
Abstract
A new hybrid material system was successfully developed from the combination of polyimide (PI), organically modified silicate (ORMOSIL), and phosphotungstic acid (HPW) from polycondensation reactions and sol–gel process. The materials were obtained in the form of flexible and free-standing films, with the formation of PI and ORMOSIL network confirmed by Fourier transform infrared. Solid-state phosphorus-31 nuclear magnetic resonance evaluated the HPW structure and confirmed its presence and also structural integrity in the materials after synthesis procedure. Thermogravimetric analysis revealed that the materials were thermally stable up to 773 K, and scanning electron microscopy images and X-ray micro-fluorescence mapping showed very good compatibility between the organic and inorganic phases with ORMOSIL improving the HPW dispersion. Furthermore, both ORMOSIL and HPW enhanced the permittivity of the materials from 2.5 to 4.3, compared to the neat PI, appointing them as potential candidates for electric and electronic applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Multifunctional and high performance materials requires accurate control of their building blocks (atoms, molecules, crystalline structure) by synthetic design, in order to achieve fully control their properties (mechanical, thermal, electrical, etc.) [1, 2]. Nevertheless, hardly a single compound will present the same level of performance when subjected to different conditions and environments. Thus, there has been increased interest in hybrid materials, consisting generally of organic and inorganic components mixed at the molecular level, with at least one of the components generated in situ during synthesis [1, 2].
Polyimides (PIs) are thermal stable and mechanical strength polymers [3]. They are of great interest for the synthesis of hybrid materials, especially by combination with Organically Modified Silicates (ORMOSIL), from polycondensation reactions and sol–gel route [4]. ORMOSIL materials are stable in very acidic and slightly basic media, and act as perfect hosts for enzymes, catalysts, photochromic compounds, etc., without degrading their properties [5–7]. Moreover, PI-ORMOSIL system provides highly intermingled and homogeneous hybrid materials, with several applications such as insulators in electronic devices [8–10]. In addition, the introduction of phosphotungstic acid (HPW) to PI-ORMOSIL system may enable its application as a high-temperature polymer electrolyte (HTPE) to be used in electrical energy storage devices [11–15]. Aerospace industry currently has a strong need for compact capacitors for power conditioning applications operating above 100 °C [16].
HPW is a thermally stable Keggin-type heteropolyoxometalate, with formula H3PW12O40, and a very strong Bronsted–Lowry acid in solid state. It has global symmetry Td, composed of 12 octahedrons MO6, organized into 4 groups of W3O13, named triads, that share vertices among themselves and with the PO4 tetrahedron in the center of the structure [17, 18]. Due to its high solubility in water and organic solvents, which causes leaching, it is usually supported on activated charcoal, ionic-exchanged resins, SiO2, or ZrO2 for solid-state applications [19–21]. However, the solubility makes this compound a suitable choice for hybrids materials formed from sol–gel process [19]. Furthermore, HPW is a low-cost choice compound that can enable electrochemical, photochromic, and photocatalytic technological platforms operating from environment temperature up to higher than 150 °C [22].
Although there are plenty of literature related to the combination of PI and ORMOSIL, to our knowledge, there are no reports of PI-ORMOSIL containing HPW. Thus, this work aims to develop and to explore the new PI-ORMOSIL-HPW hybrid material system, providing detailed composition–structure–property relationship.
Materials and methods
Materials
All reagents 4,4-Oxydiphthalic anhydride (OPDA, Sigma-Aldrich, 97 %), 4,4′-Oxydianiline (ODA, Sigma-Aldrich, 98 %), (3-aminopropyl)triethoxysilane (APTES, Sigma-Aldrich, 99 %), tetraethyl orthosilicate (TEOS, Sigma-Aldrich, 98 %), phosphotungstic acid hydrated (HPW, Sigma-Aldrich), hydrochloric acid (HCl, J.T. Baker, 36.5–38 %), and N,N-dimethylacetamide (DMAc, Sigma-Aldrich, 99 %) were used as purchased, except DMAc that was distilled and conditioned in flask containing 3 Å molecular sieves. Nitrogen (N2, White Martins, 99.99 %) and Argon (Ar, White Martins, 99.99 %) were used in the synthesis and thermal treatment, respectively.
Methods
Synthesis of the materials
ODA (1.5 mmol) was dissolved under stirring into 8 mL of DMAc, followed by addition of 1.5 mmol of ODPA, starting the formation of poly(amic acid), PAA, PI precursor. 120 min later was added APTES followed by TEOS in 1:4 (APTES:TEOS) wt% ratio, relative to the total mass of ODPA + ODA. After 10 min, 43 μmol of HPW, dissolved into 2 mL DMAc, was added, and the reaction proceeds for more 20 min when the stirring was turned off. The whole process was performed under N2 atmosphere. The solution formed was equally divided into 4 Teflon® molds and taken to a pre-heated MARCONIS’s vacuum oven for 15 h at 333 K to evaporate most part of the solvent. The free-standing PAA hybrid films were supported on alumina plates and thermal treated in a tubular furnace under Argon flow \( \left( {T_{\text{ambient}} \to 100 \;^\circ {\text{C}}/60 \;{ \hbox{min} } \to 200 \;^\circ {\text{C}}/60 \;{ \hbox{min} } \to 300 \;^\circ {\text{C}}/60 \;{ \hbox{min} } \to T_{\text{ambient}} ,{\text{all\; at}}\;3\; ^\circ {\text{C}}\;{ \hbox{min} }^{ - 1} } \right). \)
Characterization
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were collected using a SHIMADZU IRAffinity-1 spectrometer in the range from 4000 to 650 cm−1 with spectral resolution of 4 cm−1 using air as background. Thermogravimetric Analysis (TGA) were carried out under synthetic air in a SHIMADZU TG-50A equipment, in the range from 298 to 1223 K with heating rate of 10 K min−1, using platinum pans. Py-GC/MS experiments were also performed to evaluate the thermal behavior of the synthesized materials. The experiments were carried out using a CDS Analytical 5150 Pyro-probe coupled to an Agilent 7890 gas chromatograph (GC) with an Agilent 5973 N quadrupole mass spectrometer (MS) for the detection of analytes. Samples (~0.5 mg) of each compound were massed into quartz sample tubes. The sample tubes were loaded into the Pt filament coil of the pyro-probe apparatus for pyrolysis. For the determination of the products of degradation, the sample was held at an initial temperature of 60 °C for 0.5 min, and then pyrolysed at a ballistic heating rate from 60 to 1000 °C under a purge flow of helium for a total pyrolysis time of 2.0 min. The pyrolysates were continually transferred from the pyro-probe apparatus to the GC inlet using an inert transfer line (both at 300 °C) with a 10:1 split flow for the total pyrolysis runtime. Agilent DB-1 (30 m × 0.25 mm id, 0.25 microns) analytical column under constant flow of 2.0 mL min−1 with Helium carrier gas was used. The initial GC oven temperature was set at 40 °C for 2.0 min and then ramped at 10 °C min−1 to 300 °C and held for 10 min at the final temperature for a total analysis time of 38.0 min. The MS ion source and quadrupole were 230 and 150 °C, respectively. Total ion chromatograms (TIC) of the pyrolysis products were collected for each run. The collected data in the form of TIC plots were normalized to sample mass and baselined, and individual identified product peaks integrated to yield a relative assessment of the abundance of degradation products. Samples previously coated with gold using a BAL-TEC MED 020 sputter were analyzed by scanning electron microscopy (SEM) in a ZEISS LEO 440 microscope. 31P and 29Si solid-state high-resolution nuclear magnetic resonance with Magic Angle Spinning (MAS-NMR) were performed at room temperature using an INOVA Varian Unity spectrometer with magnetic field of 9.4 T. The samples were cut into pieces, dispersed in KBr, packed into Zirconia rotors of 7 mm diameter, and spun at 5 kHz. The recovery time used was 100 s, and π/2 pulses of 3.5 μs for 29Si and 4 μs for 31P were applied. The chemical shift of 29Si was referenced with solid kaolinite (-91.5 ppm, relative to TMS) and 31P with phosphoric acid 85 % (0 ppm). Impedance Spectroscopy (IS) experiments in alternating current were performed in a SOLARTRON SI 1260 impedance analyzer with 1296A Dielectric Interface coupled to a furnace, in the frequency range from 1 Hz to 1 MHz and temperature range from 298 to 423 K. A previous thermal treatment in the same temperature range was performed on the samples to avoid water interference. Wide-angle X-ray scattering (WAXS) was performed in a bench diffractometer with rotating anode X-ray source operating with Cu Kα and collector time of 120 s. The samples were set at 90° with the beam and at 150 mm from detector. The data were processed using the FIT2D software. X-ray micro-fluorescence (μ-XRF) was performed on beam line D09BXRF using the following experimental set-up: source (storage ring, 1.4 GeV); photon flux (3.9 × 1010 to 2.31 × 1011 photon s−1); polychromatic X-ray beam line (10.5 keV); elliptic shape X-ray beam (18 μm × 9 μm); CANBERRA SL30165 Si(Li) detector with energy dispersive transmission geometry with 165 eV of resolution and energy of 5.6 keV cooled by N2; micro-focalization system moving in x, y, and z directions; as well as two elliptic non-planar mirrors. The analysis was performed in 441 points measured for 5 s and the data treated with PyMca 4.4 software developed at the European Synchrotron Radiation Facility [23].
Results and discussion
Synthesized materials
Flexible and free-standing PI-ORMOSIL-HPW films were obtained from polycondensation reactions and sol–gel process, where the ORMOSIL structure was generated in situ via acid catalysis using HPW. The water present in the nanocluster structure (~11.5 wt%, about 1 HPW/21 H2O) initiated the hydrolysis and condensation process of APTES and TEOS, which was concluded during thermal treatment by using the water released from the imide ring closure. No additional water was added and as the amount of alkoxysilanes increased in the composition, the color of the materials changed from yellow to caramel red. APTES was used as coupling agent by reaction of its amino group with terminal carbonyl groups from PAA establishing NORMOSIL–CPI covalent bonds [24–27] giving rise to a class II hybrid [2]. Moreover, it was expected the ORMOSIL network to prevent HPW leaching by formation of ionic interactions [28, 29] as (≡Si–OH2)+(H2PW12O40)− and [≡Si–(CH2)3–NH3]+(H2PW12O40)−. Table 1 describes the sample formulations and respective codes of the synthesized materials.
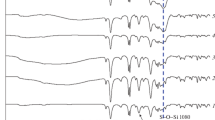
Figure 1 shows the characteristics bands of imide at 1777 and 1711 cm−1, ν as(C=O) and ν s (C=O) stretches, and at 1364 cm−1 (C–N stretch of imide ring). Amide bands from PAA at 1652 cm−1 (C=O stretch) and in 1560–1520 cm−1 range (C–N–H bending) were not observed, confirming full imidization. Other POO bands were also assigned: 1520–1420 cm−1 (breathing mode), 1596 cm−1 (–C=C– stretch), and 2900–3100 cm−1 (C–H stretch); all normal vibration modes from aromatic rings and at 1240–1270 cm−1 were assigned to C–O–C from ether bond [30–32]. The bands in 1000–1220 cm−1 region came from stretching vibrations of siloxane (Si–O–Si) bonds and around 880 cm−1 from stretching vibration of silanol (Si–OH) groups [7], indicating the formation of ORMOSIL network. HPW bands were observed at 1036 cm−1 [ν as(P–O), ν as(W–Ot) (asymmetric coupling), ν as(W–O2c2–W)], 977 cm−1 [ν as(P–O), ν as(W–Ot) (symmetric coupling)], and at 895 cm−1 [ν as(W–O2c1–W), ν as(W–O2c2–W)] [33], indicating the preservation of the HPW structure after the synthesis process.

a ATR-FTMIR spectra in the inset A highlighted the POO bands and in inset B the ORMOSIL and HPW bands and b representation of the HPW structure (Color figure online)
29Si NMR analysis, Fig. 2a, investigated the nature of the ORMOSIL network. The signals in the spectra correspond to the region of Q n and T n groups. Q 4 peak indicates predominance of fully condensed silica (SiO4) from TEOS hydrolysis and condensation, while T 3 corresponds to fully condensed sesquisiloxane with one Si–C bond, from APTES [7, 29]. The local interactions of HPW were also evaluated from 31P NMR, Fig. 2b. The peak at −15.8 ppm corresponds to PO4 in the center of the Keggin structure [21, 28, 34–36]. Synthesis conditions and interaction of HPW with ORMOSIL network could lead to the formation of lacunars resulting in a shift and a line broadening in 31P NMR spectra [20, 21]; nevertheless, there was no change on the local structure of the molecular cluster around the PO4 group. In addition, De Oliveira et al. [34] assigned a chemical shift in this region as belonging to the hexahydrated HPW.

a 29Si and b 31P MAS solid-state NMR spectra (Color figure online)
WAXS evaluated the crystallinity of the materials, and the diffractograms are shown in Fig. 3. Peaks were observed at 6.9°, 19.5°, and 29.9° (as a shoulder), corresponding to d spacing of 12.8, 4.5, and 3 Å, respectively, according to Bragg’s law [37]. Indeed, the peaks are composed by convolution of peaks according to curve fitting performed with the software WINSPEC. It was found peaks centered at 5.2°, 7.2°, 9°, 18.5°, and 31.7°. The diffraction angle at 24.4° is an amorphous halo, evidencing the semicrystalline nature of the hybrids. POO displays basically a large halo centered at 19.5°, typical of non-crystalline material. Perrin et al. [38] and Ferreira-Neto et al. [39] observed such pattern and assigned to the formation of cage-like structures formed by Polyhedral Oligomeric Silsesquioxanes (POSS) where the peaks below 10° are from distances between cages (d 1), around 20° is from diagonal of the cages (d 2), and at 30° are from distances between Si4O4 faces of the cage (d 3).

WAXS scattering intensities
μ-XRF mapping obtained for PATH1 and PATH3, Fig. 4, revealed a homogeneous distribution of silicon (Si) and tungsten (W) implying in good dispersion of ORMOSIL and HPW throughout the films as signal of good compatibility between the inorganic and organic phases. Furthermore, the images revealed an improvement of HPW dispersion in the sample with the highest Si content indicating a strong interaction between HPW and ORMOSIL, as expected. Morphological information obtained from SEM images, Fig. 5, revealed the formation of granular protuberances whose size increase with the ORMOSIL content but does not exceed 1 μm. Nevertheless, such structures are not loosely bounded to the surface, as reflection of the enhanced compatibility provided by APTES and reduced amount of water used in the hydrolysis of the silanes. Jiang et al. [40] reported that the use of stoichiometric amount of water or less increases the compatibility between the organic and inorganic phases by avoiding the formation of large silica agglomerates. Cross-section images also showed similar morphology, in addition to reveal non-porous films. The values of average thickness with respective incertitude were obtained from measurement of thickness in different regions of the analyzed area for each material. Additionally in Fig. 6, the comparison between PAT and PATH2, both prepared with same amount of alkoxysilane precursors, indicated that the introduction of HPW may also lead to more homogeneous PI-ORMOSIL materials, as can be seen by the smaller number and size of the protuberances.

Si and W μ-XRF mapping of a, c PATH1 and b, d PATH3 (Color figure online)

Topological (left) and cross-section (right) SEM images of a, d PATH1; b, e PATH2, and c, f PATH3. In image c is inserted a magnification of the observed protuberances

Topological SEM images of a PAT and b PATH2
Physical properties
Figure 7 shows the thermal stability curves obtained for the materials. All present a weight loss less than 5 wt% up to 773 K. Deeper information on the products of thermal decomposition of the materials was obtained by Py-GC/MS which revealed H2O and CO2 as the main decomposition products, Table 2. They are released from the cleavage of the imide ring, as in agreement with thermal decomposition products found by Cella [41], as well as from further condensation process of ORMOSIL and dehydration of HPW, justifying the higher amount found for PATH2. No significant levels of siloxane degradation products were detected, indicating that any volatile siloxane oligomers were either below of detection limits of the analysis and masked by the siloxane “column bleed” background or it remained confined in the condensed phase on degradation. It also seems evident that the hybrid materials such as PAT and PATH2 are somewhat more thermally stable than POO, since they do not degrade until reaching higher temperatures, and their process of thermal degradation seems to follow different steps until complete decomposition.

TG curves of the materials (Color figure online)
Based on all that was discussed so far, a model was proposed to represent the structure of the PI-ORMOSIL-HPW materials and is displayed in Fig. 8. There is an interpenetrating network (IPN) formed between PI matrix and ORMOSIL network representing the good compatibility between the organic and inorganic parts. It also illustrates the POSS structures evidenced by WAXS, as well as the HPW cluster and its hydration water.

Representation of the possible structure of PI-ORMOSIL-HPW hybrid materials. The size of the compounds is out of proportion for mere illustrative effect
Given the outstanding electrical properties of the components of PI-ORMOSIL-HPW system, such property was also evaluated. Figure 9 shows the dependence of permittivity with the temperature at 1 Hz and 1 MHz. It is observed that the ORMOSIL network increased the permittivity, further enhanced by the introduction of HPW. Regarding to ORMOSIL, according to Jeong et al. [42], the permanent dipole of the silanol group (Si–OH) is responsible for this behavior. The electrons injected from the electrodes can be trapped forming a negative charge product (SiO−), as occurs in Poole–Frenkel conduction [43]. The further increment on permittivity due to introduction of HPW was assigned to its high polarizability, which provided an impact stronger than the ORMOSIL content since PATH1 exhibited permittivity similar to PAT.

Real component of the Electrical Permittivity at a 1 Hz and b 1 MHz
Conclusions
For the first time, free-standing hybrid materials based on PI-ORMOSIL containing HPW were synthesized and their chemical and physical properties explored. FTIR and NMR analyses confirmed the formation of the ORMOSIL network, as well as revealed that the HPW structure was preserved after the synthesis procedure. The use of APTES as a coupling agent and reduced amount of water used in the hydrolysis of the silanes provided good compatibility between the organic and inorganic phases. The ORMOSIL network also improved the dispersion of HPW, leading to the formation of non-porous films and thermally stable up to 773 K. Moreover, ORMOSIL increased the permittivity of the materials which was further enhanced by HPW. Thus, the PI-ORMOSIL-HPW hybrid material system is a potential target to be further studied, aiming at the development of power energy storage devices and other electric and electronic applications.
References
Kickelbick G (2003) Concepts for the incorporation of inorganic building blocks into organic polymers on a nanoscale. Prog Polym Sci 28:83–114. doi:10.1016/S0079-6700(02)00019-9
Sanchez C, Shea KJ, Kitagawa S (2011) Hybrid materials themed issue. Chem Soc Rev 40:696–753. doi:10.1039/C0CS00136H
Liaw D-J, Wang K-L, Huang Y-C et al (2012) Advanced polyimide materials: syntheses, physical properties and applications. Prog Polym Sci 37:907–974. doi:10.1016/j.progpolymsci.2012.02.005
Morikawa A, Yamaguchi H, Kakimoto M, Imaj Y (1994) Formation of interconnected globular structure of silica phase in polyimide-silica hybrid films prepared by the sol-gel process. Chem Mater 6:913–917. doi:10.1021/cm00043a009
Tripathi VS, Kandimalla VB, Ju H (2006) Preparation of ormosil and its applications in the immobilizing biomolecules. Sensors Actuators B 114:1071–1082. doi:10.1016/j.snb.2005.07.037
Mackenzie JD, Bescher EP (1998) Structures, properties and potential applications of ormosils. J Sol Gel Sci Technol 13:371–377
Ahmad Z, Al Sagheer F, Al Arbash A, Ali AAM (2009) Synthesis and characterization of chemically cross-linked polyimide–siloxane hybrid films. J Non-Cryst Solids 355:507–517. doi:10.1016/j.jnoncrysol.2009.01.019
Min C, Wu T, Yang W, Chen C (2008) Functionalized mesoporous silica/polyimide nanocomposite thin films with improved mechanical properties and low dielectric constant. Compos Sci Technol 68:1570–1578. doi:10.1016/j.compscitech.2007.09.021
Kim Y, Kang E, Kwon YS, Korea S (1997) Electrical properties of silica-polyimide composite dielectric thin Ghns prepared via sol-gel reaction and thermal imidization. Synth Met 85:1399–1400. doi:10.1016/S0379-6779(97)80291-3
Tsai M, Whang W (2001) Low dielectric polyimide/poly (silsesquioxane)-like nanocomposite material. Polymer (Guildf) 42:4197–4207. doi:10.1016/S0379-6779(97)80291-3
Deng H, Lin L, Ji M et al (2014) Progress on the morphological control of conductive network in conductive polymer composites and the use as electroactive multifunctional materials. Prog Polym Sci 39:627–655. doi:10.1016/j.progpolymsci.2013.07.007
Gao H, Tian Q, Lian K (2010) Polyvinyl alcohol-heteropoly acid polymer electrolytes and their applications in electrochemical capacitors. Solid State Ionics 181:874–876. doi:10.1016/j.ssi.2010.05.006
Maranas JK (2012) Polymers for energy storage and delivery: polyelectrolytes for batteries and fuel cells. ACS Symp Series. doi:10.1021/bk-2012-1096
Suppes GM, Cameron CG, Freund MS (2010) A polypyrrole/phosphomolybdic acid|poly(3,4-ethylenedioxythiophene)/phosphotungstic acid asymmetric supercapacitor. J Electrochem Soc 157:A1030. doi:10.1149/1.3464802
Cuentas-Gallegos AK, Lira-Cantú M, Casañ-Pastor N, Gómez-Romero P (2005) Nanocomposite hybrid molecular materials for application in solid-state electrochemical supercapacitors. Adv Funct Mater 15:1125–1133. doi:10.1002/adfm.200400326
Venkat N, Dang TD, Bai Z et al (2010) High temperature polymer film dielectrics for aerospace power conditioning capacitor applications. Mater Sci Eng B 168:16–21. doi:10.1016/j.mseb.2009.12.038
López X, Carbó JJ, Bo C, Poblet JM (2012) Structure, properties and reactivity of polyoxometalates: a theoretical perspective. Chem Soc Rev 41:7537–7571. doi:10.1039/c2cs35168d
Coronado E, Gómez-García CJ (1998) Polyoxometalate-based molecular materials. Chem Rev 98:273–296. doi:10.1021/cr970471c
Dolbecq A, Dumas E, Mayer CR, Mialane P (2010) Hybrid organic-inorganic polyoxometalate compounds: from structural diversity to applications. Chem Rev 110:6009–6048. doi:10.1021/cr1000578
Mastikhin VM, Kulikov SM, Nosov AV et al (1990) 1H and 31P MAS NMR studies of solid heteropolyacids and H3PW12O40 supported on SiO2. J Mol Catal 60:65–70. doi:10.1016/0304-5102(90)85068-S
Micek-Ilnicka A (2009) The role of water in the catalysis on solid heteropolyacids. J Mol Catal A 308:1–14. doi:10.1016/j.molcata.2009.04.003
Katsoulis DE (1998) A survey of applications of polyoxometalates. Chem Rev 98:359–388
Solé VA, Papillon E, Cotte M et al (2007) A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra. Spectrochim Acta B 62:63–68. doi:10.1016/j.sab.2006.12.002
Chang C, Chen W (2002) Synthesis and optical properties of polyimide-silica hybrid thin films. Chem Mater 14:4242–4248
Chen Y, Iroh JO (1999) Synthesis and characterization of polyimide/silica hybrid composites. Chem Mater 11:1218–1222
Son M, Ha Y, Choi M-C et al (2008) Microstructure and properties of polyamideimide/silica hybrids compatibilized with 3-aminopropyltriethoxysilane. Eur Polym J 44:2236–2243. doi:10.1016/j.eurpolymj.2008.04.037
Son M, Han S, Han D et al (2008) Organic/inorganic hybrid composite films from polyimide and organosilica: effect of the type of organosilica precursors. Polym Bull 60:713–723. doi:10.1007/s00289-008-0904-z
Bhure MH, Kumar I, Natu AD et al (2008) Phosphotungstic acid on silica with modified acid sites as a solid catalyst for selective cleavage of tert-butyldimethylsilyl ethers. Catal Commun 9:1863–1868. doi:10.1016/j.catcom.2008.03.017
De Oliveira M, de Souza AL, Schneider J, Rodrigues-Filho UP (2011) Local structure and photochromic response in ormosils containing dodecatungstophosphoric acid. Chem Mater 23:953–963. doi:10.1021/cm1022272
Snyder RW, Thomson B, Bartges B et al (1989) FTIR studies of polyimides: thermal curing. Macromolecules 22:4166–4172. doi:10.1021/ma00201a006
Shin TJ, Lee B, Youn HS et al (2001) Time-resolved synchrotron X-ray diffraction and infrared spectroscopic studies of imidization and structural evolution in a microscaled film of PMDA-3,4′-ODA poly(amic acid). Langmuir 17:7842–7850. doi:10.1021/la0108656
Diaham S, Locatelli ML, Lebey T, Malec D (2011) Thermal imidization optimization of polyimide thin films using Fourier transform infrared spectroscopy and electrical measurements. Thin Solid Films 519:1851–1856. doi:10.1016/j.tsf.2010.10.031
Bridgeman AJ (2003) Density functional study of the vibrational frequencies of Keggin heteropolyanions. Chem Phys 287:55–69. doi:10.1016/S0301-0104(02)00978-3
De Oliveira M, Rodrigues-Filho UP, Schneider J (2014) Thermal transformations and proton species in 12-phosphotungstic acid hexahydrate studied by 1 H and 31 P solid-state nuclear magnetic resonance. J Phys Chem C 118:11573–11583. doi:10.1021/jp501887x
Essayem N, Tong YY, Jobic H, Vedrine JC (2000) Characterization of protonic sites in H 3 PW 12 O 40 and Cs 1.9 H 1.1 PW 12 O 40: a solid-state 1 H, 2 H, 31 P MAS-NMR and inelastic neutron scattering study on samples prepared under standard reaction conditions. Appl Catal A 195:109–122
Deleplanque J, Hubaut R, Bodart P et al (2009) 1H and 31P solid-state NMR of trimethylphosphine adsorbed on heteropolytungstate supported on silica. Appl Surf Sci 255:4897–4901. doi:10.1016/j.apsusc.2008.12.031
Bragg HW (1913) The reflection of X-rays by crystals. R Soc 17:43
Perrin FX (2011) Linear and branched alkyl substituted octakis(dimethylsiloxy)octasilsesquioxanes: WAXS and thermal properties. Eur Polym J 47:1370–1382. doi:10.1016/j.eurpolymj.2011.04.004
Ferreira-neto EP, Ullah S, De CF et al (2015) Preparation, characterization and photochromic behavior of phosphotungstic acid-ormosil nanocomposites. Mater Chem Phys 153:410–421
Jiang L, Wang W, Wei X et al (2007) Effects of water on the preparation, morphology, and properties of polyimide/silica nanocomposite films prepared by sol–gel process. J Appl Polym Sci 104:1579–1586. doi:10.1002/app.25692
Celia JA (1992) Degradation and stability of polyimides. Polymer Degradation 36:99–110
Jeong S, Kim D, Lee S et al (2007) Organic thin-film transistors using thin ormosil-based hybrid dielectric. Thin Solid Films 515:7701–7705. doi:10.1016/j.tsf.2006.11.143
Chisca S, Musteata VE, Sava I, Bruma M (2011) Dielectric behavior of some aromatic polyimide films. Eur Polym J 47:1186–1197. doi:10.1016/j.eurpolymj.2011.01.008
Acknowledgements
The authors acknowledge the financial support from CNPq (Grant. 142910/2010-4) and FAPESP (CEPID 2013/07793-6). This work was also performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. WAXS (proposal: GAR-14024), and μ-XRF (proposal: XAFS1-14257) data were collected in the Brazilian Synchrotron Light Source (LNLS) facilities, and we especially thank Dr. Matheus Cardoso and Dr. Carlos Alberto Pérez for their careful assistance on training on the beamlines and data manipulation. We also thank Prof. Dr. Antonio Carlos Hernandes and Technician Geraldo Frigo from Grupo de Crescimento de Cristais e Materiais Cerâmicos of the IFSC for the collaboration in the TGA analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ferreira, F.A.S., Amaral, T., Ysnaga, O.A.E. et al. Structure–property relationship of new polyimide–organically modified silicate–phosphotungstic acid hybrid material system. J Mater Sci 51, 4815–4824 (2016). https://doi.org/10.1007/s10853-016-9773-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-016-9773-2