
Abstract
The growth behavior and electronic properties of the lowest energy structures of neutral LaSin (n = 6–20) and their anions were explored by means of the ABCluster global structure searching strategy combined with the mPW2PLYP double-hybrid density functional. The results revealed that the growth behavior of the lowest energy structures of anionic LaSi − n (n = 10–20) clusters choose La-linked two silicon subclusters to La-encapsulated in silicon cages. For neutral LaSin (n = 6–20), the growth behavior of the lowest energy structures from substitutional structure to linked motifs and finally to encapsulated configurations occurs at n = 14 and 20, respectively. The simulated photoelectron spectroscopy, adiabatic electron affinities, vertical detachment energies, relative stability and HOMO–LUMO energy gaps were presented. Analyses of HOMO–LUMO energy gaps, relative stability, and chemical bonding reveal that the LaSi20− possesses ideal thermodynamic and chemical stability in a high Ih-symmetry endohedral motif, which can turn it into suitable constitutional units for cluster-assembled nanomaterials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Study of equilibrium geometries, electronic structures and properties of atomic clusters, especially silicon based on semiconductor clusters, has been an extraordinarily active area of research in the last few decades because of its importance nanoscience and nanotechnology [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. Theoretical modeling and experimental probing of silicon nanoclusters with modified new geometries show possibility of using them as nano-devices in opto-electronics, tunable lasers, etc. Especially, rare-earth modified silicon-based nanomaterals as “industrial vitamins” have been widely used in various fields such as metallurgy, military, petrochemical, glass ceramics, and new materials owing to their novel magnetic, optical and electrical properties and chemical stability [16,17,18,19,20,21,22,23,24,25,26,27,28,29]. The functional properties with a combination of good flexural strength, fracture toughness, hardness and low Schottky barrier heights make lanthanum modified silicon-based nanomaterials are ideal in devices such as substrates of the integrated circuits, host lattices for light emitting phosphors, ohmic contacts and rectifying contacts [30, 31]. The unique structures and properties of nanoclusters make they are one of the central issues in nanomaterials sciences. Therefore, it is very significant to probe the evolution of lanthanum-silicon aggregates in the transition from the molecular to condensed phase.
Only growth behavior of neutral LaSin clusters with n up to 21 has been studied by single-hybrid density functional and concluded that La atom prefers locating on the surface site of the clusters in the size range of n ≤ 10 or n ≤ 12, surrounded by Si atom with basketlike structures with 11 ≤ n ≤ 15, and completely encapsulated into fullerene cage geometries with 16 ≤ n ≤ 21 [32,33,34]. It is well known that the functional dependence on the predicted lowest-energy structure always occur for small- or medium-clusters, especially for clusters including rare earth- or transition- metal atoms. So the calculation method adopted is quite important. In light of our experience [35,36,37,38,39,40], performances of prediction structures and properties of double-hybrid density functional are better than those of pure or single-hybrid density functional for rare earth-metal atom doped silicon clusters. In this paper, the ABCluster global search technique combined with a double-hybrid density functional scheme was adopted to optimize geometries of La-doped Si 0/− n (n = 6–20) clusters with the goal of illuminating the growth behavior of the ground states, exploring the electronic properties, knowing the bonding characteristics, and furnishing important information for further studies of introducing other transition or rare-earth metals into semiconductor nanoclusters. We will see that the growth behavior of the ground states predicted in this paper is different from those reported previously [32,33,34].
Computational Details
Three techniques were adopted to search the initial geometries for LaSi 0/− n (n = 6–20) clusters. First, using the ABCluster global search technique [41] coupled with PBE functional [42] and with the 6-31G basis set for Si atoms and ECP46MWB basis set [43, 44] for La atoms, more than 300 geometries for each cluster were optimized. The detailed description of ABCluster global search technique is elsewhere [36, 38, 39]. Second, the substitutional structure schemes were adopted, in which a Si atom in the most stable structure of Sin+1 was replaced with a La atom. Third, geometries already presented in the preceding publications [20, 24] were adopted. The obtained low-lying candidate structures of the global minimum for each size were reoptimized using PBE functional in collaboration with the Stuttgart/Dresden ECPs (i.e., SDD) basis set [44, 45] for La atoms and all-electron cc-pVTZ basis set [46] for Si atoms. The vibrational frequency calculations were carried out at the same level to assure the nature of the stationary points. After accomplishment of the initial structure optimization using PBE, the low-lying isomers are again selected and reoptimiezed by means of a double hybrid mPW2PLYP functional [47] with the same basis sets. Vibrational frequency was not carried out at the mPW2PLYP level. In order to farther refine the energies, calculations of single-point energy were carried out at the mPW2PLYP level with aug-cc-pVTZ basis set [46] for Si atoms (basis set for La atoms unchanged). According to Koopmans’ theorem [48, 49], we simulated the photoelectron spectroscopy (PES) of anionic species at the mPW2PLYP level with the Multiwfn program [50]. To further understand the interaction between La atom and silicon clusters, the chemical bonding analyses in light of AdNDP (adaptive natural density partitioning) [51] were also performed. All of the calculations were implemented using the Gaussian 09 package [52].
To examine the relativistic effects and the reliability of the methods used in this work, single-point energy calculations were also executed by means of the ROCCSD(T) scheme with the aug-cc-pVTZ-DK basis set [46] for Si atoms and cc-pVTZ-DK3 basis set [53] for La atoms and the Douglas-Kroll-Hess scalar relativistic correction [54,55,56] for LaSi 0/− n (n = 1–5) small clusters. The ground-state structure and several low-lying isomers of LaSi 0/− n (n = 1–5) together with their point ground, electronic state, and relatively energies are exhibited in Figure S1 in Supplementary Information. The adiabatic electron affinity (AEA) and the vertical detachment energy (VDE) of LaSi 0/− n (n = 1–5) are also listed in Table S1 in Supplementary Information. From Figure S1 we can see that the ground state structure and energetic order of isomers evaluated by the mPW2PLYP and ROCCSD(T) schemes are identical. From Table S1 we can see that the AEAs and VDEs calculated by the mPW2PLPY scheme are in good agreement with the results predicted by the ROCCSD(T) scheme. The mean absolute deviations of mPW2PLYP result from ROCCSD(T) data are by 0.10 eV. The largest deviation is that of LaSi3−, which is off by 0.29 eV for AEA and 0.19 eV for VDE. If LaSi3− is removed, the mean absolute deviation is only 0.07 eV. All of these indicate that the results of mPW2PLYP are reliable.
Results and Discussion
The Lowest Energy Structures
The lowest energy geometries of LaSin (n = 6–20) clusters and their anions are exhibited in Figs. 1 and 2, respectively. The electronic state, average binding energies, HOMO–LUMO energy gap, and NPA charges on the La atom for the lowest energy structure are scheduled in Table 1. The corresponding low-lying structures, point ground and relative energies are exhibited in Figures S2 and S3 in Supplementary Information. For negatively charge ions, the predicted spin states of LaSi − n (1 ≤ n ≤ 20) are single excluded LaSi− and LaSi2− which is triplet as can be seen in Figure S1. For neutral clusters, the evaluated spin states are double.

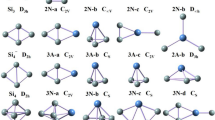
The lowest energy structures of LaSi − n (n = 6–20) and their point group

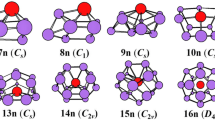
The lowest energy structures of LaSin (n = 6–20) and their point group
The growth behavior of the lowest energy structures of anionic lanthanum doped Si clusters choose linked to encapsulated configuration in light of the results of the double-hybrid mPW2PLYP scheme. For n = 6, the lowest energy structure is a pentagonal bipyramid with La atom located on the quintuple axis. For n = 7, three topological isomers (7a1, 7a2 and 7a3) compete with each other for the lowest energy structure due to the fact that their energy is nearly degenerate. Their maximum energy differences are only 0.03 eV (see Figures S2). For n = 8, the situation is similar to n = 7, three geometries (8a1, 8a2 and 8a3) are also compete with each other for the lowest energy structure. In fact the 8a1, 8a2 and 8a3 isomers can be regarded as attaching a Si atom to the 7a1, 7a2 and 7a3 geometries, respectively. The 8a2 structure is linked configuration in which La atom links to two tetrahedron Si4 subclusters. For n = 9, the lowest energy structure is a C4v-symmetry bi-face-capped tetragonal antiprism. For n = 10–19, the lowest-energy structure is linked configurations. The La atom in LaSi10− links two orthogonal trigonal bipyramid Si5 subclusters, in LaSi11− connects a trigonal bipyramid Si5 and a face-capped trigonal bipyramid Si6, in LaSi13− connects a trigonal bipyramid Si5 and a distorted bicapped octahedron Si8, in LaSi14− connects a trigonal bipyramid Si5 and a TTP (tricapped trigonal prism) motif of Si9, in LaSi15− connects an octahedron Si6 and a TTP Si9, in LaSi16− links a pentagonal bipyramid Si7 and a TTP Si9, in LaSi17− links a Si8 subcluster and a TTP Si9, in LaSi18− links to two TTP Si9, in LaSi19−, along with a Si atom, links to two TTP Si9. For n = 12, both 12a1 and 12a2 isomer compete with each other for the lowest energy structure. The energy difference between 12a1 and 12a2 is only 0.03 eV (see Figures S2). The 12a2 structure is linked configuration in which La atom links to two orthogonal face-capped trigonal bipyramid Si6 subclusters. The 12a1 geometry can be viewed as linked configuration in which La atom connects a rhombus Si4 and a Si8 subcluster. For n = 20, the lowest energy structure is encapsulated configuration with La atom located in the silicon cage.
The growth behavior of the lowest energy structures of neutral LaSin (n = 6–20) from substitutional geometries to linked structures and finally to encapsulated configurations happens at n = 14 and 20, respectively. The lowest energy structure of LaSi6 belongs to substutional structure in which a La atom was substituted for a horizontal Si atom of the lowest energy pentagonal bipyramidal structure of Si7 [2,3,4, 6]. For n = 7, similarly to its anion, three topological isomers (7n1, 7n2 and 7n3) compete with each other for the lowest energy structure. The 7n3 is substitutional structure in which a La atom was substituted for a Si atom of the lowest energy Si8 bicapped octahedron [3, 4, 6]. The energy difference between 7n3 and 7n1 is only 0.03 eV (see Figures S3). Practically the capped Si atom connected the La atom in 7n3 isomer is weakly bound and moves almost freely to the position of 7n1 geometry. The lowest energy structures 8n, 9n, 10n, 11n, 12n1, 12n2 and 13n also belong to substitutional structures, which can be viewed as the lowest energy structure of Si9, Si10, Si11, Si12, Si13 and Si14 [3,4,5,6], respectively, with a Si atom replaced by a La atom. The lowest energy structures 8n, 9n, 10n, 11n, 12n1, and 13n are different from those of their corresponding anions. The 12n2 geometry, corresponding to anionic 12a1, belongs to not only linked structure, but also subsitutional structure. The energy difference between 12n1 and 12n2 is only 0.01 eV, indicating the energy of both is degenerate. For n = 14–19, the lowest energy structures are linked configurations. And excluded LaSi16 they are similar to those of their anions. The La atom in the lowest energy structure of LaSi16 links an anomalous Si7 and an anomalous Si9 subclusters. Starting from n = 20, the lowest energy structure is encapsulated framework with La atom centered in the silicon cage. It is noted that most the lowest energy structures are different from those reported previously [32], for example n = 11–19.
Simulated PES of Anionic Clusters
PES as a powerful can examine the lowest energy structures due to the fact that the PES is susceptible to changes of the structure. Therefore, in light of theoretical generalized Koopman theorem [48, 49], we simulated the PES spectra of the lowest energy structure of the anions at the mPW2PLYP level with the Multiwfn program [50]. First, the relative energy of the orbital (ΔEn) is computed by the equation during the simulation:
Second, the first peak associated with the HOMO is set at the VDE position, and the peaks associated with the deeper orbitals are shifted to higher binding energy based on the value of − ΔEn. Third, the peaks are fitted with a Gaussian function of 0.25 eV full widths at half maximum. The simulated PES is accomplished and shown in Fig. 3. It can be seen from the simulated PES of LaSi6− that there are three major peaks located at 2.78, 3.39, and 4.56 eV in the range of ≤ 5.0 eV, respectively. For n = 7, three isomers are simulated. The simulated PES of 7a1 has two double peaks. One is located at 2.74 and 2.94 eV, and another is centered at 4.37 and 4.58 eV. The simulated PES of 7a2 shows four peaks centered at 2.51, 3.22, 3.97 and 4.80 eV, respectively. Six peaks centered at 2.51, 3.13, 3.55, 3.91, 4.15 and 4.56 eV are observed in the simulated PES of 7a3. For n = 8, three isomers are also simulated. The simulated PES of 8a1 and 8a3 has five peaks, located at 2.76, 3.05, 3.47, 4.29 and 4.64 eV and 2.74, 2.95, 3.24, 3.75 and 4.56 eV, respectively. Six peaks located at 2.91, 3.24, 3.62, 3.99, 4.30 and 4.75 eV are inspected for 8a2. For n = 9, 11, 14, and 18, they all have three peaks, locating at 3.08, 3.94 and 4.34 eV, 3.71, 4.10 and 4.96 eV, 3.60, 4.07 and 4.63 eV, and 3.65, 4.19 and 4.81 eV, respectively. For n = 10, 13, 16, and 17, they all have two peaks, centering at 3.96 and 4.33 eV, 4.32 and 4.80 eV, 3.97 and 4.36 eV, and 3.36 and 4.14 eV, respectively. For n = 12, two isomers are simulated. There are four peaks for 12a1 located at 3.14, 3.44, 3.82, and 4.27 eV, and two peaks for 12a2 centered at 3.49 and 4.46 eV, respectively. For n = 15 and 19, both have four peaks located at 3.70, 4.12, 4.49 and 4.91 eV, and 3.74, 4.26, 4.72 and 4.94 eV, respectively. Only one peak centered at 4.75 eV is observed for simulated PES of LaSi20− in the range of ≤ 5.0 eV. We expect that these simulations will give forceful motivation for experimental studies of La-doped silicon clusters and their anions.

Simulated PES spectra of the lowest energy LaSi − n (n = 6–20) clusters
Electron affinity is also a key spectral data and extremely significant for use in the chemical reaction process to determine charge transfer, bond dissociation energies and so on. AEA is defined as the energy difference in the manner [35]:
where Eneutral and Eanion are the lowest energy of the neutral and anionic clusters, respectively. And ΔEsc is an empirical structural correction factor based on the result of ScSi − n clusters [35]. The charge of a metal atom in a cluster generally inclines to be positive or slightly negative. However, it will be negatively charged when the numbers of silicon atom increase and the cluster becomes a cage structure. In light of the charge’s change, the structures of cluster are categorized into three types: no-cage, half-cage, and cage. The numerical numbers of ΔEsc are assigned to be 0.00, − 0.20 and − 0.40 eV for no-cage, half-cage and cage structures, respectively [35]. The calculated AEAs of ScSi − n with structural correction factor are in excellent agreement with the experimental data, and the average absolute error is only 0.08 eV [35]. Therefore, we took the structural correction factor into account when we calculated the AEAs of YSi − n clusters. The charges on the La atom are positive for n = 6–19 as can be seen from Table 1, while n = 20 are large negatively charged. So the lowest energy structure of LaSi − n with n = 6–19 is no-cage, but cage motif with n = 20. Because the radius of La is larger than that of Sc, the La-doped Si clusters do not appear as the half-cage like the Sc-doped Si clusters [35]. Hence, the ΔEsc is 0.00 eV for n = 6–19 and -0.40 eV for n = 20. The theoretical AEA and VDE are listed in Table 2. There are no experimental values for comparison.
Relative Stability
Both atomization energy (AE) and second energy difference (∆2E) associated with thermodynamic and relative stability have been calculated for the lowest energy structure of LaSi 0/−n (n = 6–20) clusters. The AE is the energy required for the following reactions:
The ∆2E is the energy required the following reactions:
The evaluated AE and ∆2E are pictured in Fig. 4. It can be seen from Fig. 4a that (1) the AE values of negatively charged ions are all larger than those of their neutrals due to the fact that the LaSin is open-shell clusters, while LaSi − n is closed-shell clusters. The closed shell configurations minimize the electronic repulsions according to the Pauli exclusion principle. The higher data of the AE, the more stable the clusters. The most stable structure is anionic LaSi20−. (2) The cluster size distributions starting from n = 12 for LaSi − n anions and n = 14 for LaSin neutral display even–odd alternations. The clusters with an odd numbers of silicon atoms are less stable than those with an even numbers. However, for small clusters, LaSi9− is obviously more stable than its adjacent clusters. These consequences are distinctly reproduced in Fig. 4b due to the fact that the ∆2E are a susceptive measure for relative stability.

Size dependences of a atomization energy (AE), b second energy difference (∆2E), and c HOMO–LUMO energy gap for the lowest energy LaSi − n (n = 6–20) clusters
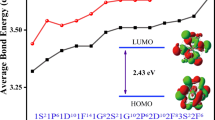
To some extent, HOMO–LUMO energy gap (Egap) can be viewed as a significant criterion to reflect the chemical reactivity, especially for a substance containing rare earth elements which sometimes possess novel photochemical sensitivity. The larger the Egap, the weaker the chemical reactivity. The Egap of LaSi 0/− n (n = 6–20) clusters are drawn in Fig. 4c. There are no the odd–even oscillation behavior for Egap. For LaSi − n , the Egap of n = 10,11 and 13 is relatively large and range from 4.34 to 4.51 eV. For n = 20, the Egap is the smallest, and by 2.42 eV. For neutral LaSin, the Egap for n = 14 and 17 is relatively large and range from 4.10 to 4.20 eV. The smallest Egap is 2.31 eV of LaSi20 cluster. The Egap of anion YSi20− is very close to that of the corresponding neutral, showing that an extra electron does not change its chemical reactivity, but improves its thermodynamic stability.
Chemical Bonding Analysis
To gain an in-depth understanding of the ideal thermodynamic stability and chemical reactivity of anionic LaSi20− as mentioned above, the AdNDP method [51] is employed to perform quantitative insight into the nature of the bonding between the La atom and Si clusters. From Fig. 5 we can see that the chemical bonding of 84 valence electrons can be divided to two types: 2c–2e and 6c–2e. The Si20 outer fullerene cages are characterized by 30 2c–2e localized σ Si–Si bonds with 1.95 |e| in each bond. The 12 delocalized 6c–2e bonds with 1.96 |e| in each bond are responsible for the conjugation between the central La atom and the Si20 outer Fullerene shell and stabilize the encapsulated LaSi20− species. In addition, the valence electron filling of 84 magic number electrons for LaSi20− is described as 1S21P61D101F141G82S21G102P62D103S22F14 in terms of spherical jellium model and the molecular orbital energies, which similar to that of superatom also stabilizes it.

AdNDP analysis of the LaSi20− cluster. ON stands for the occupation number
Conclusions
The growth behavior and electronic properties of the lowest energy structures of neutral LaSin (n = 6–20) and their anions have been explored by means of the ABCluster structure searching method combined with the mPW2PLYP double-hybrid density functional. The results revealed that the growth behavior of the lowest energy structures of anionic LaSi − n (n = 10–20) clusters choose La-linked two silicon subclusters to La-encapsulated in silicon cages. For neutral LaSin (n = 6–20), the growth behavior of the lowest energy structures from substitutional structure to linked motifs and finally to encapsulated configurations occurs at n = 14 and 20, respectively. The simulated photoelectron spectroscopy, adiabatic electron affinities, vertical detachment energies, relative stability and HOMO–LUMO energy gaps were presented. Analyses of HOMO–LUMO energy gaps, relative stability, and chemical bonding reveal that the LaSi20− possesses faultless thermodynamic and chemical stability in a high Ih-symmetry endohedral motif, which can turn it into suitable constitutional units for cluster-assembled nanomaterials.
References
M. F. Jarrold (1991). Science 252, 1085–1092.
E. C. Honea, A. Ogura, D. R. Peale, C. Félix, C. A. Murray, K. Raghavachari, W. O. Sprenger, M. F. Jarrold, and W. L. Brown (1999). J. Chem. Phys. 110, 12161–12172.
I. Vasiliev, S. Öğüt, and J. R. Chelikowsky (1997). Phys. Rev. Lett. 78, 4805–4808.
S. Nigam, C. Majumder, and S. K. Kulshreshtha (2006). J. Chem. Phys. 125, 074303.
X. L. Zhu, X. C. Zeng, and Y. A. Lei (2004). J. Chem. Phys. 120, 8985–8995.
J. C. Yang, W. G. Xu, and W. S. Xiao (2005). J. Mol. Struct. Theochem. 719, 89–102.
L. P. Ding, F. H. Zhang, Y. S. Zhu, C. Lu, X. Y. Kuang, J. Lv, and P. Shao (2015). Sci. Rep. 5, 15951.
W. G. Sun, J. J. Wang, C. Lu, X. X. Xia, X. Y. Kuang, and A. Hermann (2017). Inorg. Chem. 56, 1241–1248.
Y. Y. Jin, Y. H. Tian, X. Y. Kuang, C. Z. Zhang, C. Lu, J. J. Wang, J. Lv, L. P. Ding, and M. Ju (2015). J. Phys. Chem. A 119, 6738–6745.
Y. Y. Jin, G. Maroulis, X. Y. Kuang, L. P. Ding, C. Lu, J. J. Wang, J. Lv, C. Z. Zhang, and M. Ju (2015). Phys. Chem. Chem. Phys. 17, 13590–13597.
X. D. Xing, A. Hermann, X. Y. Kuang, M. Ju, C. Lu, Y. Y. Jin, X. X. Xia, and G. Maroulis (2016). Sci. Rep. 6, 19656.
B. L. Chen, W. G. Sun, X. Y. Kuang, C. Lu, X. X. Xia, H. X. Shi, and G. Maroulis (2018). Inorg. Chem. 57, 343–350.
P. Shao, B. L. Chen, L. P. Ding, D. B. Luo, C. Lu, and X. Y. Kuang (2017). Phys. Chem. Chem. Phys. 19, 25289–25297.
M. Ju, J. Lv, X. Y. Kuang, L. P. Ding, C. Lu, J. J. Wang, Y. Y. Jin, and G. Maroulis (2015). RSC Adv. 5, 6560–6570.
L. P. Ding, P. Shao, C. Lu, F. H. Zhang, L. Ding, and T. L. Yuan (2016). Phys. Chem. Chem. Phys. 18, 23296–23303.
M. Ohara, K. Koyasu, A. Nakajima, and K. Kaya (2003). Chem. Phys. Lett. 371, 490–497.
K. Koyasu, J. Atobe, S. Furuse, and A. Nakajima (2008). J. Chem. Phys. 129, 214301.
A. Grubisic, Y. J. Ko, H. P. Wang, and K. H. Bowen (2009). J. Am. Chem. Soc. 131, 10783–10790.
A. J. Kenyon (2005). Semicond. Sci. Technol. 20, R65–R84.
H. Tsunoyama, M. Shibuta, M. Nakaya, T. Eguchi, and A. Nakajima (2018). Acc. Chem. Res. 51, 1735–1745.
X. X. Xia, A. Hermann, X. Y. Kuang, Y. Y. Jin, C. Lu, and X. D. Xing (2016). J. Phys. Chem. C 120, 677–684.
R. N. Zhao, Y. H. Yuan, J. G. Han, and Y. H. Duan (2014). RSC Adv. 4, 59331–59337.
R. N. Zhao and J. G. Han (2014). RSC Adv. 4, 64410–64418.
T. G. Liu, W. Q. Zhang, and Y. L. Li (2014). Front. Phys. 9, 210–218.
C. G. Li, L. J. Pan, P. Shao, L. P. Ding, H. T. Feng, D. B. Luo, and B. Liu (2015). Theor. Chem. Acc. 134, 34.
J. C. Yang, J. Wang, and Y. R. Hao (2015). Theor. Chem. Acc. 134, 81.
X. H. Xie, D. S. Hao, Y. M. Liu, and J. C. Yang (2015). Comput. Theor. Chem. 1074, 1–8.
X. H. Xie, D. S. Hao, and J. C. Yang (2015). Chem. Phys. 461, 11–19.
R. N. Zhao, R. Chen, Y. H. Yuan, F. Gu, and J. G. Han (2016). J. Chem. Sci. 128, 365–371.
Z. Lenčéš, L. Benco, J. Madejová, Y. Zhou, L. Kipsová, and K. Hirao (2008). J. Eur. Ceram. Soc. 28, 1917–1922.
H. Yu, L. L. Wang, J. L. Everaert, Y. L. Jiang, D. Mocuta, N. Horiguchi, N. Collaert, and K. D. Meyer (2017). IEEE Electron Device Lett. 38, 843–846.
Q. Peng and J. Shen (2008). J. Chem. Phys. 128, 084711.
T. T. Cao, X. J. Feng, L. X. Zhao, X. Liang, Y. M. Lei, and Y. H. Luo (2008). Eur. Phys. J. D 49, 343–351.
D. B. Zhang and J. Shen (2004). J. Chem. Phys. 120, 5104–5109.
Y. M. Liu, J. C. Yang, and L. Cheng (2018). Inorg. Chem. 57, 12934–12940.
S. He and J. C. Yang (2017). Theor. Chem. Acc. 136, 93.
H. M. Ning, Y. S. Gu, L. Cheng, and J. C. Yang (2018). Chin. J. Struct. Chem. 37, 854–870.
J. C. Yang, Y. T. Feng, X. H. Xie, H. W. Wu, and Y. M. Liu (2016). Theor. Chem. Acc. 135, 204.
Y. T. Feng, J. C. Yang, and Y. M. Liu (2016). Theor. Chem. Acc. 135, 258.
L. Y. Hou, J. C. Yang, and Y. M. Liu (2017). J. Mol. Model. 23, 117.
J. Zhang and M. Dolg (2015). Phys. Chem. Chem. Phys. 17, 24173–24181.
J. P. Perdew, K. Burke, and M. Ernzerhof (1996). Phys. Rev. Lett. 77, 3865–3868.
M. Dolg, H. Stoll, and H. Preuss (1993). Theor. Chim. Acta. 85, 441–450.
M. Dolg, H. Stoll, A. Savin, and H. Preuss (1989). Theor. Chim. Acta. 75, 173–194.
X. Y. Cao and M. Dolg (2001). J. Chem. Phys. 115, 7348–7355.
D. E. Woon and T. H. D. Jr (1993). J. Chem. Phys. 98, 1358–1371.
T. Schwabe and S. Grimme (2006). Phys. Chem. Chem. Phys. 8, 4398–4401.
J. Akola, M. Manninen, H. Häkkinen, U. Landman, X. Li, and L. S. Wang (1999). Phys. Rev. B 60, R11297.
D. J. Tozer and N. C. Handy (1998). J. Chem. Phys. 109, 10180–10189.
T. Lu and F. W. Chen (2012). J. Comput. Chem. 33, 580–592.
D. Y. Zubarev and A. I. Boldyrev (2008). Phys. Chem. Chem. Phys. 10, 5207–5217.
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox Gaussian 09, Revision C.01 (Gaussian Inc, Wallingford, CT, 2010).
Q. Lu and K. A. Peterson (2016). J. Chem. Phys. 145, 054111.
B. A. Hess (1985). Phys. Rev. A 32, 756–763.
B. A. Hess (1986). Phys. Rev. A 33, 3742–3748.
G. Jansen and B. A. Hess (1989). Phys. Rev. A 39, 6016–6017.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21863007 and 21263010), by Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (Gran No. NMGIRT-A1603), and by Natural Science Foundation of Inner Mongolia (Grant No. 2016MS0307).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, Y., Liu, Y., Li, S. et al. Theoretical Study on the Growth Behavior and Photoelectron Spectroscopy of Lanthanum-Doped Silicon Clusters LaSi 0/− n (n = 6–20). J Clust Sci 30, 789–796 (2019). https://doi.org/10.1007/s10876-019-01541-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-019-01541-8