
Abstract
We have investigated the influence of ruthenium doping on magnetic and magnetocaloric properties of Pr0.67Ca0.33Mn1 − x RuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) compounds. X-ray results revealed that the samples have an orthorhombic structure with Pnma space group. The magnetization analysis also showed that Ru doping destroys CO antiferromagnetic and simultaneously inducing ferromagnetism. Variation of lattice parameters and magnetization measurements revealed the existence of mixed valence states of Ru3+ and Ru4+. With increasing Ru content, a slight increase in Curie temperature and magnetization values was observed until x = 0.15. At further Ru doping rates, a considerable decrease was observed in Curie temperature and saturation magnetization. Such a behavior at high Ru concentration rates in saturation magnetization and Curie temperature was attributed to a smaller magnetic moment of Ru4+ than that of Mn3+ and Mn4+ and increase of AFM interaction between the Ru3+– Mn3+ and Ru3+– Mn4+ pairs. The decrease in |ΔSm| was attributed to the second-order magnetic phase transition and the reduction in saturation magnetization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The magnetic cooling studies based upon the magnetocaloric effect (MCE) have become one of the most attractive research areas in previous years due to high energy saving and environmentally friendly compared to traditional gas-compression based refrigeration [1]. In this context, numerous studies on MCE for different kinds of materials have been published in the previous few decades [2,3,4,5]. The previous studies have shown that magnetocaloric materials must possess some important properties for effective use of magnetic cooling technology such as sufficiently high magnetic entropy change at very low magnetic fields, a large working temperature range near room temperature, low cost and easy preparation methods [6]. Among the various potential magnetocaloric materials, due to its outstanding features such as the high magnetization, practical preparation methods, low price, good chemical stability, tunable Curie temperature (TC); the A-site and B-site doped mixed-valence manganites (RE1 − xAxMn1 − yByO3) have been reported as promising candidates in magnetic cooling applications [7].
Among the various manganites, Pr1 − xCaxMnO3 series has attracted great attention because of exhibiting fairly rich and extraordinary magnetic and transport properties. In particular, Pr0.67Ca0.33MnO3 alloy has come to the fore with its charge-order (CO) state, ferromagnetic (FM) and antiferromagnetic (AFM) coexistence, metamagnetic transition and phase separation (PS) features [8,9,10,11,12]. Previous studies about Pr0.67Ca0.33MnO3 have shown that these materials exhibit a charge-order transition at TCO (200–230 K) [8, 9] and then followed by an antiferromagnetic transition at TN [10]. At relatively low temperatures (T < 120 K), a ferromagnetic (FM) metallic phase begins to develop in the antiferromagnetic environment. The coexistence of FM and AFM phases is called PS state [11]. Below the Curie temperature (TC < 60 K), the FM phase becomes more pronounced. [9]. In previous studies, it has been shown that the above-mentioned properties of Pr0.67Ca0.33MnO3 are very sensitive to the magnetic and electrical field, large enough pressure and optical excitations [13]. Rather than these external effects, the most spectacular effect, especially on the CO-AFM state of Pr1 − xCaxMnO3, has been achieved by doping of the Mn-sites with various other transition metals (Co, Cr, Ni, Mg, Al, Sc, Ti, Ga, Zn, In, Sn, Fe, Ir) [14,15,16]. In recent years, among the various dopands, Ru has been realized to be one of the most effectual dopant causing important changes in magnetotransport properties [17,18,19,20,21]. In this context, magnetic and transport properties of many Ru doped manganites such as Pr0.6Ca0.4Mn1 − xRuxO3 [18], Pr0.4Ca0.6Mn1 − xRuxO3 [19], Pr0.5Ca0.5Mn1 − xRuxO3 [20], Pr1 − xCaxMn1 − yRuyO3 [21] and other type of CO-insulating manganites [22, 23] have been investigated in detail. In these studies, it has been reported that a small amount of Ru doping causes the breakdown of the CO-AFM state and the emergence of the FM state, resulting in an increase in the Curie temperature (TC).
However, nearly all of the studies on the Ru doping effect in Pr1 − xCaxMnO3 type manganites are about magnetic and transport properties [18, 19, 21]. Although the magnetocaloric properties of Pr1 − xCaxMnO3 type manganites have been studied sufficiently [24], the studies on the effect of Ru doping on magnetocaloric properties of Pr1 − xCaxMnO3 type manganites are quite limited [20]. Therefore, in the present work, the structural, magnetic and magnetocaloric properties of Ru doped Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) compounds have been studied in detail.
2 Experimental
Polycrystalline samples of Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) were obtained by a standard solid-state reaction method using stoichiometric mixtures of Pr6O11, CaCO3, MnO and RuO2. Pre-sintering was done at 800 °C for 12 h. The powders mixed with grinding were given a disc shape using a press. The disk-shaped specimens were first sintered in air at 1200 °C for 24 h. The last sintering process was actualized at 1350 °C in the air for 24 h. With a cooling rate of 3 °C/min, all samples were cooled to room temperature. The structural analyzes were made by using X-ray diffraction and a scanning electron microscope at room temperature. The magnetic measurements were performed using a Q-3398 (Cryogenic) magnetometer.
3 Results and discussion
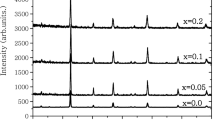
Figure 1a shows the X-ray diffraction spectrum of the Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) samples. X-ray diffraction patterns have shown that all the samples have an orthorhombic structure with Pnma space group. The lattice parameters were obtained by comparing the results from the simulation of the Rietveld enhancement method using the GSAS-II software with the experimental X-ray diffraction patterns. Vertical bars represent the positions of the Bragg reflections. The Rietveld refinement showed an excellent agreement between the calculated and observed patterns. This agreement again confirms that the samples have a single phase. The variation of lattice parameters (a, b and c) and the unit cell volume (Vcell) as a function of Ru concentration (x) are presented in Fig. 2. It has observed that the lattice parameters (a, b, c) and as a consequence cell volume (Vcell) increases almost linearly with increasing Ru content. One of the most curious subjects in Ru-containing manganite has been the determination of the valence states of Ru ions. Although many studies have been performed on the effect of Ru substitution for Mn in manganites, there is still no complete unanimity about the exact valence states of Ru ions [18,19,20,21,22,23]. Considering the ionic radii, it is clearly seen that the most pertinent valence states of Ru should be Ru3+ (0.68 Å), Ru4+ (0.62 Å) and Ru5+ (0.565 Å) [25] in order to preserve the perovskite structure. Well, which one or which ones of them exist in present structure. Maignan et al. have proposed the presence of Ru4+ and Ru5+ ions in Pr0.5Sr0.5Mn1 − xRuxO3 compounds [17]. In X-ray photoelectron spectrum (XPS) analysis of La0.7Sr0.3Mn0.9Ru0.1O3 sample, Krishnan and Ju have claimed that Ru3+ and Ru4+ coexist in the structure [26]. In addition, Sahu and Manoharan [27] found a clear evidence of Ru5+ ion in the La0.6Pb0.4Mn1 − xRuxO3 alloy by using X-ray absorption measurements. However, another X-ray study on the La1 − xNaxMn1–yRuyO3+δ alloys has verified that Ru5+ ion is not present in this alloy [28]. Lakshmi et al. [29, 30] has conducted studies on the valence state of Ru in the La0.67Ca0.33Mn1 − xRuxO3 alloy and concluded that Ru3+ and Ru4+ states should exist in the alloy.

a X-ray diffraction patterns of the Pr.607Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) compounds at at room temperature, b and c the Rietveld refinement for x = 0.1 and 0.3 samples

Concentration dependence of lattice parameters and the unit-cell volume of the Pr.607Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) compounds
The variation of lattice parameters with Ru content gives very important clues about the valence state of Ru. From the variation of the lattice parameters in Ru doped samples, an estimate can be made indirectly about the valence state of Ru ions. If the ionic radii of Ru4+/5+ (Ru4+ = 0.62 Å and Ru5+ = 0.565 Å) and Mn3+/4+ (Mn3+ = 0.645 Å and Mn4+ = 0.53 Å) are considered, the presence of Ru4+/5+ is expected to introduce a decrease in the lattice parameters. However, the considerable increase in lattice parameters of present Ru doped samples (Fig. 2), eliminates the presence of Ru5+ in the structure and points to other two possibilities. In this case, the most likely possibility is the presence of either Ru3+ or Ru4+ where the ionic radius of Ru3+ (0.68 Å) and Ru4+ (0.62 Å) are large to cause such an increase in lattice parameters. Here, three distinct possibilities can be suggested about the valence state of Ru in examining samples. These possibilities are only Ru3+, only Ru4+ and mixed-state of Ru3+ and Ru4+ respectively. All the suggested possibilities could cause the expected enlargement in lattice parameters. It would not be very persuasive to make an estimate the valence states of Ru in the examined samples by taking into account the change in lattice parameters alone. Therefore, the valence state of Ru in the present compound will be re-examined in detail, taking into account magnetic measurements. However, inferences from magnetization measurements, as will be discussed in detail later on, do not favor the presence of Ru3+ or Ru4+ alone. This strengthens the possibility of the existence of Ru3+ and Ru4+ mixed valence states in present compounds.
Figure 3 shows the typical SEM micrographs for Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.2, 0.3, 0.5) samples with a magnification of 10,000. It is clearly seen that SEM pictures point out a smooth and homogeneous polycrystalline structure. The mean particle size is in the range of 6–14 µm for undoped sample (x = 0). The grain size is found to significantly decrease to 0.5–2 µm for x = 0.5 with increased Ru content. Lakshmi et al. [29, 30] have also reported a alike reduction in particle size with Ru content for La0.67Ca0.33Mn1 − xRuxO3 (0 ≤ x < 0.1) samples.

SEM micrographs of the Pr.607Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.2, 0.3, 0.5) compounds
Figure 4 shows the temperature dependence of magnetization for Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) samples. The data were taken under an applied magnetic field of 0.1 T. The magnetization curves in Fig. 4 for the parent compound (x = 0) attract attention with their different behavior from the others. It is clearly seen that the parent compound exhibits multiple magnetic transitions. With the decrease of temperature from 275 K, the magnetization gradually increases and then slightly decreases around the TCO = 210 K. As marked in Fig. 4, observed this small broad peak on the magnetization curve has been attributed to existence of CO state. Towards to lower temperatures, a marked anomaly occurred at 167 K as a very weak kink in the magnetization curve indicates the start of the AFM ordering at that temperature. Towards to lower temperatures, the increase in magnetization curves refers the increase of FM phase. From the magnetization examinations, it has been inferred that that ferromagnetic and charge-ordered AFM phases exist at the same time and ferromagnetic phase grows up in the charge-ordered AFM media. For a magnetic field H = 0.1 T, a more apparent FM phase becomes emerge at TC = 45 K. All the magnetization curves for Ru doped samples (x = 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) show a transition from paramagnetic to ferromagnetic at their Curie temperature (TC) and no evidence of CO transition is observed in the magnetization curves. The signatures of antiferromagnetic ordering completely disappear the magnetization measurements have revealed that the Ru substitution in Pr0.67Ca0.33Mn1 − xRuxO3 manganites demolishes the charge-ordered AFM phase and simultaneously inducing ferromagnetism and metallicity. The Curie temperature, Tc, generally determined by the temperature of the minimum value for |dM/dT|. The Curie temperatures were determined to be 45 K, 98 K, 167 K, 185 K, 182 K, 171 K and 142 K for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5 respectively. The variation of TC with increasing Ru content was given inset in Fig. 4. As can be seen, TC first increases from 45 K for x = 0 to 185 K for x = 0.15 and then decreases back to 142 K as x increases to 0.5. The similar behavior of TC with increasing dopant (x) has also been observed in some other Ru doped perovskites such as Pr1 − xCaxMn1 − xRuxO3 (0 ≤ x ≤ 0.6) [21], Pr0.8Na0.2Mn1 − xRuxO3 (0 ≤ x ≤ 0.2) [22] and La0.72 − xCa0.31+xMn1 − xRuxO3 (0 ≤ x ≤ 0.3) [31]. Wang et al. [21] has concluded that that the FM state has replaced by the weak AFM state or paramagnetic state in the high x-range. Ji et al. [22] have asserted that in pristine sample, the CO and antiferromagnetism competes with double-exchange (DE) induced ferromagnetism and at low concentrations of Ru leads to the TC enhancement. They have also deduced that at larger concentration rates the DE interaction is weakening again due to the over compensated density of the hole.

The temperature dependence of magnetization for Pr.607Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) compounds. The inset presents the variation of TC with Ru content
Figure 5 shows the magnetic field dependence of magnetization for Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.3, 0.5) at various temperatures. As can be seen in Fig. 5, the magnetization isotherms for the pristine sample (x = 0) show paramagnetic behavior above the CO transition temperature (TCO = 210 K). At lower temperatures than that of CO transition, the magnetization shows a rapid increase at above 4 T magnetic field. As the temperature comes near to the Curie point (TC = 45 K), magnetization jumps continue, but the critical magnetic field that causes these jumps nearly reduces to 1 T. Well below the Curie temperature (T < 20 K), the magnetization curves display almost ferromagnetic behavior. The unconventional step-like behavior in magnetization isotherms has been attributed to partly repression of charge-ordered AFM state under an applied field. This situation in general called as metamagnetic transition and interpreted based on the scenario of PS [8,9,10,11]. At low temperatures, due to the contention between the FM and AFM-CO phases, the sample gets into a strongly blocked regime. At that point, if the magnetic field reaches up the critical value, the ferromagnetic phase could cope the antiferromagnetic phase and consequently a sudden increase is observed in magnetization curves. Interestingly, metamagnetic transitions could be observed, albeit slightly, in the magnetization curves of the x = 0.05 sample. This means that the CO-AFM state is still dominance, albeit slightly weakened. As can be seen in Fig. 5, in higher Ru doped samples (x = 0.1, 0.3, 0.5), the signatures of antiferromagnetic ordering completely disappear. The magnetization curves for all samples exhibit typical FM behavior. At low temperatures (T < TC), the magnetization curves increase rapidly and reach the maximum value even at lower magnetic fields. At high temperatures (T > TC), the magnetization isotherms show a linear tendency with the magnetic field as in fully paramagnetic materials. The field dependence of magnetization for Pr0.67Ca0.33Mn1 − xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) samples at 5 K and at magnetic field interval of − 7 to 7 T also given in Fig. 5. The saturation magnetization is determined to be 80.58, 85.78, 81.49, 75.24, 73.92, 69.37 and 38.44 emu/g for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5 samples, respectively. It is clearly seen that the saturation magnetization first increases 80.58 emu/g for x = 0 to 85.78 emu/g for x = 0.05 and then monotonically decreases with increasing Ru content. Wang et al. [21] has observed the same trend in saturation magnetization for Pr1 − xCaxMn1 − xRuxO3 (0 − ≤ x ≤ 0.6). Such a behavior at high Ru concentration rates in saturation magnetization is attributed to a smaller magnetic moment of Ru4+ than that of Mn3+ and Mn4+ and relatively weaker Ru–Ru interactions.

Isothermal magnetization curves at various temperatures for Pr.607Ca0.33Mn1–xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) and the field dependence of the magnetization at 5 K in the range of − 7 to 7 T applied magneticfield
In order to check the accuracy of our above prediction about the probable valency states of ruthenium, the effective (µeff), saturation (µS) and theoretical (µt) magnetic moments per formula unit have been determined and compared each other. By using the Curie–Weiss law, a detailed analysis of the inverse magnetic susceptibility in the paramagnetic region has been carried out. The variation of inverse magnetic susceptibility (H/M) with temperature for present samples at a 0.1 T magnetic field is given in Fig. 6a. All the curves have fitted with the Curie–Weiss law \(\chi (M/H)=C/(T-{\theta }_{C})\), where θC is called as the paramagnetic Curie temperature and C is the Curie constant. In that calculation, θC was determined to be 40.41, 110.23, 186.59, 192.21, 186.44, 184.33 and 159.07 K for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5, respectively. The calculated θC values are a little bit higher than their Curie temperatures. As can be seen from the results, the positive value of θC point outs the FM interactions. In some previous studies, the divergence between θC and TC values have been attributed to broad magnetic transition [32] or magnetic in homogeneities [33]. The Curie constant (C) was calculated to be 0.0257, 0.0210, 0.0177, 0.0149, 0.0134, 0.0120 and 0.0088 emu K/g. Oe for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5, respectively. The effective magnetic moment was determined using \(C = N_{A} \,\mu_{eff}^{2} /3\,k_{B}\). μeff was calculated to be 3.75, 3.39, 3.11, 2.86, 2.70, 2.32 and 1.45μB for the x = 0, 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5 samples, respectively. Figure 6b shows the variation of μeff with increasing Ru content.

a Temperature dependence of the inverse susceptibility (H/M) for the x = x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 samples. The solid lines correspond to the Curie–Weiss law fit to the susceptibility data. b The variation of saturation magnetic moment (μs), effective magnetic moment (μeff) and calculated magnetic moments (μt) in the case of Ru3+, Ru4+ and Ru3+/Ru4+ as a function of Ru concentration (x)
Determining the saturation magnetization as saturation magnetic moments µS (µB/f.u) will provide the magnetic moments could be compared with each other. The saturation magnetic moments could be determined by using the following equation:
where N is Avogadro’s number, μB is the Bohr magneton, MS is the saturation magnetization and W is the molecular weight of the Pr0.67Ca0.33Mn1−xRuxO3 compounds. µS was calculated to be 3.035, 3.298, 2.928, 2.784, 2.547, 2.234, 1.306 μB for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 and illustrated in Fig. 6b. In order to confirm the accuracy of valence states attributed to Ru in examining samples, the magnetic moments per formula unit have been calculated theoretically in the case of possible Ru3+, Ru4+, and Ru3+/Ru4+ valence states in the compounds. The chemical formulas for Ru3+, Ru4+ and Ru3+/Ru4+- substituted compounds can be illustrated by the following equations:
All the above-mentioned chemical formulas lead to a ferromagnetic moment as follows equations:
where \({\mu }_{{\mathrm{Mn}}^{3+}}=4{\mu }_{B}\), \({\mu }_{{\mathrm{Mn}}^{4+}}=3{\mu }_{B}\), \({\mu }_{{\mathrm{Ru}}^{3+}}=3{\mu }_{B}\) and \({\mu }_{{\mathrm{Ru}}^{4+}}=2{\mu }_{B}\). In addition to μeff and μs, the variation of μt with Ru content is illustrated in Fig. 6b with open symbols. As seen in Fig. 6b, while the calculated μt values for Ru3+/Ru4+ are nearly matched the experimentally determined μeff and μS, for Ru3+ and Ru4+ deviate from experimental results. The magnetization results presented in Fig. 6b support the existence of mixed-valence state of Ru3+ and Ru4+ in the considered samples. Furthermore, the obtained results confirm the previously suggested estimates about valence states of Ru. What has been done regarding the valence state of Ru in this study is just an estimation. Despite the variation of the lattice parameters and magnetization measurements give important clues about the valence state of Ru, it is still needed further detailed investigations such as X-ray photoelectron spectroscopy (XPS) [26].
A magnetic entropy change associated with MCE in a certain temperature interval and at an applied magnetic field is generally calculated using magnetization curves (Fig. 5) and the following equation:
In determining the magnetic entropy change, ΔSm, from isothermal magnetization curves, generally an equivalent approximation (Eq. 9) of the integral in Eq. 8 is used.
The magnetic entropy change in pristine (x = 0) sample has been investigated in detail in previous studies [8, 9]. Therefore, the results for x = 0 sample were not given here. The calculated magnetic entropy changes for x = 0.05, 0.1, 0.2, 0.3 and 0.5 samples are given in Fig. 7. The maximum value of |ΔSm| and the temperature value at which |ΔSm| is maximum tend to an increment with increasing magnetic field. Figure 7 also shows the |ΔSm| for x = 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 samples at 5 T magnetic field Fig. 7 clearly shows the decrease in the peak value of the |ΔSm| with increasing Ru concentration. Peak values of |ΔSm| were determined to be decreased from 3.91 J/kg.K for x = 0.05 to 1.37 J/kg.K for x = 0.5 at 5 T magnetic field. The variation of |ΔSm| is in agreement with that of previous Ru doped Pr0.5Ca0.5Mn1−xRux (x = 0.03, 0.05, 0.1) [20] and Nd0.5Ca0.5Mn1−yRuyO3 (y = 0, 0.03, 0.05, 0.1) [23] manganites. In doped manganites, the variation in the peak values of |ΔSm| is generally interpreted as depending the magnetization value and the type of magnetic phase transition, which are the basic parameters that determine the value of the magnetic entropy change of a sample. As discussed above, the saturation magnetization decreased from 85.78 emu/g for x = 0.05 to 38.44 emu/g for x = 0.5 which is one of the clear evidence of the decrease in |ΔSm|. In order to fully understand the change in |ΔSm| with Ru doping, the type of the magnetic phase transition must also be clarified.

Magnetic entropy change for x = 0.05, 0.1, 0.2, 0.3, 0.5 samples at various magnetic fields and magnetic entropy change for x = 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5 samples at 5 T magnetic field
In order for a magnetic material to be considered an effective magnetic coolant, it is not sufficient to simply have a large |ΔSm| value. The temperature interval in which |ΔSm| maintains the maximum value are also very significant. In this case, one of the accepted criteria to appreciate refrigeration efficiency is the relative cooling power (RCP). It is described as the peak value of |ΔSm| and the temperature of full width at half maximum ( =|ΔSm|. δTFWHM) and determines the value of heat transfer from a hot area to a cold area in a thermodynamic cycle. The estimated RCP values (at 5 T magnetic field change) are 343.68, 441.99, 290.69, 207.82, 213.39 and 130.12 J/kg for the x = 0.05, 0.1, 0.15, 0.2, 0.3 and 0.5, respectively. The calculated RCP values of present samples are comparable or larger than those of other Ru-doped manganites. For Pr0.5Ca0.5Mn1−xRuxO3, the RCP was reported to increase from 284.9 J/kg for x = 0.03 to 303.6 J/kg for x = 0.1 [20]. For Nd0.5Ca0.5Mn1−yRuyO3 (y = 0, 0.03, 0.05 and 0.10), the RCP values were reported to decrease from 268 J/kg for x = 0.03 to 234 J/kg for x = 0.1 [23]. Such a high RCP value of magnetocaloric materials is usually attributed to the rather high δTFWHM values due to the second-order magnetic phase transition.
So as to sure the nature of the phase transition of examining materials, it must be clarified by using various techniques. It is seen that many techniques are used in the literature in determining the nature of phase transitions [6]. The slope of the Arrott plots indicates whether a phase transition is in first or second-order. In accordance with the Banerjee criterion [34], the positive slope of Arrott curves, points out a second-order phase transition. Otherwise, if curves near the transition temperature show a negative slope, then the magnetic transition is first-order. Figure 8 shows the Arrott plots for Pr0.67Ca0.33Mn1−xRuxO3 (x = 0, 0.05, 0.1, 0.2, 0.5) samples. The negative slopes of M2 versus H/M curves for pristine (x = 0) sample are a clear indication of a first-order phase transition from ferromagnetic to paramagnetic state. Interestingly, the Arrott plots for x = 0.05 exhibits an almost mixed behavior of first and second-order magnetic phase transition. To clarify the nature of the phase transition whether it is in first-order or second-order especially for x = 0.05 sample, further detailed investigation is needed. For further Ru concentration rates (x ≥ 0.1), the positive slope of Arrott plots reveals that the magnetic phase transition has changed from first-order to second-order with increasing Ru content. The first and second-order character of phase transition for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 samples are clearly seen in Arrott plots (Fig. 8) given only at their Curie temperatures. More detailed study of phase transition nature near the Curie temperature has been carried out using the Landau theory. The magnetic free energy F (M, T) can be expanded as a power of the magnetization M, neglecting higher order contributions as;

Arrott plots for for x = 0, 0.05, 0.1, 0.2 and 0.5 samples at various temperatures and Arrott plots for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 at their transition temperatures
Here, c1 (T), c3 (T) and c5 (T) are denoting the Landau coefficients and the sign of the c3 (T) defines the type of the magnetic phase transition. While in the case of c3 (T) < 0 a first-order phase transition is expected, in the case of c3 (T) > 0, a second-order phase transition is expected. From the condition of ∂F (M, T)/∂M = 0, we obtain;
The Landau coefficients were determined by fitting Eq. 11a to magnetization isotherms (Fig. 5). Another possibility is to fit the Eq. 11b to Arrott plots (Fig. 8), the determined Landau coefficients are shown in Fig. 9 for Pr0.67Ca0.33Mn1−xRuxO3 (x = 0.05, 0.1, 0.2, 0.5) compounds. The positive values of c3 (T) for Ru doped samples at transition temperatures indicate that the magnetic phase transitions are in second-order.

The temperature dependence of c1 (T.g/emu), c3 (T.g3/emu3) and c5 (T.g5/emu5) for x = 0.05, 0.1, 0.2, 0.5 samples
By taking the derivative of the free energy with respect to the temperature, ΔSm is obtained as;
Using the value of c1 (T), c3 (T) and c5 (T) coefficients (Fig. 9) and their derivative in Eq. 12, the ΔSm was calculated theoretically under a magnetic field change of 1 T for x = 0.05, 0.1, 0.2 and 0.5 samples. The calculated and experimental magnetic entropy changes are shown in Fig. 10 for x = 0.05, 0.1, 0.2 and 0.5 samples at 1 T magnetic field. While the agreement between experimental and theoretical data at low Ru concentrations (x = 0.05 and 0.1 were given in Fig. 10) is at a satisfactory level, a small deviation, especially at lower temperatures, between experimental ant theoretical data for the further Ru doping levels (x = 0.2 and 0.5 were given in Fig. 10) is clearly seen. The agreement between theoretical and experimental results are generally attributed to the magnetoelastic coupling (interaction between the magnetization and the strain of a magnetic material) and electron interaction have strong influence in determining the magnetocaloric effect of examining materials [35]. In the case of small deviations between experimental and theoretical results as in x = 0.2 and 0.5 samples, it has been concluded that the magnetoelastic coupling and electron interaction do not contribute directly to the magnetic entropy. This small deviation observed between the experimental and theoretical data is generally attributed to the existence of mixed phases especially at lower temperatures [36,37,38,39,40,41].

Experimental and theoretical magnetic entropy change for x = 0.05, 0.1, 0.2, 0.5 samples at 1 T magnetic field
Another technique most commonly used to determine the order of magnetic phase transitions is to investigate the field dependence of the ΔSm using the relation expressed as:
where C is a constant and n is the exponent related to the magnetic state of samples [42, 43]. Determining the values of the exponent n at particular temperatures gives important clue about the type of the phase transition in the present compounds. The Curie–Weiss law foresees that the value of n will approach to n = 2 in the paramagnetic phase above the Curie temperature. In ferromagnetic phase (well below the transition temperature), n shows a trend approaching n = 1 [43]. Near the Curie temperature, the variation of n is completely depends on the type of phase transition. At the transition temperature (T = TC), n exceeding 2 (n > 2) is attributed to a first-order phase transition [44]. At a given temperature and magnetic field, the exponent n is determined using logarithmic derivative of ΔSm (T, H) [42]:
The determined n values for the Pr0.67Ca0.33Mn1−xRuxO3 (x = 0, 0.05 and 0.5) compounds are given in Fig. 11 as a function of temperature. For all Ru doped samples, exponent n shows a minimum at transition temperature. In pristine sample (x = 0), the exponent n exhibits a different behavior at the transition temperature. Figure 11 shows that exponent n has a maximum that exceeds 2 at the transition temperature for pristine sample. The variation of n for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 at 5 T magnetic field is also given in Fig. 11. It is clearly seen that the variation of n with temperature for pristine sample is quite different from that of Ru doped samples. While the variation of n in pristine sample exhibits a first-order magnetic phase transition behavior, the Ru doped samples display a second-order phase transition behavior.

Temperature dependence of the exponent n at vatious magnetic fields for x = 0, 0.2 and 0.5 samples and the variation of exponent n as a function of temperature for x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5 samples only at 5 T magnetic field
4 Conclusions
We have investigated the influence of Ru doping on structural, magnetic and magnetocaloric properties of Pr0.67Ca0.33Mn1−xRuxO3 (x = 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5) compounds. The standard solid-state process prepared the polycrystalline samples. X-ray results pointed out that all the samples crystallize in an orthorhombic structure in Pnma space group. The small broad peak observed on the magnetization curve for the parent compound (x = 0) at TCO = 210 K was attributed to the existence of antiferromagnetic charge ordered state. The magnetization measurements also showed that Ru doping destroys CO antiferromagnetism and simultaneously inducing ferromagnetism and metallicity. Variation of lattice parameters and magnetization measurements revealed the existence of mixed valence states of Ru3+ and Ru4+. With increasing Ru content, a slight increase in Curie temperature and saturation magnetization were observed up to x = 0.15. But for the further Ru doping, a considerable decrease was observed in Curie temperature and saturation magnetization. Such a behavior at high Ru concentration rates in saturation magnetization and Curie temperature was attributed to a smaller magnetic moment of Ru4+ than that of Mn3+ and Mn4+ and increase of AFM interaction between the Ru3+– Mn3+ and Ru3+– Mn4+ pairs. Variation of lattice parameters and magnetization measurements revealed the existence of mixed valence states of Ru3+ and Ru4+. The decrease in |ΔSm| (from 3.91 J/kg.K for x = 0.05 to 1.37 J/kg.K for x = 0.5 at 5 T) was ascribed to the second-order magnetic phase transition and the reduction in saturation magnetization. The larger RCP value was reported to be 441.99 J/kg for x = 0.1 sample at 5 T magnetic field change. Such a high RCP value was attributed to the rather high δTFWHM values due to the second-order magnetic phase transition. The relatively large value of the RCP, the tunable Curie temperature by Ru substitution make these compounds attractive candidates for magnetic refrigeration applications just below room temperature. In this study, the structural, magnetic and magnetocaloric properties of the samples prepared in terms of cooling technology were examined for the first time by us and their suitability for magnetic cooling technology was revealed.
Availability of data
The data that support the findings of this study are available from the corresponding author on request.
References
K.A. Gschneidner Jr., V.K. Pecharsky, A.O. Tsokol, Recent developments in magnetocaloric materials. Rep. Prog. Phys. 68, 1479–1539 (2005)
V.K. Pecharsky, K.A. Gschneidner, Giant magnetocaloric effect in Gd-5(Si2Ge2). Phys. Rev. Lett. 78, 4494–4497 (1997)
H. Wada, Y. Tanabe, Giant magnetocaloric effect of MnAs1–xSbx. Appl. Phys. Lett. 79, 3302–3304 (2001)
O. Tegus, E. Bruck, K.H. Buschow, F.R. de Boer, Transition-metal-based magnetic refrigerants for room-temperature applications. Nature 415, 150–152 (2002)
F.W. Wang, X.X. Zhang, F.X. Hu, Large magnetic entropy change in TbAl2 and (Tb0.4Gd0.6)Al-2. Appl. Phys. Lett. 77, 1360–1362 (2000)
V. Franco, J.M. Borrego, A. Conde, Influence of Co addition on the magnetocaloric effect of FeCoSiAlGaPCB amorphous alloys. App. Phys. Lett. 88, 132509 (2006)
H. Gencer, V.S. Kolat, T. Izgi, S. Atalay, Magnetocaloric Effect in Perovskite Manganites (Materials Research Forum LLC, 2020), pp.18–37
A.M. Gomes, F. Garcia, A.P. Guimaraes, M.S. Reis, V.S. Amaral, P.B. Tavares, Magnetocaloric effect of the (Pr, Ca)MnO3 manganite at low temperatures. J. Magn. Magn. Mater. 290–291, 694–696 (2005)
M. Pektas, H. Gencer, T. Izgi, V.S. Kolat, S. Atalay, Field induced unusual magnetic behavior at low temperature in Pr0.67Ca0.33MnO3. Acta Phys. Polonica A 125, 217–219 (2014)
S. Hebert, V. Hardy, A. Maignan, R. Mahendiran, M. Hervieu, C. Martin, B. Raveau, Magnetic-field-induced step-like transitions in Mn-site doped manganites. J. Solid State Chem. 165, 6–11 (2002)
F. Rivadulla, M.A.L. Quintela, L.E. Hueso, C. Jardon, A. Fondado, J. Rivas, M.T. Causa, R.D. Sanchez, Strong ferro-antiferromagnetic competition and charge ordering in Pr0.67Ca0.33MnO3. Solid State Commun. 110, 179–183 (1999)
R. Mahendiran, A. Maignan, S. Hebert, C. Martin, M. Hervieu, B. Raveau, J.F. Mitchell, P. Schiffer, Ultrasharp magnetization steps in perovskite manganites. Phys. Rev. Lett. 89, 286602 (2002)
S. Majumdar, T. Elovaara, H. Huhtinen, S. Granroth, P. Paturi, Crystal asymmetry and low-angle grain boundary governed persistent photoinduced magnetization in small bandwidth manganites. J. Appl. Phys. 113, 063906 (2013)
B. Raveau, A. Maignan, C. Martin, Insulator-metal transition induced by Cr and Co doping in Pr0.5Ca0.5MnO3. J. Solid State Chem. 130, 162–166 (1997)
S. Hebert, A. Maignan, V. Hardy, C. Martin, M. Hervieu, B. Raveau, Avalanche like field dependent magnetization of Mn-site doped charge-ordered manganites. Solid State Commun. 122, 335–340 (2002)
A. Maignan, F. Damay, C. Martin, B. Raveau, Nickel-induced metal-insulator transition in the small a cation manganites Ln (0.5) Ca (0.5) MnO (3). Mater. Res. Bull. 32, 965–972 (1997)
A. Maignan, C. Martin, M. Hervieu, B. Raveau, Ru doping of the A-type antiferromagnetic Pr0.5Sr0.5MnO3: conversion to a metallic ferromagnetic. J. Appl. Phys. 89, 500–5003 (2001)
C.A. Lambert, M. Gervais, M. Zaghrioui, S. Roger, F. Gervais, N. Raimboux, P. Simon, Temperature dependence of phase separation and magnetic anisotropy by electron spin resonance in Pr0.6Ca0.4Mn0.9Ru0.1O3. Eur. Phys. J. B 47, 207–211 (2005)
B. Raveau, A. Maignan, C. Martin, M. Hervieu, Ru doping of perovskite manganites: an effective route to ferromagnetism, metallicity. CMR J. Superconductivity 14, 217–229 (2001)
V.S. Kumar, R. Mahendiran, B. Raveau, Effect of Ru-doping on magnetocaloric effect in Pr based charge ordered manganites. IEEE Trans. Magnetics 46, 1652–1655 (2010)
Y.L. Wang, M.F. Liu, Y.L. Xie, Z.B. Yan, S. Dong, J.M. Liu, Manipulating the ferromagnetism in narrow-bandwidth Pr1−xCaxMnO3 (0<=x<=0.6) by means of the Mn-Ru t(2g) ferromagnetic super-exchanges. J. App. Phys. 118, 123901(1–10) (2015)
J.T. Ji, Y. Li, T.S. Zhao, Y.W. Du, The effect of Ru doping on the alkali-doped manganite Pr0.8Na0.2MnO3. J. Mag. Mag. Mater. 322, 3857–3861 (2010)
C. Krishnamoorthi, S.K. Barik, R. Mahendiran, Effect of Ru-substitution on electrical and magnetocaloric properties of Nd0.5Ca0.5MnO3. Solid State Commun. 151, 107–111 (2011)
A.M. Gomes, F. Garcia, A.P. Guimarães, M.S. Reis, V.S. Amaral, Field-tuned magnetocaloric effect in metamagnetic manganite system. Appl. Phys. Lett. 85, 4974–4976 (2004)
R.D. Shannon, C.T. Prewitt, Acta Cryst. Sec. A. 32, 751 (1976)
K.M. Krishnan, H.L. Ju, Role of stoichiometry and structure in colossal magnetoresistive La1–xSrxMn1–yRuyO3+δ. Phys. Rev. B 60, 14793–14803 (1999)
R.K. Sahu, S.S. Manoharan, A Zener pair effect in lanthanum rutheno manganite. J. Appl. Phys. 91, 7517 (2002)
L. Malavasi, M.C. Mozzati, E.D. Tullio, C. Tealdi, G. Flor, Redox behavior of Ru-doped La1−xNaxMnO3+δ manganites. Phys. Rev. B 71, 174435 (2005)
L.S. Lakshmi, V. Sridharana, D.V. Natarajana, S. Chandra, V.S. Sastrya, T.S. Radhakrishnana, P. Pandianb, R.J. Joseyphusb, A. Narayanasam, Possible magnetic phase separation in Ru-doped La0.67Ca0.33MnO3. J. Mag. Mag. Mater. 257, 195–205 (2003)
L.S. Lakshmi, V. Sridharan, A.A. Sukumar, M. Kamruddin, V.S. Sastry, V.S. Raju, Double MITs and magnetoresistance: an intrinsic feature of Ru substituted La0.67Ca0.33MnO3. J. Phys.: Condens. Matter 18, 4427–4442 (2006)
M.F. Liu, Z.Z. Du, Y.L. Xie, X. Li, Z.B. Yan, J.M. Liu, Unusual ferromagnetism enhancement in ferromagnetically optimal manganite La0.7–yCa0.3+yMn1–yRuyO3 (0<=y<0.3): the role of Mn-Ru t(2g) super-exchange. Sci. Rep. 4, 9922 (2015)
S.K. Srivastava, B. Samantaray, T. Bora, S. Ravi, Magnetic and electrical properties of Mn-substituted (La0.85Ag0.15) CoO3 compounds. J. Magn. Magn. Mat. 474, 605–612 (2019)
M.E. Hagary, Y.A. Shoker, S. Mohammad, A.M. Moustafa, A. Abd El-Aal, H. Michor, M. Reissner, G. Hilscher, A.A. Ramadan, Structural and magnetic properties of polycrystalline La0.77Sr0.23Mn1−xCuxO3 (0≤ x≤ 0.5) manganites. J. Alloys Compd. 468, 47–53 (2009)
B.K. Banerjee, Phys. Lett. 12, 16–17 (1964)
S. Das, T.K. Dey, Magnetocaloric effect in potassium doped lanthanum manganite perovskites prepared by a pyrophoric method. J. Phys.: Condens. Matter 18, 7629–7641 (2006)
R. Cherif, E.K. Hlil, M. Ellouze, F. Elhalouani, S. Obbade, Study of magnetic and magnetocaloric properties of La0.6Pr0.1Ba0.3MnO3 and La0.6Pr0.1Ba0.3Mn0.9Fe0.1O3 perovskite-type manganese oxides. J. Mater. Sci. 49, 8244–8251 (2014)
O. Karaagac, H. Köckar, The effects of temperature and reaction time on the formation of manganese ferrite nanoparticles synthesized by hydrothermal method. J. Mater. Sci.: Mater. Electron. 31, 2567–2574 (2020)
O. Karaagac, B. Bilir, H. Kockar, Superparamagnetic cobalt ferrite nanoparticles, effect of temperature and base concentration. J. Supercond. Nov. Magn. 28, 1021–1027 (2015)
O. Karaagac, S. Atmaca, H. Kockar, A facile method to synthesize nickel ferrite nanoparticles: parameter effect. J. Supercond. Nov. Magn. 30, 2359–2369 (2017)
C. Hasirci, O. Karaagac, H. Köckar, Superparamagnetic zinc ferrite: a correlation between high magnetizations and nanoparticle sizes as a function of reaction time via hydrothermal process. J. Magn. Magn. Mater. 474, 282–286 (2019)
F. Ozel, H. Kockar, S. Beyaz, O. Karaagac, T. Tanrisever, Superparamagnetic iron oxide nanoparticles: effect of iron oleate precursors obtained with a simple way. J. Mater. Sci.: Mater. Electron. 24, 3073–3080 (2013)
V. Franco, J.S. Blazquez, B. Ingale, A. Conde, The magnetocaloric effect and magnetic refrigeration near room temperature: materials and models. Annu. Rev. Mater. Res. 42, 305–342 (2012)
M. Pektas, T. Izgi, H. Gencer, S. Atalay, V.S. Kolat, N. Bayri, Effects of Ru substitution on the structural, magnetic and magnetocaloric properties of Pr0.68Ca0.22Sr0.1Mn1−xRuxO3(x=0, 0.05, 0.1 and 0.2) compounds. J. Mater. Sci.: Mater. Electron. 31, 15731–15741 (2020)
J.Y. Law, V. Franco, L.M.M. Ramírez, A. Conde, D.Y. Karpenkov, I. Radulov, K.P. Skokov, O. Gutfleisch, A quantitative criterion for determining the order of magnetic phase transitions using the magnetocaloric effect. Nat. Commun. 9, 2680 (2018)
Acknowledgements
This work was supported by Inonu University with project number FBA-2021-2778.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest. This article does not contain any studies involving animal or human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kanıkırmızı, T., İzgi, T., Bayri, N. et al. Structural, magnetic and magnetocaloric properties of Ru doped Pr0.67Ca0.33Mn1 − xRuxO3 manganites. J Mater Sci: Mater Electron 33, 21778–21795 (2022). https://doi.org/10.1007/s10854-022-08966-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-022-08966-4