
Abstract
Six new dioxadiaza macrocyclic Schiff bases with 18-, 19-, 20- and 22-membered rings have been synthesized from diamine and dialdehyde building blocks. An X-ray diffraction study and molecular modelling (at the HF/6-31G(d,p) level of theory) have been performed to determine their molecular geometries and structural configurations. According to the theoretical studies the most stable conformers of the macrocycles are those, in which the lone pair electrons localized on the nitrogen atoms are exo-oriented, and those on the oxygen atoms are endo-oriented with respect to the cavity. In order to obtain more proper metal ion receptors with all lone pair electrons directed into the cavity, two Schiff base macrocycles have been reduced to the corresponding diamines, of which one could be isolated only as a very stable oxaazacyclophane borane adduct.
Graphical Abstract
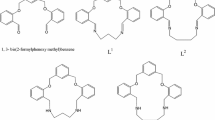
Six dioxadiaza macrocyclic Schiff bases with 18-, 19-, 20- and 22-membered rings were synthesized and their molecular geometries and structural configurations were determined by X-ray diffraction and molecular modelling. In addition, two of these macrocycles were reduced to the corresponding diamines, of which one could be isolated only as a very stable oxaazacyclophane borane adduct.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The interest in exploring new metal ion complexes with macrocycles containing nitrogen and oxygen donor atoms has been increasing, because they result very attractive for supramolecular and coordination chemistry. Particularly important are systems that are related to bioactive molecules such as metalloporphyrins, corrins and antibiotics [1, 2].
A convenient route for the construction of polyoxaaza macrocycles is Schiff base condensation, a method that has been developed already for the synthesis of one of the first macrocycles employed for metal complexation. Varying the number, position and orientation of the donor atom lone pair electrons within the polyoxaaza macrocycles as well as the cavity size, highly selective receptors for a variety of metal ions can be designed [2]. On the other hand, through the proper choice of building blocks for the formation of the Schiff base macrocycles it is possible to obtain compounds with attractive and sophisticated structures, which could be employed for creating new materials such as chemical sensors, catalysts, liquid crystals, and pharmaceutical substances [3–5].
Many synthetic protocols for the preparation of tetra- [6–11], penta- [12–32], hexa-, and polydentate Schiff base macrocyclic compounds involve metal ions as templating agents that orient all or part of the ligand donor atoms into an optimal conformation for the ring closure [1, 2, 33–41]. However, the template synthetic method can have the disadvantage that stable metal-complexed macrocycles are generated, thus making the preparation of the metal-free ligand difficult. Therefore, during the last two decades, considerable effort has been made to develop metal-free methods for the synthesis of macrocycles [2, 42], in particular to overcome the common problem that the desired product is obtained in very low yield.
We are interested in new synthetic [43–45] and semi-synthetic [46–50] macrocycles containing oxygen and nitrogen atoms in order to study their coordinating capacity towards metal cations as well as their ability to act as hosts for neutral and charged guest atoms and molecules. We report herein on the synthesis and structural characterization of six new Schiff base dioxadiaza macrocycles and some of their hydrogen-reduced amine analogues.
Experimental section
All reagents for synthesis were purchased from commercial suppliers and used without further purification. NMR spectra were recorded on 400 MHz Bruker AVANCE 400 and Varian UNITY INOVA spectrometers. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane and J values are given in Hertz. Mass spectra were recorded on a high resolution Jeol MStation 700 mass spectrometer using the FAB+ technique. Infrared spectra were recorded on a Perkin-Elmer Spectrum GX FTIR spectrophotometer. Melting points are uncorrected.
X-ray crystallography
Compound 3 was crystallized from ethanol and the oxaazacyclophane-borane adduct 10 from chloroform. X-ray diffraction studies were performed at 293(2) K on a BRUKER-AXS APEX diffractometer with a CCD area detector (MoK α , λ = 0.71073 Å, monochromator: graphite). Frames were collected via ω/ϕ-rotation at 10 s per frame (SMART) [51]. The measured intensities were reduced to F 2 and corrected for absorption with SADABS (SAINT-NT) [52]. Structure solution, refinement, and data output were carried out with the SHELXTL-NT software package [53]. Non-hydrogen atoms were refined anisotropically, while hydrogen atoms were placed in geometrically calculated positions using a riding model. The N–H and B–H hydrogen atoms in compound 10 have been localized by difference Fourier maps. The crystals of compound 10 were weak and contained solvent molecules (CHCl3), thus giving somewhat elevated R values.
Details of the crystal structure determination and solution refinement of 3 (C28H22N2O2): colourless rectangular prism with dimensions 0.52 × 0.20 × 0.16 mm3, M = 418.48 g mol−1, orthorhombic, P2 1 2 1 2 1 , a = 9.4764(13), b = 10.7696(14), c = 21.506(3) Å, V = 2,194.8(5) Å3, Z = 4, ρ calc = 1.266 mg/m3, μ = 0.080 mm−1, 2430 independent reflections measured, 1719 reflections observed with I ≥ 2σ(I), parameters refined 289, R = 0.078, R w = 0.1338, goodness of fit = 1.158. Details of the crystal structure determination and solution refinement of 10 (C28H40B2N2O2 · CHCl3): [50] colourless rectangular prism with dimensions 0.35 × 0.23 × 0.18 mm3, M = 577.61 g mol−1, triclinic, P-1, a = 10.3152(10), b = 10.9207(11), c = 15.6942(16) Å, α = 110.262(2), β = 98.274(2), γ = 101.512(2), V = 1,580.9(3) Å3, Z = 2, ρ calc = 1.213 mg/m3, μ = 0.318 mm−1, 5548 independent reflections measured, 3824 reflections observed with I ≥ 2σ(I), parameters refined 367, R = 0.1228, R w = 0.2416, goodness of fit = 1.253.
Crystallographic data for 3 and 10 have been deposited at the Cambridge Crystallographic Data Center as CCDC No. 663938 and No. 650762 respectively. Copies of the data can be obtained free of charge upon application to CCDC, 12 Union Road, Cambridge CB2 1EZ, U.K. (e-mail: deposit@ccdc.cam.ac.uk).
Computational methods
The geometry of each stationary point was fully optimized using the Gaussian 03 software package [54] with the double zeta 6-31G(d,p) basis set. All stationary points were characterized as minima as proved by an analysis of the harmonic vibrational frequencies, using analytical second derivatives. All of the structures were visualized with the Chemcraft 1.5 program [55].
Dialdehyde 1
This compound was prepared by a modified procedure to that reported previously [56, 57] A solution of salicylaldehyde (1.00 g, 8 mmol) in anhydrous DMF (5 mL) was added to a suspension of K2CO3 (in excess) in the same solvent (25 mL). The suspension was kept under nitrogen and stirred at 80 °C during 30 min. Then, a solution of α,α′-dibromo-m-xylene (1.08 g, 4 mmol) in anhydrous DMF (5 mL) was added slowly to the mixture and the resulting suspension was then stirred at 80 °C. The reaction was monitored by TLC (heptane:acetone 60:40) until a single product was observed and the starting material disappeared. The suspension was filtered in order to remove the potassium salt. The filtrate was evaporated in vacuum to dryness and acetone was added until a precipitate appeared. A light-brown solid was obtained after filtration, acetone washing, and drying in vacuum. 
Yield: 95%; m.p. 112–114° (from acetone); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 10.56 (s, 2 H, H-1), 7.86 ppm (dd, 3 J(H–H) = 7.6 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.57 ppm (d, 3 J(H–H) = 7.6 Hz, 2 H; H-10), 7.54 ppm (dt, 4 J(H–H) = 1.6 Hz, 3 J(H–H) = 8 Hz, 2 H; H-5), 7.45 ppm (t, 3 J(H–H) = 7.6 Hz, 1 H; H-11), 7.46 ppm (s, 1 H; H-12), 7.08 ppm (t, 3 J(H–H) = 7.6 Hz, 2 H; H-4), 7.05 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 5.23 ppm (s, 4 H; H-8); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 189.8 (C-1), 161.0 (C-7), 136.9 (C-9), 136.1 (C-10), 129.4 (C-12), 128.9 (C-3), 127.3 (C-11), 126.2 (C-5), 125.3 (C-2), 121.3 (C-6), 113.1 (C-4), 70.4 (C-8); IR (KBr, cm−1): ν = 3014 (w), 2935 (w), 2878 (m), 2765 (w), 1668 (s), 1600 (s), 1482 (s), 1459 (s), 1403 (m), 1369 (m), 1303 (s), 1288 (s), 1233 (s), 1185 (m), 1164 (m), 1104 (m), 1008 (s), 898 (m), 846 (m), 821 (m), 790 (m), 773 (m), 711 (s); FAB-MS: m/z (%) 347 ([M+], 25), 307 (21), 289 (13), 225 (45), 224 (13), 197 (16), 195 (11), 154 (100), 136 (64), 121 (9), 106 (6); Elem anal. Calcd for C22H18O4 (346.38): C, 76.29; H, 5.24; O, 18.48; found: C, 75.96; H, 5.24.
Dialdehyde 2
Compound 2 was prepared in a similar manner to that described for dialdehyde 1 using α,α′-dibromo-m-xylene (1.08 g, 4 mmol) and salicylaldehyde (1.00 g, 8 mmol). The product was obtained as white crystals in higher yield (see below) to that reported previously in the literature (84%) [57]. 
Yield: 98%; m.p. 188–190 °C (from acetone); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 10.57 (s, 2 H; H-1), 7.87 ppm (dd, 3 J(H–H) = 8.0 Hz, 4 J(H–H) = 1.8 Hz, 2 H; H-3), 7.56 ppm (dt, 3 J(H–H) = 8.0 Hz, 4 J(H–H) = 1.8 Hz, 2 H; H-5), 7.50 ppm (s, 4 H, H-10), 7.07 ppm (t, 3 J(H–H) = 8.0 Hz, 2 H; H-4), 7.05 ppm (d, 3 J(H–H) = 8.0 Hz, 2 H; H-6), 5.24 ppm (s, 4 H; H-8); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 189.9 (C-1), 161.1 (C-7), 136.4 (C-9), 136.1 (C-5), 128.8 (C-3), 127.9 (C-10), 125.4 (C-2), 121.3 (C-4), 113.2 (C-6), 70.3 (C-8); IR (KBr, cm−1): ν = 2936 (w), 2850 (m), 2763 (m), 1687 (s), 1599 (s), 1488 (s), 1458 (s), 1401 (m), 1376 (s), 1307 (s), 1286 (s), 1242 (s), 1191 (s), 1164 (s), 1102 (s), 1009 (s), 994 (s), 866 (m), 851 (m), 803 (m), 779 (s); FAB-MS: m/z (%) 345 ([M+], 28), 329 (1), 307 (22), 289 (14), 273 (2), 260 (2), 242 (2), 225 (9), 219 (5), 192 (100), 165 (4). Elem anal. Calcd for C22H18O4 (346.38): C, 76.29; H, 5.24; O, 18.48; found: C, 76.10; H, 5.24.
Macrocycle 3
Compound 3 was synthesized from 1 (0.16 g, 0.46 mmol) and 1,3-benzenediamine (0.05 g, 0.46 mmol). The reaction mixture was stirred in ethanol under high pressure during 24 h at 70 °C. A considerable amount of yellow crystals appeared during the reaction, which were removed by filtration and then washed with ethanol and dried in vacuum. 
Yield: 31%; m.p. 311–314 °C (from ethanol); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 9.07 (s, 2 H; H-1), 8.22 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.94 ppm (s, 1 H, H-12), 7.49 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-5), 7.35 ppm (m, 3 H, H-10, H-11), 7.21 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-15), 7.17 ppm (t, 3 J(H–H) = 8 Hz, 1 H; H-16), 7.12 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 7.10 ppm (t, 3 J(H–H) = 8 Hz, 2 H; H-4), 6.90 ppm (t, 4 J(H–H) = 2 Hz, 1 H; H-13), 5.09 ppm (s, 4 H, H-8); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 158.5 (C-1), 157.2 (C-7), 153.6 (C-14), 136.6 (C-9), 132.7 (C-5), 128.9 (C-16), 128.8 (C-3), 128.4 (C-11), 127.0 (C-10), 124.3 (C-12), 121.5 (C-4), 121.2 (C-15), 112.1 (C-2), 112.0 (C-13), 108.2 (C-6), 71.2 (C-8); IR (KBr, cm−1): ν = 3063 (w), 3006 (w), 2928 (w), 2876 (w), 1615 (s), 1599 (s), 1574 (s), 1478 (s), 1456 (s), 1368 (s), 1299 (s), 1265 (s), 1226 (s), 1162 (s), 1145 (m), 11034 (s), 1006 (s), 964 (m), 864 (m), 763 (s); FAB-MS: m/z (%) 419 ([M+], 15), 391 (7), 346 (5), 341 (5), 329 (3), 307 (100), 289 (38), 273 (7), 260 (5), 235 (5), 219 (9). Elem anal. Calcd for C28H22N2O2 (418.49): C, 80.35; H, 5.30; N, 6.69; O, 7.66; found: C, 80.45; H, 5.29; N, 6.92.
Macrocycle 4
Compound 4 was prepared in an analogous manner to that described for macrocycle 3, using 1 (0.16 g, 0.46 mmol) and 1,3-propanediamine (0.342 g, 0.46 mmol). The product was obtained as a white solid. 
Yield: 39%; m.p. 95–197 °C (from ethanol); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 8.83 (s, 2 H; H-1), 8.02 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.92 ppm (s, 1 H; H-12), 7.38 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-5), 7.28 ppm (m, 3 H; H-10 and H-11), 7.03 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 7.01 ppm (t, 3 J(H–H) = 8 Hz, 2 H; H-4), 5.20 ppm (s, 4 H; H-8), 3.59 ppm (t, 4 H; H-13), 2.25 ppm (m, 2 H; H-14); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 161.8 (C-1), 157.0 (C-7), 137.5 (C-9), 131.8 (C-5), 128.2 (C-3), 127.6 (C-11), 126.0 (C-12), 125.0 (C-10), 124.0 (C-2), 121.2 (C-4), 112.0 (C-6), 69.3 (C-8), 57.2 (C-13), 29.7 (C-14); IR (KBr, cm−1): ν = 3680 (w), 3654 (w), 3446 (m), 2911 (w), 2886 (w), 1640 (s), 1598 (s), 1489 (m), 1449 (s), 1371 (m), 1290 (m), 1241 (s), 1044 (m), 752 (m); FAB-MS: m/z (%) 385 ([M+], 100), 369 (2), 341 (2), 309 (9), 307 (57), 289 (26), 273 (5), 260 (3), 242 (3), 235 (4), 219 (5), 207 (2), 202 (1). Elem anal. Calcd for C25H24N2O2 (384.47): C, 78.08; H, 6.29; N, 7.29; O, 8.34; found: C, 78.48; H, 6.29; N, 7.27.
Macrocycle 5
Compound 5 was prepared in an analogous manner to that described for macrocycle 3, using 1 (0.16 g, 0.46 mmol) and 1,4-butanediamine (0.037 g, 0.46 mmol). The product was obtained as a pale yellow solid. 
Yield: 44%; m.p. 97–102 °C (from ethanol); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 8.80 (s, 2 H; H-1), 7.97 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.75 ppm (s, 1 H; H-12), 7.41 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-5), 7.31 ppm (m, 3 H; H-10 and H-11), 6.97 ppm (t, 3 J(H–H) = 8 Hz, 2 H; H-4), 6.93 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 5.16 ppm (s, 4 H; H-8), 3.67 ppm (t, 4 H; H-13), 1.26 ppm (q, 4 H; H-14); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 157.6 (C-1), 156.7 (C-7), 137.6 (C-9), 131.6 (C-5), 128.4 (C-11), 127.3 (C-3), 126.5 (C-12), 125.3 (C-10), 124.7 (C-2), 121.1 (C-4), 111.9 (C-6), 69.5 (C-8), 60.5 (C-13), 27.0 (C-14); IR (KBr, cm−1): ν = 3331 (w), 3029 (w), 2938 (s), 2839 (s), 1638 (s), 1599 (s), 1581 (m), 1488 (s), 1456 (s), 1372 (s), 1342 (m), 1298 (s), 1286 (s), 1242 (s), 1156 (s), 1111 (m), 1017 (s), 999 (s), 789 (m), 749 (s); FAB-MS: m/z (%) 399 ([M+], 7), 329 (29), 277 (22), 222 (8), 208 (6), 174 (37), 172 (24), 132 (45), 104 (100), 78 (54), 77 (28), 65 (6).
Macrocycle 6
Compound 6 was prepared in an analogous manner to that described for compound 3, using 2 (0.16 g, 0.46 mmol) and 1,3-benzenediamine (0.05 g, 0.46 mmol). The product was obtained as a yellow solid. 
Yield: 53%; m.p. 161–164 °C (from ethanol); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 8.64 (s, 2 H; H-1), 8.10 (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.48 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-5), 7.34 ppm (t, 3 J(H–H) = 8 Hz, 1 H; H-14), 7.33 (t, 3 J(H–H) = 8 Hz, 2 H; H-4), 7.24 ppm (s, 4 H; H-10), 7.15 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-13), 7.10 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 6.51 ppm (t, 4 J(H–H) = 2 Hz, 1 H; H-11), 5.14 ppm (s, 4 H; H-8); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 159.9 (C-1), 156.6 (C-7), 153.0 (C-12), 137.4 (C-9), 132.9 (C-5), 129.6 (C-10), 129.3 (C-14), 127.5 (C-3), 126.0 (C-4), 122.9 (C-13), 121.9 (C-11), 114.7 (C-2), 105.9 (C-6), 73.6 (C-8); IR (KBr, cm−1): ν = 3057 (w), 3034 (w), 2942 (w), 2878 (w), 1614 (s), 1598 (s), 1573 (s), 1483 (s), 1452 (s), 1420 (m), 1360 (s), 1261 (s), 1160 (s), 1138 (m), 1100 (s), 1038 (m), 985 (s), 955 (s), 893 (m), 855 (m), 748 (s); FAB-MS: m/z (%) 419 ([M+], 100), 418 (16), 314 (3), 307 (83), 289 (42), 230 (9), 219 (12), 207 (40), 190 (14); Elem anal. Calcd for C28H22N2O2 (418.49): C, 80.36; H, 5.30; N, 6.69; O, 7.65; found: C, 79.96; H, 5.26; N, 6.78.
Macrocycle 7
Compound 7 was prepared in an analogous manner to that described for compound 3 using 2 (0.16 g, 0.46 mmol) and 1,4-butanediamine (0.037 g, 0.46 mmol). The product was obtained as a white solid. 
Yield: 30%; m.p. 175–178 °C (from ethanol); NMR characterization studies could not be performed because of insolubility of the compound in most common solvents; IR (KBr, cm−1): ν = 3066 (w), 3027 (w), 2930 (s), 2854 (m), 2830 (m), 1644 (s), 1598 (s), 1577 (m), 1518 (m), 1438 (s), 1456 (s), 1374 (s), 1340 (m), 1298 (s), 1281 (s), 1250 (s), 1234 (s), 1160 (m), 1036 (s), 1008 (s), 750 (s); FAB-MS: m/z (%) 399 ([M+], 31), 391 (4), 371 (3), 345 (6), 307 (100), 289 (52), 273 (9), 235 (10), 196 (4), 192 (12), 165 (10). Elem anal. Calcd. for C26H26N2O2 (398.5): C, 78.36; H, 6.58; N, 7.03; O, 8.03; found: C, 78.32; H, 6.68; N, 7.09.
Macrocycle 8
Compound 8 was prepared in an analogous manner to that described for compound 3 using 2 (0.16 g, 0.46 mmol) and 1,6-hexanediamine (0.053 g, 0.46 mmol). The product was obtained as a white powder. 
Yield: 86%; m.p. 164–166 °C (from ethanol); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 8.72 (s, 2 H; H-1), 7.82 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.43 ppm (s, 4 H; H-10), 7.38 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-5), 7.04 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 7.01 ppm (t, 3 J(H–H) = 8 Hz, 2 H; H-4), 5.16 ppm (s, 4 H; H-8), 3.61 ppm (t, 4 H; H-11), 1.71 ppm (m, 4 H; H-12) 1.38 ppm (m, 4 H; H-13); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 157.9 (C-1), 156.7 (C-7), 131.6 (C-9), 127.8 (C-5), 127.7 (C-3), 127.5 (C-10), 126.1 (C-2), 121.5 (C-4), 113.1 (C-6), 70.3 (C-8), 61.1 (C-11), 29.6 (C-12), 26.2 (C-13); IR (KBr, cm−1): ν = 3398 (w), 3071 (w), 3034 (w), 2926 (m), 2880 (m), 2824 (m), 1911 (w), 1802 (w), 1688 (w), 1633 (s), 1598 (s), 1579 (s), 1519 (w), 1486 (s), 1452 (s), 1371 (s), 1296 (s), 1284 (s), 1239 (s), 1158 (s), 1105 (s), 1042 (s), 1003 (s), 799 (s), 749 (s); FAB-MS: m/z (%) 427 ([M+], 100), 426 (9), 322 (3), 307 (18), 289 (12), 273 (2), 257 (2), 242 (2), 222 (5), 208 (7), 204 (12), 202 (7), 165 (4). Elem anal. Calcd for C28H30N2O2 (426.55): C, 78.84; H, 7.09; N, 6.57; O, 7.50; found: C, 78.49; H, 6.99; N, 6.72.
Macrocycle 9
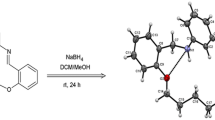
The diamine 9 was synthesized from the reaction of Schiff base 3 (0.050 g, 0.11 mmol) and LiBH4 (0.005 g, 0.11 mmol) in dry THF (10 mL) as solvent. The mixture was first stirred at 60 °C during 7.5 h and then overnight at room temperature. Then, a little amount of water was added, the solvents were evaporated in vacuum and a chloroform-water extraction was performed. A white solid was recovered after the organic phase had been evaporated. 
Yield: 84%; m.p. 210–212 °C (from chloroform); 1H NMR (400 MHz, DMSO-d 6, 25 °C, TMS): δ = 7.64 (s, 1 H; H-12), 7.43 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-3), 7.29 ppm (m, 3 H; H-10 and H-11), 7.09 ppm (dt, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-5), 6.93 ppm (t, 3 J(H–H) = 8 Hz, 1 H; H-16), 6.87 ppm (t, 3 J(H–H) = 8 Hz, 2 H; H-4), 6.78 ppm (d, 3 J(H–H) = 8 Hz, 2 H; H-6), 6.07 ppm (dd, 3 J(H–H) = 8 Hz, 4 J(H–H) = 2 Hz, 2 H; H-15), 5.85 ppm (t, 4 J(H–H) = 2, 1 H; H-13), 5.18 ppm (s, 4 H; H-8), 4.33 ppm (s, 4 H; H-1), 1.55 ppm (s, 2 H; N–H); 13C NMR (400 MHz, DMSO-d 6, 25 °C; δ, ppm): δ = 155.9 (C-7), 149.2 (C-14), 138.1 (C-9), 129.5 (C-16), 128.7 (C-11), 128.6 (C-3), 127.9 (C-5), 127.8 (C-2), 126.5 (C-12), 125.2 (C-10), 120.9 (C-4), 111.9 (C-6), 103.5 (C-15), 97.2 (C-13), 69.6 (C-8), 43.0 (C-1); IR (KBr, cm−1): ν = 3433 (s), 3034 (w), 2917 (m), 1612 (s), 1590 (s), 1519 (s), 1486 (s), 1458 (s), 1361 (m), 1306 (s), 1219 (s), 1155 (m), 1062 (m), 1035 (s), 810 (s), 924 (m), 810 (s), 778 (s), 755 (s); FAB-MS: m/z (%) 422 ([M+], 20), 420 (17), 401 (48), 341 (33), 327 (59), 281 (100), 267 (41), 207 (74), 191 (45).
Oxaazacyclophane borane adduct 10
Details of the synthesis and characterization of this compound were recently reported by our group [50]. 
Results and discussion
Following the two-step protocol shown in Scheme 1, the Schiff base macrocycles 3–8 were synthesized. Reaction of salicylaldehyde with α,α′-dibromo-m-xylene and α,α′-dibromo-p-xylene gave the corresponding dialdehydes 1 and 2, which were then condensed with diamines having a nitrogen separation proper for the ring closure. In the case of the m-xylylene dialdehyde derivative, 1,3-benzenediamine, 1,3-propanediamine and 1,4-butanediamine were chosen, while for the p-xylylene dialdehyde derivative 1,3-benzenediamine, 1,4-butanediamine and 1,6-hexanediamine have been employed, in order to obtain a series of compounds that might be suitable for the complexation of at least one metal ion within the cavity. In such a way dioxadiaza macrocycles having ring sizes varying from 18 to 22 members have been obtained (18 for 3 and 4, 19 for 5 and 6, 20 for 7 and 22 for 8).

Synthetic protocol used for the synthesis of compounds 1–8
The formation of the Schiff base compounds could be evidenced by the IR spectra that showed intense bands at ν = 1615, 1640, 1638, 1614, 1644 and 1633 cm−1 for compounds 3–8, respectively, which correspond to the stretching vibration of the C=N bonds. The 1H NMR spectra showed single signals for the protons corresponding to the –HC=N– group at 9.07, 8.83, 8.80, 8.64 and 8.72 ppm for compounds 3–6 and 8, respectively. Compound 7 is insoluble in most of the common organic solvents and could therefore not be characterized via this spectroscopic method. In the 13C NMR spectra, the signal corresponding to the imine group was observed at δ = 158.5, 161.8, 157.6, 159.9 and 157.9 ppm for 3–6 and 8, respectively. FAB+ mass spectrometry gave peaks at m/z = 419, 385, 399, 419, 399 and 427 that correspond to the [M + H]+ ions of compounds 3–8, respectively.
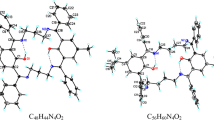
Since the new Schiff bases contain two nitrogen and two oxygen donor atoms, a theoretical study was carried out to evaluate the orientation of the lone pair electrons on the donor atoms in order to know if the synthesized compounds are suitable to coordinate metal atoms. We explored all possibilities taking into account that the imine C=N bond can have E and Z configuration, and found that the E isomers have lower energy when compared to the Z isomers. There are two different configurations for the E isomers, which differ in the orientation of the lone pair electrons located at the nitrogen atoms. The lowest energy was found for those structures where the lone pair electrons on the nitrogen atoms are oriented towards the periphery of the macrocycle (exo), while the lone pair electrons of the oxygen atoms are oriented into the cavity (endo) (Fig. 1). Structures 3, 4 and 6 belong to the point group C s , while 5, 7 and 8 belong to the point group C2.

Geometry-optimized molecular structures for compounds 3–8 using ab initio calculations at the HF/6-31G(d,p) level of theory
This is supported by the X-ray structure of compound 3. From Fig. 2 it can be seen that the orientation of the donor atoms is in agreement with the calculated structure shown in Fig. 1.

Perspective view of the molecular structure of macrocycle 3
Apparently, in this arrangement the annular tension in the central heterocycle is minimized and there is an attractive interaction between the imine hydrogen atoms and the oxygen atoms within the macrocycle. The NBO (Natural Bond Orbitals) analysis suggests, for the case of structure 3, that the lone pair electrons of the oxygen atoms are delocalized mainly into the aromatic ring of the salicylidene fragment: n O → π*C=Carom (Fig. 3a). The stabilization energy E(2) for this interaction is 39.04 kcal/mol. The C–H⋯O intraannular interaction is much smaller, E(2) = 0.94 kcal/mol, and results from a delocalization of the n O → σ*H–C type (Fig. 3b).

Orthogonal NBOs for the oxygen lone pair at the HF/6-31G(d,p) level of theory: a nπO interacting with the π*C=Carom antibond, and b n O → σ*H–C
The theoretical study discussed before suggests that the lone pair electrons on the nitrogen atoms are exo-oriented, which reduces the possibility for coordination to a metal atom located within the cavity. However, an option to achieve the reorientation of the nitrogen lone pair electrons is reduction of the C=N bond. In order to explore this possibility, Schiff bases 3 and 8 were reduced with LiBH4 to the corresponding diamines. While reduction of compound 3 gave the expected free diamine 9 in good yield (84%), the reduction of 8 provided the oxaazacyclophane-borane adduct 10 that shows considerable stability in air and towards hydrolysis [50]. Both compounds have been characterized by spectroscopic methods (IR, 1H and 13C NMR) and FAB+ mass spectrometry. In the case of compound 10 it was also possible to establish the molecular structure by X-ray crystallography (Fig. 4). As we can see from Fig. 4, in this case at least one nitrogen lone pair electron can be directed into the cavity.

Perspective view of the molecular structure of compound 10 [50]
Conclusions
The above discussion has shown that Schiff base macrocyclic structures can be formed easily in two steps of synthesis, which then can be reduced in order to increase the affinity toward metal atoms. Diimine 8 showed a different behavior in the reaction with LiBH4, when compared to its analogue 3, and resulted in the formation of a very stable oxaazacyclophane-borane adduct.
Preliminary experiments with metal salts, such as copper, nickel and zinc perchlorate, have shown that some of the macrocyclic hosts described herein are capable of metal complexation. On the other hand, it is important to emphasize that diamines prepared from the corresponding Schiff bases could also serve as receptors for anions, in particular if the nitrogen atoms are protonated. We are currently working on complexation studies of the macrocyclic hosts and on the preparation of further macrocyclic Schiff bases and their hydrogen-reduced amine analogues with improved structures and additional binding sites, in order to generate hosts with a more specific capability for guest complexation in organic and aqueous media.
References
Radecka-Paryzek, W., Patroniak, V., Lisowski, J.: Metal complexes of polyaza and polyoxaaza Schiff base macrocycles. J. Coord. Chem. Rev. 249, 2156–2175 (2005)
Borisova, N.E., Reshetova, M.D., Ustynyuk, Y.A.: Metal-free methods in the synthesis of macrocyclic Schiff bases. Chem. Rev. 107, 46–79 (2007) (references therein)
Barba, V., Villamil, R., Luna, R., Godoy-Alcántar, C., Höpfl, H., Beltran, H.I., Zamudio-Rivera, L.S., Santillan, R., Farfan, N.: Boron macrocycles having a calix-like shape. Synthesis, characterization, X-ray analysis, and inclusion properties. Inorg. Chem. 45, 2553–2561 (2006)
Ma, C.T.L., MacLachlan, M.J.: Supramolecular assembly and coordination-assisted deaggregation of multimetallic macrocycles. Angew. Chem., Int. Ed. 44, 4178–4182 (2005)
Chand, D.K., Bharadwaj, P.K.: Synthesis of designed hetero-polytopic cryptands through Schiff base condensation. Tetrahedron 53, 10517–10522 (1997)
Radecka-Paryzek, W.: The Scandium(III) ion as a template for the synthesis of hexaaza quadridentate macrocyclic ligand. Inorg. Chim. Acta. 35, L349–L350 (1979)
Radecka-Paryzek, W.: Magnesium(II) and zinc(II) complexes containing a 14-membered hexaaza macrocyclic ligand. Inorg. Chim. Acta. 34, 5–8 (1979)
Radecka-Paryzek, W.: The synthesis and characterization of the macrocyclic and ring-opened complexes formed in the reaction of the lanthanides with 2,6-diacetylpyridine and hydrazine. Inorg. Chim. Acta. 52, 261–268 (1981)
Radecka-Paryzek, W.: The yttrium ion as a template in the synthesis of the macrocyclic and acyclic complexes formed in the reactions with 2,6-diacetylpyridine and hydrazine. Polyhedron 8, 1217–1221 (1989)
Radecka-Paryzek, W., Luks, E.: The dioxouranium(VI) ion as a template for the synthesis of hexaaza tetradentate macrocyclic ligand. Polyhedron 13, 899–901 (1994)
Radecka-Paryzek, W., Patroniak-Krzyminiewska, V.: The template synthesis and characterization of the new macrocyclic Schiff-base complexes of scandium (III) and yttrium (III) ions. Pol. J. Chem. 69(1), 1–4 (1995)
Bollinger, J.E., Roundhill, D.M.: Complexation of the uranyl ion with the aminomethylenediphosphonates MAMDP and AMDP. Inorg. Chem. 33, 6421–6424 (1994)
Chrisstoffels, L.A.L., De Jong, F.F., Reinhoudt, D.N.: Facilitated transport of salts by neutral anion carriers. Chem. Eur. J. 6, 1376–1385 (2000)
Harrowfield, J.: Calixarenes as potential uranophiles: What are the consequences of ring size variation? Gaz. Chim. Ital. 127, 663–671 (1997)
Kemp, T.J., Read, P.A.: Fast atom bombardment studies of amino acid and peptide complexes of the uranyl ion: ligand attachment, dissociation and fragmentation. Inorg. Chim. Acta. 241, 105–110 (1996)
Kuroki, J., Ishihara, K., Hanaki, N., Ohara, S., Yamamoto, H.: Metal-templated macrolactamization of triamino and tetramino esters. Facile synthesis of macrocyclic spermidine and spermine alkaloids, (S)-(+)-dihydroperiphylline, (±)-buchnerine, (±)-verbacine, (±)-verbaskine, and (±)-verbascenine. Bull. Chem. Soc. Jpn. 71, 1221–1230 (1998)
Nijenhuis, W., Van Doorn, A.R., Reichwain, A.M., De Jong, F., Reinhoudt, D.N.: Urea transport by macrocyclic carriers through a supported liquid membrane. J. Am. Chem. Soc. 113, 3607–3608 (1991)
Patroniak, V., Kubicki, M., Mondry, A., Lisowski, J., Radecka-Paryzek, W.: Pentaaza macrocyclic ytterbium(III) complex and solvent controlled supramolecular self-assembly of its dimeric μ-2:2 peroxo-bridged derivatives. Dalton Trans. 3295–3304 (2004)
Patroniak-Krzyminiewska, V., Litkowska, H., Radecka-Paryzek, W.: Metal-template synthesis and characterization of a nitrogen–oxygen donor Schiff base macrocyclic system. Monatsh. Chem. 130, 243–247 (1998)
Radecka-Paryzek, W., Litkowska, H.: Rare earth macrocyclic complexes derived from spermine. J. Alloys Compd. 300–301, 435–438 (2000)
Radecka-Paryzek, W., Patroniak, V., Kubicki, M.: The template synthesis and characterization of pentaaza macrocyclic complexes of rare earth elements. The crystal structure of the 2,14-dimethyl-3,6,10,13,19-pentaazabicyclo[13.3.1]nonadeca-1(19),2,13,15,17-pentaene-dichlorolutetium(III)perchlorate. Polyhedron 22, 2773–2779 (2003)
Radecka-Paryzek, W., Patroniak, V., Kubicki, M.: First example of template synthesis of pentaaza macrocyclic ytterbium(III) complex and solvent-controlled supramolecular self-assembly of its dimeric μ-η2:η2 peroxo-bridged derivative. Inorg. Chem. Commun. 7, 455–458 (2004)
Radecka-Paryzek, W., Patroniak-Krzyminiewska, V.: Yttrium(III) complexes of pentadentate Schiff base macrocyclic ligands with N3O2 and N5 set of donor atoms. Polyhedron 14, 2059–2062 (1995)
Radecka-Paryzek, W., Patroniak-Krzyminiewska, V.: Azaoxa macrocyclic and acyclic complexes of lanthanides. Collect. Czech. Chem. Commun. 63, 363–370 (1998)
Radecka-Paryzek, W., Patroniak-Krzyminiewska, V., Litkowska, H.: The template synthesis and characterization of the yttrium and lanthanide complexes of new 19-membered pentadentate azaoxa macrocycle. Polyhedron 17, 1477–1480 (1998)
Shinkai, S., Shiramama, Y., Satoh, H., Manabe, O., Arimura, T., Fujimoto, K., Matsuda, T.: Selective extraction and transport of UO2 2+ with calixarene-based uranophiles. J. Chem. Soc., Perkin Trans. 2, 1167–1171 (1989)
Stradling, G.N.: Decorporation of actinides: a review of recent research. J. Alloys Compd. 271, 72–77 (1998)
Tabushi, I., Yoshizawa, A.: Kinetic investigation of uranyl-uranophile complexation. 1. Macrocyclic kinetic effect and macrocyclic protection effect. Inorg. Chem. 25, 1541–1546 (1986)
Tarafder, M.T.H., Ali, M.A., Wee, D.J., Azahari, K., Silong, S., Crouse, K.A.: Complexes of a tridentate ONS Schiff base. Synthesis and biological properties. Trans. Met. Chem. 25, 456–460 (2000)
Thuery, P., Nierlich, M., Souley, B., Asfari, Z., Vicens, J.: Complexation of a hexameric uranium(VI) cluster by p-benzylcalix[7]arene. J. Chem. Soc., Dalton Trans. 2589–2594 (1999)
Castelli, V.V.A., Cort, A.D., Mandolini, L., Reinhoudt, D.N., Schiaffino, L.: Catalysis of the addition of benzenethiol to 2-cyclohexen-1-ones by uranyl-salophen complexes: a catalytic metallocleft with high substrate specificity. Chem. Eur. J. 6, 1193–1198 (2000)
Youhnovski, N., Drandarov, K., Guggisberg, A., Hesse, M.: Macrocyclic spermine alkaloids from verbascum: isolation, structure elucidation, and syntheses of the (E/Z)-isomeric pairs (S)-verbasikrine/(S)-isoverbasikrine and (S)-verbamekrine/(S)-isoverbamekrine. Helv. Chim. Acta. 82, 1185–1194 (1999)
Alexander, V.: Design and synthesis of macrocyclic ligands and their complexes of lanthanides and actinides. Chem. Rev. 95, 273–342 (1995)
Brooker, S.: Complexes of thiophenolate-containing Schiff-base macrocycles and their amine analogues. Coord. Chem. Rev. 222, 33–56 (2001)
Brooker, S.: Some copper and cobalt complexes of Schiff-base macrocycles containing pyridazine head units. Eur. J. Inorg. Chem. 41, 2535–2547 (2002)
Collinson, S.R., Fenton, D.E.: Metal complexes of bibaracchial Schiff base macrocycles. Coord. Chem. Rev. 148, 19–40 (1996)
Curtis, N.F.: In: McCleverty, J.A., Meyer, J.T. (eds.) Comprehensive Coordination Chemistry II, vol. 1, Chap. 20. Elsevier, Pergamon, Oxford, UK; San Diego, CA, USA (2004)
Fenton, D.E., Vigato, P.A.: Macrocyclic Schiff base complexes of lanthanides and actinides. Chem. Soc. Rev. 17, 69–90 (1988)
Hernández-Molina, R., Mederos, A.: In: McCleverty, J.A., Meyer, J.T. (eds.) Comprehensive Coordination Chemistry II, vol. 1, Chap. 19. Elsevier, Pergamon, Oxford, UK; San Diego, CA, USA (2004)
Okawa, H., Furutachi, H., Fenton, D.E.: Heterodinuclear metal complexes of phenol-based compartmental macrocycles. Chem. Rev. 174, 51–75 (1998)
Vallarino, L.M.J.: In: Gschneidner, K.A. Jr., Eyring, L. (eds.) Handbook on the Physics and Chemistry of Rare Earths, vol. 15, Chap. 104. Elsevier, Amsterdam (1991)
Singh, N., Hundal, M.S., Hundal, G., Martinez-Ripoll, M.: Zinc templated synthesis—a route to get metal ion free tripodal ligands and Lariat Coronands, containing Schiff bases. Tetrahedron 61, 7796–7806 (2005)
Höpfl, H., Farfán, N.: New macrocyclic oligoboronates. J. Organomet. Chem. 547, 71–77 (1997)
Höpfl, H., Sánchez, M., Barba, V., Farfán, N., Rojas, S., Santillán, R.: Synthesis and study of monomeric and dimeric boronates by spectroscopic methods and X-ray crystallography. Inorg. Chem. 37, 1679–1692 (1998)
Barba, V., Höpfl, H., Farfán, N., Santillan, R., Beltran, H.I., Zamudio-Rivera, L.S.: Boron–nitrogen macrocycles: a new generation of calix[3]arenes. Chem. Commun. 2834–2835 (2004)
Lara, K.O., Godoy-Alcántar, C., Eliseev, A.V., Yatsimirsky, A.K.: Recognition of α-amino acids derivatives by N,N′-dibenzylated S,S-(+)-tetrandrine. Org. Biomol. Chem. 2, 1712–1718 (2004)
Lara, K.O., Godoy-Alcántar, C., Eliseev, A.V., Yatsimirsky, A.K.: Ester cleavage by S,S-(+)-tetrandrine derivative bearing thiol groups. Arkivoc vi, 293–306 (2005)
Moreno-Corral, R.A., Lara, K.O.: Complexation studies of nucleotides by tetrandrine derivatives bearing anthraquinone and acridine groups. Supramol. Chem. 20(4), 427–435 (2008)
Wong-Molina, A., Lara, K.O., Sánchez, M., Burboa, M.G., Gutiérrez-Millán, L.E., Marín, J.L., Valdez, M.A.: Interaction of calf thymus DNA with a cationic tetrandrine derivative at the air–water interface. J. Biomed. Nanotechnol. 4(1), 52–61 (2008)
Reyes-Márquez, V., Lara, K.O., Sánchez, M., Gálvez-Ruiz, J.C.: Unconventional hydrogen and dihydrogen bonded supramolecular array of a 2,6-dioxa-9,16-diaza-1,3(1,2),4(1,4)-tribenzenacycloheptadecaphane-borane adduct. Arkivoc v, 115–123 (2008)
SMART: Bruker molecular analysis research tool, version 5.618. Bruker AXS, Madison, WI (2000)
SAINT + NT, version 6.04. Bruker AXS, Madison, WI (2001)
SHELXTL-NT, version 6.10. Bruker AXS, Madison, WI (2000)
Gaussian 03, Revision C.02, Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, Jr., J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C., Pople, J.A. Gaussian, Inc., Wallingford, CT (2004)
Zhurko, G. A.: Chemcraft (2008). http://www.chemcraftprog.com
Reddy, D., Chandrashekar, T.K.: Short-chain basket handle porphyrins: synthesis and characterisation. J. Chem. Soc., Dalton Trans. 4, 619–625 (1992)
Kim, J., Lindoy, L.F., Ahn, T.-H., Choi, G.-S.: New N2O2- and N2O3-macrocycle ligands incorporating p-xylyl groups. Synth. Commun. 34, 3653–3659 (2004)
Acknowledgments
This work was supported by Programa de Mejoramiento del Profesorado de la Secretaría de Educación Pública (PROMEP) and Consejo Nacional de Ciencia y Tecnología (CONACyT) under grant CB-54675. Viviana Reyes-Márquez thanks CONACyT for a postgraduate scholarship. We thank Dr. Juan Carlos Gálvez-Ruiz for helpful discussions on the oxaazacyclophane-borane adduct.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reyes-Márquez, V., Sánchez, M., Höpfl, H. et al. Synthesis and structural characterization of 18-, 19-, 20- and 22-membered Schiff base macrocycles. J Incl Phenom Macrocycl Chem 65, 305–315 (2009). https://doi.org/10.1007/s10847-009-9588-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-009-9588-8