
Abstract
Two new phenol-based metal-free macrocyclic Schiff bases, cyclo-bis{2-[benz(N-propan-1,3-diyl)imidoyl][6-benzimidoyl][4-methyl]phenol} and cyclo-bis{2-[benz(N-butan-1,4-diyl)imidoyl][6-benzimidoyl][4-tert-butyl]phenol} have been synthesized and their structures determined by single crystal X-ray crystallography. The DFT geometry optimization calculations were performed to compare experimental and theoretical results. A comparison of the dihedral angles between mean planes of the central phenolato rings and peripheral phenyl rings in the crystal with the DFT theoretical calculations has been included for each molecule. Electronic transitions have been predicted by DFT molecular orbital calculations and compared with experimental absorption spectral data.
Graphic Abstract
A one-pot synthesis, crystal structure and theoretical calculations of 20- and 22-membered macrocyclic ligands are reported.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Macrocyclic Schiff bases have potential applications as models for protein metal binding sites, synthetic ionophores, magnetic exchange phenomena, therapeutic reagents, cyclic antibiotics, host–guest interaction and in phase transfer catalysis [1, 2]. A large variety of [1 + 1] and [2 + 2] macrocyclic ligands have been synthesised by a template procedure in the presence of suitable metal ions [3]. There are various reports of metal-free macrocycles [4,5,6] available in the literature involving cyclodextrines [7,8,9,10,11,12], shape persistent macrocycles [13,14,15,16,17,18,19,20] and crown ethers, spherands and cryptands [21,22,23,24,25,26]. We have previously reported the synthesis and crystal structure of a neutral tetraiminodiphenol macrocycle with a C2 lateral chain, cyclo-bis{2-[benz(N-ethan-1,3-diyl)imidoyl][6-benzimidoyl] [4-methyl]phenol}, cyclo-[LMeH2] (n = 2) (I) [27]. As part of our study on phenol-based Schiff bases [28,29,30,31,32], we herein report the synthesis and crystal structures of cyclo-bis{2-[benz(N-propan-1,3-diyl)imidoyl][6-benzimidoyl][4-methyl]phenol}, Cyclo-[LMeH2] (n = 3) (II) and cyclo-bis{2-[benz(N-butan-1,4-diyl)imidoyl][6-benzimidoyl][4-tert-butyl]phenol}, Cyclo-[Lt-BuH2] (n = 4) (III), and theoretical calculations of I, II and III (Scheme 1) with C2, C3 and C4 lateral chains.

Schematic diagrams of structures I, II and III
Experimental
Synthesis of Metal-Free Macrocyclic Ligands
The metal-free macrocyclic Schiff base ligands were synthesised by the Schiff base condensation of 4-R-2,6-dibenzoyl phenol (where R = Me, But) with propane-1,3-diamine/butane-1,4-diamine in dry THF under inert atmosphere. The synthetic scheme for (II) and (III) is given in Scheme 2. The detailed synthetic procedure and characteristics of the macrocyclic ligands are given below.

Synthesis of II and III
Cyclo-bis{2-[benz(N-propan-1,3-diyl)imidoyl][6-benzimidoyl][4-methyl]phenol}
Cyclo-[LMeH2] (II)
To a solution of 4-methyl-2,6-dibenzoylphenol (0.632 g, 2.00 mmol) in dry THF (30 mL), a solution of propane-1,3-diamine (0.158 g, 2.00 mmol) in dry THF (30 mL) was added over approximately 30 min via a dropping funnel. The solution turned yellow while it was constantly stirred for 2 h at room temperature. The resulting solution was allowed to stand for one week, during which yellow crystals of II formed. The crystals were filtered off, washed with cold ethanol and dried in air. Yield: 60 mg, 60%; mp 478 K.
Anal Calc for (II), C48H44N4O2 (%): C, 81.33; H, 6.26; N, 7.90. Found: C, 81.60; H, 6.15; N, 8.12. ESI–MS: m/z: 709 [MH]+. 1H NMR (300 MHz, CDCl3) δ 1.73 (s, 4H, mid-CH2); δ 2.21 (s, 6H, CH3); δ 3.29 (m, 8H, NCH2); δ 6.61–7.85 (m, 24H, C6H2 & C6H5); δ 16.27 (s br, 2H, OH). 13C NMR (75 MHz, CDCl3): δ 20.4 (CH3); δ 48.9 (CH2); 119.4, 124.1, 125.0, 127.8, 128.2, 129.4, 131.4, 133.0, 133.8, 134.2, 136.6, 137.9, 139.4 (aromatic carbon); 160.4 (C···O); 174.7 (C=N). UV–Vis (nm) (ε/L mol−1 cm−1) (CH3CN): 429 (217), 378 (3564), 254 (4785).
Cyclo-bis{2-[benz(N-butan-1,4-diyl)imidoyl][6-benzimidoyl][4-tert-butyl]phenol}
Cyclo-[Lt-BuH2] (III)
To a solution of 4-tert-butyl-2,6-dibenzoylphenol (0.716 g, 2.00 mmol) in dry THF (30 mL), a solution of butane-1,4-diamine (0.172 g, 2.00 mmol) in dry THF (30 mL) was added over approximately 30 min via a dropping funnel. The solution turned yellow and was stirred for 2 h at room temperature. The resulting solution was allowed to stand for 1 week, during which yellow crystals of III formed. The crystals were filtered off, washed with cold ethanol and dried in air. Yield: 80 mg, 65%; mp 483 K.
Anal Calc for (III), C56H60N4O2 (%): C, 81.91; H, 7.37; N, 6.82. Found: C, 82.20; H, 7.20; N, 6.60. ESI-MS: m/z: 820 [MH]+. 1H NMR (300 MHz, CDCl3) δ 1.29 (s, 18H, CH3); 1.72 (s, 8H, mid-CH2); δ 3.56 (m, 8H, NCH2); δ 6.81–7.65 (m, 24H, C6H2 & C6H5); δ 16.30 (s br, 2H, OH). 13C NMR (75 MHz, CDCl3) δ 31.2 (Me); δ 34.4 (CMe3); δ 30.4, 48.5 (CH2); 124.4, 128.7, 129.6, 131.0, 133.0, 133.5, 134.6, 138.2, 139.0 (aromatic carbon); 164.6 (C···O); 175.6 (C=N). UV–Vis (nm) (ε/L mol−1 cm−1) (CH3CN): 430 (220), 380 (3600), 258 (4800).
X-ray Structure Determination
Diffraction data were collected with a Rigaku Oxford Diffraction Gemini Ruby CCD diffractometer using graphite monochromated, Mo-Kα radiation (wavelength 0.710732 Å) for II and Cu-Kα radiation (wavelength 1.54178 Å) for III at 123 K. Absorption corrections were made by multi-scan and analytical methods using the CrysAlisPro software [33]. The structures were solved by direct methods using SHELXT [34] and all of the non-hydrogen atoms were refined anisotropically by full-matrix least-squares on F2 using SHELXL2018 [35]. Hydrogen atoms were placed in their calculated positions and then refined using the riding model. The programs SHELXTL and XP were used for graphics [36]. X-ray data collection and structure refinements are given in Table 1.
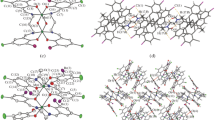
ORTEP diagrams [37] of II and III are displayed in Figs. 1 and 2 while the crystal packing is shown in Figs. 3 and 4, respectively. Geometrically optimized structures of the molecules for I, II and III are shown in Fig. 5. Bond lengths and bond angles are all within expected ranges [38]. Hydrogen bonds and weak intermolecular interactions for II and III are listed in Table 2.

ORTEP diagram for II, C48H44N4O2, showing the atom numbering scheme with 30% probability ellipsoids. Dashed lines indicate O–H···N hydrogen bonds generating S(6) ring motifs

ORTEP diagram for III, C56H60N4O2, showing the atom numbering scheme with 30% probability ellipsoids. Dashed lines indicate both O–H···N (IIIA) and N–H···O (IIIB) intramolecular hydrogen bonds showing tautomers

Packing diagram for II, C48H44N4O2 viewed along the c axis showing intramolecular O–H···N hydrogen bonds

Packing diagram for III, C56H60N4O2, viewed along the b axis showing both O–H···N and N–H···O intramolecular hydrogen bonds

Optimized Geometries for I, II and III
Computational Details
Theoretical DFT molecular orbital calculations (WebMo Pro [39] with the Gaussian-03 program package [40]) employing the B3LYP (Becke three parameter Lee–Yang–Parr exchange correlation functional), which combines the hybrid exchange functional of Becke [41] with the gradient correlation functional of Lee, Yang and Parr [42] and the 6-31 G(d) basis set [43] were performed for II and III. No solvent corrections were made in these calculations. Starting geometries were taken from X-ray refinement data. The optimized results in the free molecule state are, therefore, compared to those in the crystalline state. Experimentally determined oscillator strengths (f) were determined by use of an equation relating them to the molar decadic absorption coefficient (ε) (f = 4.32 × 10−9·εmax·∆ɷ1/2) [44,45,46]. The molar decadic absorption coefficient measures the intensity of the optical absorption at a given wavelength. Deconvolution of the spectra to obtain the εmax and ∆ɷ1/2 values was carried out by the Origin program [47]. Discrepancies between the experimental and calculated band centres and band intensities exist. All calculations were using default convergence criteria.
Density Functional Theory (DFT) Calculations
Bond angles and bond distances observed in the crystal structure match well those obtained by the geometry optimized DFT calculations at the B3LYP 6-31G(d) level. That is, the differences between the two values are within normal ranges and are generally consistent with bond lengths and angles for similar types of compounds. In addition, a comparison of the angles between the mean planes of the central phenolato and peripheral phenyl rings in the crystal and with the DFT geometry optimized calculation have been included in a discussion of the structural aspects for each molecule. From a DFT molecular orbital calculation for each compound, surface plots for the two highest occupied molecular orbitals (HOMO, HOMO−1) and three lowest unoccupied molecular orbitals (LUMO, LUMO+1, LUMO+2) are displayed to provide a visual evidence of the molecular orbitals involved in the spectroscopic electronic energy transitions examined. Based on correlation of the energies of these HOMO–LUMO frontier surfaces to the UV–Vis absorption spectra, electronic excitation transition predications are suggested.
Results and Discussion
Structural Study of (II) and (III)
In (II) and (III), the 20-membered and 22-membered macrocyclic ligands, respectively are located on a crystallographic inversion centre with half of the molecule in the asymmetric unit (Figs. 1, 2). The phenolic hydrogen forms a hydrogen bond with donor–acceptor distances of 2.53 Å and 2.50 Å to the imine nitrogen (Table 2) in II and III, respectively. The aliphatic methylene diimine groups in II are in a gauche conformation as shown by the N–C–C–C/C–C–C–N torsion angles of ca. 66°. The mean plane of the central phenolato ring (C1–C6) makes dihedral angles of 82.58(5)° and 72.88(5)° II and 82.05(15)° and 73.97(15)° III, respectively, with the peripheral phenyl rings (C9–C14) and (C19–C24) II and (C8–C13) and (C19–C24) III. Both the peripheral phenyl rings are inclined by an angle of 24.55(5)° and 18.02 (16)° in II and III, respectively. The mean phenolic C–O distance is slightly longer in II [1.3435(17) Å] than in III [1.335(4) Å].
In II, the molecular conformation is stabilised by classical intramolecular O–H···N hydrogen bonds generating an S(6) ring motif (Fig. 3). In addition, weak intermolecular C–H···O interactions involving the methylene chain and a phenolato oxygen atom (Table 2) [48] are found to influence the crystal packing.
In III, the macrocyclic ligand possesses both OH and NH tautomeric character in its molecular structure and is stabilized by both O–H···N and N–H···O classical intramolecular hydrogen bonds (Fig. 4, Table 2). The molecules are further linked by weak C–H···O and C–H···π interactions forming a three-dimensional network.
The observation of tautomeric character in (III) (Scheme 3) may be predicted due to the presence of tert-butyl groups that are more electron releasing than the methyl groups in (I) and (II).

Tautomeric form of (III)
Theoretical Study of I, II and III
The synthesis and structure of I was previously reported [27]. However, DFT calculations were performed to enable comparisons between I, II and III.
DFT Optimized Geometries
DFT geometry optimization calculations for I, II and III (Fig. 5) resulted in bond lengths and bond angles similar to those observed from experimental data and any small differences were unremarkable. The largest discrepancies in the bond length of III were in the range of 0.049 to 0.064 Å while the largest differences between the experimental and theoretical values for bond angles were between 4.50° and 6.75°. The dihedral angle between the mean planes of the central phenolato ring and peripheral phenyl rings are calculated to be 86.0(7)° and 76.4(4)° in I and 66.3(1)° and 67.1(9)° in II, slightly more twisted from those observed in the crystal structure with 82.99(8)° and 88.20(8)° for I and 82.58(5)° and 72.88(5)° for II. The calculated dihedral angle between the mean planes of the peripheral phenyl rings are 17.5(1)° I and 11.8(6)° II, compared the experimentally obtained values of 17.36(8)° and 24.55(5)°, respectively. In III, the dihedral angles between the mean planes of the central phenolato ring (C1–C6) and peripheral phenyl rings (C8–C13 and C19–C24) calculated to be 81.1(1)° and 73.4(3)°, are almost same as those observed in the crystal structure at 82.05(15)° and 73.97(15)°, respectively. The computed dihedral angle between the mean planes of the two peripheral phenyl rings is 26.1(1)°, an increase of 8.08(1)° compared to the experimental result. These changes support the suggestion that weak C–H···O intermolecular interactions involving the methylene chain and phenolato oxygen atom in concert with intramolecular O–H···N and N–H···O hydrogen bonds and weak C–H···π interactions involving the tert-butyl group and benzimidoyl ring all play a role in the crystal packing of the molecule (Table 2).
Electronic Absorption Spectra and DFT Molecular Orbital Calculations
Calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals for I, II and III are shown in Fig. 6 and Table 3.

Calculated frontier molecular orbitals for I, II and III
In the HOMO−1 and HOMO for I, II and III, the electronic clouds are distributed primarily on both central phenolato rings. In the LUMO and LUMO+1 the electronic clouds are delocalized primarily on the central phenolato ring and imino group of the benzimidoyl ring for I and II and on the imido nitrogen on both sides in III. In the LUMO+2 they are dispersed primarily on the imido nitrogen and benzimidoyl rings of the opposite side in I and II while in III they are located only the benzimidoyl ring of one side of the ring.
The observed experimental absorption spectra show three band envelopes with λmax values located at 419, 368 and 250 nm I, 429, 378 and 254 nm II and 430, 380 and 258 nm III, respectively. Electronic transitions are generally paired between the various molecular orbitals of the ground and excited states corresponding to these three band envelopes as indicated in Table 3. Therefore, the first absorption band envelope at 419–430 nm is assigned to contributions primarily from HOMO → LUMO and HOMO−1 → LUMO. The second absorption band at 368–380 nm is assigned to overlapping contributions from HOMO → LUMO+1 and HOMO−1 → LUMO+1. The third absorption band at 250–258 nm is assigned to overlapping contributions from HOMO → LUMO+2 and HOMO−1 → LUMO+2.
It is evident further that electron transitions among frontier molecular orbitals are corresponding to the transitions of phenolate oxygen to phenyl ring (PhO− → Ph), n → π* (C=N) and π → π* (phenyl ring) transitions.
In the present investigation, the HOMO–LUMO gap: 4.67 eV (I) > 4.54 eV (II) > 3.76 eV (III) is well correlated with an increasing methylene chain in (I), (II) and (III).
Summary and Conclusions
The crystal and molecular structure of two metal-free tetraiminodiphenol macrocyclic ligands containing C3 and C4 lateral chains, were determined. The frontier molecular orbitals of tetraiminodiphenols with the C2, C3 and C4 lateral chain were studied with density functional theory (DFT-B3LYP 6-31 G(d)) geometry optimization and molecular orbital calculations. Correlation between the calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals to the electronic excitation transitions from the absorption spectrum of each compound were determined. In each compound, the DFT molecular orbital calculation, supported by a geometry optimization calculation confirmed that the excitation energies of the surfaces of the frontier molecular orbitals from the HOMO−1 and HOMO to LUMO, LUMO+1 and LUMO+2 electronic excitations closely match the λmax values of the absorption spectra in overlapping contributions from three excitations within each band envelope. Further, the decreasing HOMO–LUMO energy gap correlates very well with the number of methylene spacers in the diamine fragment of the Schiff base. Because of the presence of tert-butyl groups in III which are more electron releasing then the methyl groups in I and II, the former appears to be tautomer. In the crystal structures of two compounds, it has been determined that hydrogen bonds and/or weak C—H···O and C—H···π intermolecular interactions play a role in the crystal packing of each molecule. This is consistent changes in the mean planes between the rings within the asymmetric unit comparing the crystal structures and to the molecular structures obtained from density functional theory (DFT) geometry optimization calculations.
Supporting Information
X-ray crystallographic files, in CIF format, for the structure determination of (II) (1844201) and (III) (1844202) have been deposited with the Cambridge Crystallographic Data Centre, CCDC:26091. Copies of this information may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ (fax: +44-1223-336033; email: deposit@ccdc.cam.uk or at http://www.ccdc.cam.ac.uk).
References
Alexander V (1995) Chem Rev 95:273–342
Chu Z, Huang W, Wang L, Gou S (2008) Polyhedron 27:1079–1092
Tamburini S, Vigato PA (2004) Coord Chem Rev 248:1717–2128
Tian Y, Tong J, Frenzen G (1999) Sun J-Yu. J Org Chem 64:1442–1446
Atkins AJ, Black D, Blake AJ, Becerra AM, Parsons S, Ramirez LK, Schroder M (1996) Chem Commun 4:457–464
Tian YQ, Tong J (1997) Chin Chem Lett 8:107–110
Biwer A, Antranikian G, Heinzle E (2002) Appl Microbiol Biotechnol 59:609–617
Del Valle EMM (2010) Process Biochem 31:1033–1046
Thatiparti TR, Shoffstall AJ, Von Recum HA (2010) Biomaterials 31:2335–2347
Marcolino VA, Zanin GM, Durrant LR, Benassi MDT, Matioli GJ (2011) Agric Food Chem 59:3348–3357
De Oliveira V E, Almeida EWC, Castro HV, Edwards HGM, Dos Santos HF, de Oliveira LFC (2011) J Phys Chem A 115:8511–8519
Brusseau ML, Wang X (1997) Wang W -Z. Environ Sci Technol 31:1087–1092
Hoger S (2004) Chem Eur J 10:1320–1329
Zhang W, Moore JS (2005) J Am Chem Soc 127:11863–11870
Zang L, Che Y, Moore JS (2008) Acc Chem Res 41:1596–1608
Akine S, Taniguchi T, Nabeshima T (2001) Tetrahedron Lett 42:8861–8864
Gallant AJ, MacLachlan MJ (2003) Angew Chem Int Ed 42:5307–5310
Hui JK-H, MacLachlan MJ (2006) Chem Commun 23:2480–2482
Guieu S, Crane AK, MacLachlan MJ (2011) Chem Commun 47:1169–1171
Chen Z, Guieu S, White NG, Lelj F, MacLachlan MJ (2016) Chem Eur J 22:17657–17672
Pedersen CJ (1988) Angew Chem Int Ed Engl 27:1021–1027
Pedersen CJ (1967) J Am Chem Soc 86:7017–7036
Dietrich B, Lehn JM, Sauvage JP (1969) Tetrahedron Lett 10:2885–2888
Lehn J (1988) Angew Chem Int Ed Engl 27:89–112
Cram DJ, Kaneda T, Helgeson RC, Lein GM (1979) J Am Chem Soc 101:6752–6754
Cram DJ (1986) Angew Chem Int Ed Engl 25:1039–1134
Asatkar AK, Verma VK, Jain TA, Singh R, Gupta SK, Butcher RJ (2011) Acta Crystallogr E67:o2724–o2725
Gupta SK, Anjana C, Butcher RJ, Sen N (2010) Acta Crystallogr E66:m1531–m1532
Gupta SK, Anjana C, Sen N, Jasinski JP, Golen JA (2012) J Chem Crystallogr 42:960–967
Ganaie JA, Kumar J, Butcher RJ, Jasinski JP, Gupta SK (2016) J Chem Crystallogr 46:93–104
Gupta SK, Anjana C, Sen N, Butcher RJ, Jasinski JP, Golen JA (2015) Polyhedron 89:219–231
Gupta SK, Sen N, Ganaie JA, Butcher RJ, Jasinski JP (2017) J Coord Chem 70:3147–3170
Rigaku Oxford Diffraction (2015) CrysAlis Pro. The Woodlands
Sheldrick GM (2015) Acta Crystallogr A 71:3–8
Sheldrick GM (2015) Acta Crystallogr C 71:3–8
Sheldrick GM (2008) Acta Crystallogr A 64:112–118
Johnson CK (1976) ORTEP II. Report ORNL-5138. Oak Ridge National Laboratory, Oak Ridge
Allen FH, Kennard O, Watson DG, Brammer L, Orpen A, Taylor RJ (1987) Chem Soc Perkin Trans 2:S1–S19
Schmidt JR, Polik WF (2007) WebMO Pro, version 8.0.01e; WebMO, LLc: Holland. http://www.webmo.net
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision D.01. Gaussian Inc., Wallingford
Becke AD (1998) Phys Rev A 38:3098
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Hehre WJ, Random L, Schleyer PR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Pearl GM, Zerner MC, Broo A, McKelvey J (1998) J Comput Chem 19:781–796
Holland JP, Barnard PJ, Bayly SR, Dilworth JR, Green JC (2009) Inorg Chim Acta 362:402–406
Guillaumont D, Nakamura S (2000) Dyes Pigm 46:85–92
Origin 8.0. Origin Lab (2007) Origin Lab Corporation, Northampton
Bernstein J, Davis RE, Shimoni L, Chang NL (1995) Angew Chem Int Ed Engl 34:1555–1573
Acknowledgements
Financial support from the Government of India through the Department of Science and Technology [Project no. SR/S1/IC-38/2007] and the University Grants Commission [Project no. F.37-500/2009 (SR)] is gratefully acknowledged. RJB wishes to acknowledge the National Science Foundation for funds to purchase the diffractometer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ganaie, J.A., Sen, N., Butcher, R.J. et al. Synthesis, Crystal Structures, Density Functional Theory (DFT) Calculations and Molecular Orbital Calculations of Two New Metal-Free Macrocyclic Schiff Bases Derived from 2,6-Dibenzoyl-4-alkylphenol and Diamines. J Chem Crystallogr 50, 400–409 (2020). https://doi.org/10.1007/s10870-019-00812-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-019-00812-6