J. M. Sun, Y. Z. Gu, and Y. X. Wang (2009). Density-functional study of structural, electronic, and magnetic properties of the EuSin (n = 1–13) clusters. J. Chem. Phys. 131, 114–312.
T. G. Liu, G. F. Zhao, and Y. X. Wang (2011). Structural, electronic and magnetic properties of GdSin (n = 1–17) clusters: a density functional study. Phys. Lett. A. 375, 1120–1127.
M. R. Nimlos, B. L. Harding, and G. B. Ellison (1987). The electronic states of Si2 and Si−2 as revealed by photoelectron spectroscopy. J. Chem. Phys. 87, 5116.
K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure, Constants of Diatomic Molecules, vol. IV (Van Nostrand Reinhold, New York, 1979).
Y. R. Zhao, Y. Q. Xu, P. Chen, Y. Q. Yuan, Y. Qian, and Q. Li (2021). Structural and electronic properties of medium-sized beryllium doped magnesium BeMgn clusters and their anions. Results Phys. 26, 104341.
T. Lu and F. W. Chen (2012). Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592.
T. Lu and F. W. Chen (2011). Calculation of molecular orbital composition. Acta. Chim. Sin. 69, 2393–2406.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 11804212), Youth Talent Invitation Scheme of Shaanxi Association for science and technology (Nos. 20180506 and 20190506) and the Shaanxi University of Science & Technology Key Research Grant (Nos. 2016BJ-01 and BJ15-07 ).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships which have or could be perceived to have influenced the work reported in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shao, P., Zhao, ZL., Zhang, H. et al. Probing the Structures, Stabilities and Electronic Properties of Neutral and Anionic PrSinλ (n = 1–9, λ = 0, − 1) Clusters: Comparison with Pure Silicon Clusters. J Clust Sci 33, 2723–2733 (2022). https://doi.org/10.1007/s10876-021-02188-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-021-02188-0