
Abstract
Imitation gold electroplating of a citric acid system was studied using citric acid as the main complexing agent, copper sulphate and sodium stannate as the main salts and NaOH as the buffering agent in the electroplating solution. The effects of different electroplating solutions on the microtopography, composition, phase structure and colour of the electroplated layer were evaluated by SEM, EDS, XRD and photo analysis. Different electroplating solutions were analysed and compared by electrochemical analysis, UV–Vis, FTIR and NMR spectroscopy to analyse the mechanism of the electrochemical reaction. The results showed that at pH 9.5, copper and tin ions exist in the form of Cu2Cit2H-24− and Sn(OH)62−, respectively, in the solution. The cathode had only a single deposition peak at − 1.3 V vs. Hg|HgO. Therefore, the above two complex ions can be reduced and codeposited at the same voltage to form Cu, [Cu, Sn], Cu6Sn5, Cu10Sn3 and Cu4O3 phases. A gold-coloured, dense, low-Sn binary Cu–Sn alloy coating containing 13.72% Sn was finally obtained. These results provide a theoretical basis for the electrodeposition of Cu–Sn alloys.
Graphic abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cu–Sn plating will appear in silver-white, gold, copper and black tones and is used for surface finishing of ornaments. Amongst them, the imitation gold plating Cu–Sn alloy is mainly decorated in gold colour. Imitation gold plating is the most widely used method in the industry for electroplating copper–tin (Cu–Sn) alloys, and its cyanide electroplating system was the earliest to be industrialised [1]. The plating solution is stable and easy to maintain. However, toxic cyanide can pollute the environment and harm operators. As people’s environmental awareness has gradually increased, electroplated Cu–Sn alloys have gradually turned to a non-cyanide era. In recent years, the non-cyanide electroplating system in the industry has mainly included phosphate [2], gluconate [3], 1-hydroxyethylidene-1,1-diphosphonic acid [4], EDTA [5], sorbitol [6, 7], the mesylate system [8] and the citrate system [9]. The characteristics of each system are as follows: in a pyrophosphate solution [10] system, Meng et al. deposited a Cu–Sn alloy coating from a pyrophosphate-based electrolyte by electrodeposition methods of DC, PC and PRC. The frequency and duty cycle remarkably affected the hydrogen permeation behaviour during electrodeposition, and the degree of hydrogen permeation increased as the average deposition current density of the electrodeposition process increased. In a gluconate solution system, Survila et al. [3] explained the reaction mechanism of the complexing agent and the metal ion and analysed the mechanism and kinetics of the cathodic reaction process by electrochemical testing of the plating solution. Barbano et al. [5] studied the process conditions for an EDTA system to achieve Cu–Sn codeposition under acidic conditions. Electrochemical analysis and other methods were used to compare the effects of different halogens on the reduction of metal ions; Cu–Sn deposits with different metal contents were obtained. Almeida et al. [11] studied the electroplating alloy of a sorbitol system and found that the colour of the coating was affected by changing the proportion of the main salt and the content of the complexing agent. Volov et al. [12] studied the electrodeposition of Cu–Sn alloy thin films in acidic copper sulphate and tin sulphate electrolytes. The content of Sn in the Cu–Sn coating was found to be a function of the applied current density, rotational speed and Sn ion concentration.
The citric acid system is the most environmentally friendly plating solution, and its cost is low. The plating solution in the citric acid system is simple, stable and easy to maintain. The drug is not toxic, and the process is simple [13, 14]. Therefore, in imitation gold plating, the citric acid system has broad application prospects [15, 16]. Heidari et al. [17] found that Cu and Sn ions formed complexes with citric acid at different pH values, and Cu–Sn alloy codeposition was achieved using boric acid and hexadecyltrimethylammonium bromide as additives. Gougaud et al. [18] compared sodium citrate and tartaric acid as complexing agents to electroplate low-tin copper–zinc–tin alloys in weakly acidic systems. Meudre et al. [19] found that in acid Cu–Sn electrolytes, the presence of gelatine affected the crystal size and morphology of the Cu–Sn deposits. The codeposition of Cu and Sn was facilitated by the closer deposition potentials of Cu and Sn in the presence of gelatine on bare Pt with an improved morphology, specifically exhibiting far less dendritic growth.
Until now, although many different cyanide-free plating systems and their improvement measures have been reported, only the effects are explained. The mechanism of action has not been elaborated, which restricts the further development of imitation gold plating research. In this work, the citric acid system was used to investigate the effects of different electroplating solutions on the layer properties and mechanism during the non-cyanide electroplating of Cu–Sn alloy. The electrode reaction was studied by cyclic voltammetry (CV). The structure and surface morphology of the alloy layers were characterised by XRD, EDS and SEM. The properties of the electroplating solution were analysed by UV–Vis and IR spectroscopy. Finally, the mechanism of each substance in the electroplating solution was analysed. The results not only provide guidance on the electroplating of Cu–Sn alloy in citric acid system but also provide reference for other electroplating alloy systems.
2 Experiment
2.1 Reagents
The order in which the substances are dissolved in the electroplating solution is important. 17.5 g of C6H8O7·H2O (≥ 99.5%, Tianjin Zhiyuan Chemical Reagent Co.) was dissolved in 60 mL of deionized water, and after stirring and dissolution, the solution showed no colour. C6H8O7·H2O (17.5 g) was dissolved in 60 mL of deionized water, and the solution was observed to be colourless after thorough stirring. After CuSO4·5H2O (0.15 mol, ≥ 99.0%, Tianjin Zhiyuan Chemical Reagent Co.) was added thereto and stirred until it was completely dissolved, the solution exhibited a light blue colour. Further, 10 g of NaOH (≥ 96.0%, Tianjin Beichen Fangzheng Reagent Factory) was added to the solution, and the blue colour gradually deepened as the amount of the added NaOH increased. After complete dissolution and cooling, 0.3 mL of H3PO4 (≥ 85%, Tianjin Beichen Fangzheng Reagent Factory) was added, and the mixture was thoroughly stirred to form blue solution A. Then, 0.08 mol of Na2SnO3·3H2O (≥ 98%, Tianjin Guangfu Fine Chemical Research Institute) was added in 15 mL of deionized water until it was completely dissolved. H2O2 ((0.06 mL, ≥ 30%, Tianjin Beichen Fangzheng Reagent Factory) was added to form colourless solution B. Solution B was slowly added to Solution A until the mixed solution became clear and transparent blue. The solution was added to nearly 100 mL, and the pH of the solution was adjusted to 9.5 with solid NaOH. Finally, the volume was increased to 100 mL to form dark blue electroplating solution BR. Electroplating solution BR was mainly composed of 0.15 mol L−1 CuSO4·5H2O, 0.08 mol L−1 Na2SnO3·3H2O, 0.83 mol L−1 C6H8O7·H2O, 3.0 mL L−1 H3PO4, 0.6 mL L−1 H2O2 and 2.5 mol L−1 NaOH. Na2SnO3·3H2O was hardly soluble in water but was easily soluble in alkaline solution; thus, it should be added to alkaline solution when dissolved. Solution BR was very stable and did not precipitate when left for half a month. However, after an extended time, a few precipitates formed at the bottom of the solution, and the layer obtained by electroplating exhibited a black colour. Water was purified using a water purification system (PALL Cascada II I 30, USA). All reagents were of analytical grade.
2.2 Electrochemical tests
Electrochemical tests were conducted using PARSTAT PMC1000 electrochemical workstation. A three-electrode system composed of the reference electrode, which is a Hg|HgO electrode, the working electrode (WE) (Φ = 10 mm, area = 78.5 mm2), which is made of 304 stainless steel, and the counter electrode (CE) (Φ = 10 mm, area = 78.5 mm2), which is made of Cn0.999. The material of WE and CE are commonly used in the electrocoating experiments. All the electrodes were finely polished and washed [20, 21]. To prevent fluctuation of the electrode area during testing, except for the effective area of the electrode, the rest was sealed by an insulating polymer. The scanning speed of the cyclic voltammetry was 50 mV/s.
2.3 Electroplating experiment
The electrolysis system consisted of an electrocoating bath, a cathode plate, and an anode plate. The electrocoating bath’s effective volume was 100 mL. The cathode was made of 304 stainless steel plates with dimensions 30 mm × 70 mm × 1.0 mm and whose single-side effective area was 12.0 cm2. The anode was made of Cn0.999 with dimensions 30 mm × 70 mm × 1.0 mm and whose single-side effective area was 12.0 cm2. During the electrodeposition process, the temperature was maintained at 293–303 K; and the cathodic current density and electrolysis time were 0.4 A·dm−2 and 5 min, respectively. Intelligent constant direct-current power (WJY-30 V/10 A) was used as the power source for electrolysis.
2.4 Characterisation
A HITACHI SU8010, field emission environmental SEM–EDS was used for surface investigation and feature detection. Images and spectra were collected at 20 kV accelerating voltage. The phase analysis of the coatings was carried out by Bruker D2 Phaser X-ray diffraction instrument (XRD) (Rigaku, Japan). Ultraviolet visible (UV–Vis) spectroscopy was recorded on a TU-1901 UV–Vis spectrophotometer (Beijing Persee, China). Fourier transform infrared spectroscopy (FTIR) was recorded using a Magna 550II FTIR spectrometer (Nicolet, USA). Nuclear magnetic resonance (NMR) was recorded with an Avance III 400 M (Bruker, Germany). The macroscopic morphology of the coatings was recorded by an optical camera (Canon A590 IS).
3 Results and discussion
3.1 Effects of electroplating solutions on the electrochemical reaction of the electrode
The effects of electroplating solutions on the electrode interface were analysed through the cyclic voltammetry (CV) curves in Fig. 1. In Fig. 1a, the black line was obtained from low to high potential in the forward scanning, and the peak B(0.6 V vs. Hg|HgO) means the dissolution of anode materials. The red line was acquired from high to low potential in the reserve scanning, and the peak A (− 1.3 V vs. Hg|HgO) represents the deposition of the cathode materials. Thus this curve shows that peak A attributed to the codeposition of Cu and Sn [22,23,24,25,26].

Effects of the concentration of the substances on electrochemical reaction by CV curves. a BR, b Na2SnO3·3H2O, c CuSO4·5H2O, d C6H8O7·H2O, e H3PO4, f H2O2 and g pH
Figure 1b shows the influence of Na2SnO3·3H2O concentration on electrochemistry. With the increasing concentration of Na2SnO3·3H2O, the potential of the cathode deposition peak A was basically unchanged and only the peak height changed. When the concentration of Na2SnO3·3H2O was 0.08 mol L−1, the deposition peak B reached the maximum, indicating that the amount of cathode deposition material reached the maximum at this time. In this process, the potential of the dissolution peak B changed, and the peak value changed. It showed that the concentration of Na2SnO3·3H2O affected the dissolution of anode material.
Figure 1c shows the influence of CuSO4·5H2O concentration on electrochemistry. With the increase of CuSO4·5H2O concentration, the height of cathodic deposition peak A at − 1.3 V vs Hg|HgO gradually increased. It shows that the CuSO4·5H2O concentration increased, and the amount of cathode deposition material increased. The dissolution peak B gradually decreased as the concentration of CuSO4·5H2O increased, and the peak gradually increased, indicating that the greater the concentration of CuSO4·5H2O, the greater the dissolution rate of the anode material.
Figure 1d shows the influence of the content of complexing agent C6H8O7·H2O on electrochemistry. As the content of C6H8O7·H2O increased from 0.69 to 1.30 moL L−1, the potential of the cathode deposition peak A did not change, but the peak value gradually increased. This shows that the concentration of C6H8O7·H2O had no effect on the deposition reaction. Whilst the dissolution peak B potential increased, the peak value gradually increased.
Figure 1e shows the influence of H3PO4 content on electrochemistry. With the increase of H3PO4 content, the peak value of deposition peak A and the potential basically did not change. The potential of the dissolution peak B increased, and the peak first increased and then decreased. This shows that the concentration of H3PO4 does not affect the cathode deposition reaction.
Figure 1f shows the effect of H2O2 content on electrochemistry. Increasing the content of H2O2, the cathodic deposition peak A potential and peak value had no change. The peak shape of peak B changed, indicating that the increase of H2O2 content had an effect on the dissolution of anode material.
Figure 1g shows the influence of different pH values on electrochemistry. As the pH increases, the potential and peak value of the cathode deposition peak A changed. When the pH reached 13, a new cathodic peak C appeared at − 1.3 V vs Hg|HgO, which indicated that the pH affects the electrochemical reaction. Combining the analysis of Fig. 3f and Reference S1, it can be seen that Cu4O3 was produced in the coating at this time. When the pH continued to increase, the dissolution peak B potential first increased and then decreased. This shows that pH not only affected the dissolution of anode material but also affected the deposition of cathode material.
3.2 Effects of different electroplating solutions on the microtopography of the coating
The effects of each component on the plated sheet were analysed taking SEM images at different multiples on the coatings (Fig. 2). When the electroplating solution did not contain Na2SnO3·3H2O as the main salt, the coating consisted of particles of varying sizes from 0.2 to 0.5 µm with an uneven surface (Fig. 2a). When the electroplating solution did not contain CuSO4·5H2O, nearly no substance appeared on the surface of the coating (Fig. 2b). When the electroplating solution contained only 0.69 mol L−1 C6H8O7·H2O as the complexing agent, the coating was flat, and the crystal was dense, but the local coating had a hollow structure (Fig. 2c). When the electroplating solution did not contain H3PO4, the coating crystallised unevenly and locally had a small amount of particles and pore structure (Fig. 2d). When the electroplating solution did not contain H2O2, the surface of the coating consisted of a large amount of particles with sizes in the range of 0.5–0.8 µm and it was not flat (Fig. 2e). The coating particles at pH 6.35 were significantly larger than those at pH of 9.5 (Fig. 2f and g). When the pH was 9.5, the coating was dense, and no large particles were attached to the surface. When the pH was 13, the surface of the coating consisted of 0.2–0.4 µm particles, and the crystalline particles were mostly rough with a spherical shape (Fig. 2h).

Effects of the substances on the SEM and EDS results. a1 a2 and a3: [Na2SnO3·3H2O] = 0 mol L−1. b1, b2 and b3: [CuSO4·5H2O] = 0 mol L−1. c1, c2 and c3: [C6H8O7·H2O] = 0.69 mol L−1. d1, d2 and d3: [H3PO4] = 0 mol L−1. e1, e2 and e3: [H2O2] = 0 mol L−1. f1, f2 and f3: pH 6.35. g1, g2 and g3 pH 9.5(BR). h1, h2 and h3 pH 13.0
EDS analysis revealed the effect of different electroplating solutions on the composition of the coating. Table 1 shows that the coating consisted mainly of Cu, Sn, Fe, C, O, Si, Cr, Ni and Mn, and the Fe, C, O, Si, Cr, Ni and Mn were mainly derived from the composition of the substrate. To study the ratio of Cu and Sn in the coating, the values of Cu/(Cu + Sn) and Sn/(Cu + Sn) (wt%) are summarised in Table 1. When the electroplating solution did not contain Na2SnO3·3H2O, the elements of the substrate were negligible, and the coating was mainly composed of 100.0% Cu/(Cu + Sn). This finding was consistent with the purple-red colour shown in Fig. S1. When the electroplating solution did not contain CuSO4·5H2O, deposition of Cu or Sn in the coating was not detected; hence, the coating exhibited a ground colour of the stainless steel substrate. When the electroplating solution contained only 0.69 mol L−1 C6H8O7·H2O and did not contain H3PO4 or H2O2, and the Cu/Sn ratio in the coating was approximately 87/13. Given that the electroplating solutions of c, d and e shown in Table 1 contain 0.15 mol L−1 CuSO4·5H2O and 0.08 mol L−1 Na2SnO3·3H2O, the coatings contain Cu and Sn, and the Cu/Sn ratio was almost the same. This conclusion was consistent with the photographs of the three groups, C6H8O7·H2O, H3PO4 and H2O2, in the supporting literature of Fig. S1. Figure S1 shows that the amount of C6H8O7·H2O, H3PO4 and H2O2 has little effect on the composition of the coating; hence, the colour of the coating did not change much. When the pH changed from 6.35 to 13 in Table 1f, g and h, the Cu/Sn ratio in the coating changed very significantly. When the pH was 9.5, the contents of Cu and Sn in the coating were 86.28% and 13.72%, respectively. Under this condition, the content of Sn was the highest, and the colour of the coating should be golden yellow. This result was consistent with the golden yellow colour shown in Fig. S1.
3.3 Effects of electroplating solutions on the phase structure of the coatings
Figure 3 shows the XRD patterns of the coatings obtained by using different concentration of substances in the electroplating solutions. The results were compared with the expected patterns provided by the Joint Committee on Powder Diffraction Standards (JCPDS), which revealed the presence of Cu (JCPDS 04-0836), [Cu, Sn] (JCPDS 44-1477), Cu6Sn5 (JCPDS 45–1488), Cu10Sn3 (JCPDS 26-0564), Cu4O3 (JCPDS 49-1380) and other crystal structures [26,27,28,29].

Effects of the concentration of the substances in electroplating solutions on the XRD results. a Na2SnO3·3H2O, b CuSO4·5H2O, c C6H8O7·H2O, d H3PO4, e H2O2 and f pH. Note: The substances not mentioned are the same as that in the electrolyte BR
When the concentration of Na2SnO3·3H2O was 0 mol L−1, only the absorption peak of Cu was contained in the coating (Fig. 3a). This was consistent with the results of supporting literature in Fig.S1, so the plating colour was purple-red. As the concentration increased, the coating contained the crystal forms of Cu, [Cu, Sn], Cu6Sn5 and Cu10Sn3. The coating was mainly Cu phase, and the 2θ positions of the diffraction peaks were 43.297° (111), 50.433° (200), 74.130° (220) and 89.937° (311). It contained the [Cu, Sn] phase, and the 2θ positions of the [Cu, Sn] diffraction peaks were 43.472°, 50.077° (002), 73.195° (022) and 94.378°. The 2θ positions of the Cu6Sn5 diffraction peak was 42.972° (132), 44.782° (024), 48.243° (133) and 74.381° (− 733). The 2θ values of the Cu10Sn3 diffraction peak was 42.738° (300), 44.209° (212), 49.902° (220), 64.526° (115), 74.539° (323) and 81.253° (421). As the concentration of Na2SnO3·3H2O increased, the position of the diffraction peaks did not change, but the intensity changed. Therefore, as the concentration of Na2SnO3·3H2O increased, the content of each component in the coating changed. Hence, as the concentration of Na2SnO3·3H2O increased, a golden yellow colour appeared in Fig. S1.
When the concentration of CuSO4·5H2O was 0 mol L−1, nearly no coating was observed on the surface of the substrate (Fig. 3b). The diffraction peak of the stainless steel substrate only was composed of Fe, and its diffraction peak was obviously weak, in which the result was consistent with that of Fig.S1. When the concentration was between 0.075 and 0.35 mol L−1, the coating contained diffraction peaks, such as Cu3Sn, Cu, [Cu, Sn] and Cu10Sn3. The intensity of the diffraction peak changed, but the position of the diffraction peak did not change. Therefore, as the concentration of CuSO4·5H2O increased, the content of each component in the coating changed. Thus, the conclusions drawn from Fig. 3a and b explained why the colours of the previous photographs in Fig. S1 have changed.
The effects of the concentration changes of C6H8O7·H2O, H3PO4 and H2O2 in the electroplating solution on the coating are revealed in Fig. 3c–e, respectively. As the concentration changed, the shape and position of the diffraction peaks remained basically unchanged. Thus, the composition of the electroplated coating did not change much. Figure 3c–e shows Cu, [Cu, Sn] and Cu6Sn5 phases. These conclusions can explain the cause of the coating colour change in Fig. S1. However, with the increase of the concentration of H3PO4, the Cu10Sn3 diffraction peak appeared in the coating, and the peak became increasingly obvious, indicating that the composition of the alloy changed.
The peak shape of the diffraction peak changed obviously with the change in the pH of the electroplating solution (Fig. 3f). When the pH increased from 6.35 to 7.5, the diffraction peaks of Cu, [Cu, Sn] and Cu6Sn5 obviously increased in the coating. When the pH of the solution increased to 13, the Cu4O3 peak appeared in the diffraction peak of the coating, and the 2θ positions of the diffraction peaks were 36.342° (004), 43.960° (220) and 75.513° (422). Hence, the composition of the alloy had changed. These conclusions were consistent with the blackened photographs of the coatings.
The above conclusions combined with the results of the electrochemical analysis in 3.1 can be drawn: the main salt, complexing agent, H3PO4 and H2O2 components and their concentrations only change the composition content of the coating alloy. Only the change of pH value affected the electrode reaction, thereby generating new components in the coating.
3.4 Effects of electroplating solutions on UV–Vis and FTIR
To investigate the complexation reaction mechanism of each component in the experiment, UV–Vis and FTIR analyses were performed on the electroplating solutions of different components. First, the effect of each component in the electroplating solution on the UV–Vis spectra was studied. As seen from Fig. 4a and c, the absorption peak for the electroplating solution was at 194 nm when the solution did not contain any main salt. When the electroplating solution did not contain CuSO4·5H2O, the absorption peak was at 199 nm. When the solution did not contain Na2SnO3·3H2O, the absorption peak was at 192 nm. Solution BR had an absorption peak at 196 nm when it contained the above two main salts. Therefore, Na2SnO3·3H2O and CuSO4·5H2O in the electroplating solution resulted in a redshift and a blueshift, respectively, in the ultraviolet absorption peak. In addition, as seen by comparing the results at different solution pH values of 6.35, 9.5 and 13.0, the absorption peak redshifted with an increase in pH, and the redshift was the most obvious when the pH reached 13.

Effects of electroplating solutions on UV–Vis. a and b UV of the electrolytes, c and d wavelength shift of peaks for a and b, respectively. b Effect of the concentration of each substance in the BR on the absorption peak when it is maximised
Second, the effects of the concentration of each component in the electroplating solution on the absorption peak are analysed in Fig. 4b and d. Maximum concentrations of CuSO4·5H2O, Na2SnO3·3H2O, C6H8O7·H2O, H3PO4 and H2O2 were added to plating solution BR. With the increase in the concentrations of CuSO4·5H2O and Na2SnO3·3H2O, the absorption peak was blueshifted. No influence was observed on the absorption peak when the concentration of the complexing agent C6H8O7·H2O was increased. When the concentrations of H3PO4 and H2O2 were increased, the absorption peaks were all blueshifted to 195 nm with little effect. In summary, the analysis shows that the UV absorption peak was affected mainly by the concentration of the main salts and the pH.
According to the literature [30], the weak peak at 800 cm−1 was caused by the in-plane bending vibration of PO43− (Fig. 5a). The peak at 850 cm−1 was caused by the deformation vibration of the C–H in the methylene group, and the weak peak at 960 cm−1 was caused by the symmetric stretching vibration of PO43−. When the content of H3PO4 was maximised, the peak intensity at 960 cm−1 was significantly increased (Fig. 5b). The absorption peak at 1150 cm−1 was caused by the stretching vibration of the C–O bond of the carbon chain in citric acid and the –OH moiety of the tertiary alcohol. When the composition of the solution was different, the intensity of the absorption peak at this point was different. The absorption peak at 1150 cm−1 for the sample without CuSO4·5H2O was weaker than the peak for the sample without any main salt, and the absorption peak at 1150 cm−1 for the sample without Na2SnO3·3H2O was stronger than the peak for the sample without any main salt. The absorption peak in the spectrum of the BR solution at this point was weaker than the peak for the sample without any main salt. These findings indicate that citric acid is an excellent complexing agent for Cu2+ and that the complexation reaction differs when the solution components are different.

Effects of electroplating solutions on FTIR. a FTIR of the electrolyte with different main salts, and b FTIR at the maximum concentration of each component in the BR. Their components are: BR; BR with 0.20 M CuSO4·5H2O; BR with 0.22 M NaSnO3·3H2O; BR with 0.47 M C6H8O7·H2O; BR with 44.3 mL/L H3PO4; BR with 3.0 mL/L H2O2
The intensity of the absorption peak at 1150 cm−1 was decreased at pH values of 6.35, 9.5 and 13.0, indicating that the alkaline strength of the solution was enhanced, hydrolysis of citric acid in the solution was promoted and the complexation was different. Figure 5b shows the infrared spectrum of the BR solution and the maximum content of each component. Compared with the BR solution, when the concentrations of CuSO4·5H2O and Na2SnO3·3H2O reached a maximum, the intensity of the absorption peak at 1150 cm−1 was enhanced. When the concentration of CuSO4·5H2O was the largest, the absorption peak intensity at 1150 cm−1 reached a maximum, indicating that the concentration of the main salt had an effect on the complexation reaction. When the concentrations of C6H8O7·H2O and H2O2 reached a maximum, the intensity of the absorption peak at 1150 cm−1 hardly changed. This result indicates that the concentrations of C6H8O7·H2O and H2O2 had little effect on the complexation reaction, whilst the increase in the concentration of H3PO4 had a great influence.
The peak at 1250 cm−1 was attributed to the stretching vibration of C–C in citric acid. In Fig. 5b, when the content of C6H8O7·H2O reached a maximum, the peak intensity at this point was also the strongest [31]. The peak at 1375 cm−1 was caused by the symmetric stretching vibration of the C–O bond in COO−. The peak at 1610 cm−1 was caused by the asymmetric stretching vibration of the C–O bond of the carboxylate ion COO−. The absorption peaks at these two sites were only slightly affected by the composition and content of the electroplating solution.
3.5 Effects of electroplating solutions on NMR
To investigate the effects of electroplating solutions on the NMR results, the electroplating solution was prepared by replacing the deionised water with a deuterated reagent, and the H spectrum of the nuclear magnetic resonance was tested, as shown in Fig. 6. The chemical shift of the test absorption peaks is summarised in Table 2. According to the literature, peak #1 at a chemical shift of 4.72 ppm was the absorption peak of the deuterated reagent [32, 33].

Effects of electroplating solutions on NMR. a 0.83 mol L−1 C6H8O7·H2O, pH 9.5; b (a) + 0.15 mol L−1 CuSO4·5H2O; c (a) + 0.08 mol L−1 Na2SnO3·3H2O; d (c) + 0.15 mol L−1 CuSO4·5H2O; e (d) + 3.0 mol L−1 H3PO4; f (e) + 0.6 mol L−1 H2O2
Figure 6a shows the H spectrum of 0.83 mol L−1 C6H8O7·H2O dissolved in the deuterated reagent. At pH 9.5, four chemical environments in C6H8O7 were observed: H in crystal water H2O, H in the HOOC– group, H in the HO–C– group and H in the –CH2– group. Amongst them, the H in crystal water H2O, the HOOC– group and the HO–C– group were replaced with D from D2O; hence, almost no peak was observed, and the spectrum mainly consisted of an absorption peak at 4.72 ppm. Theoretically, two –CH2– groups should contribute to one peak. However, given that the magnetic environments were not equivalent, the two chemical shifts for the H atoms on one C were different, and thus, two peaks appeared. At the same time, the two peaks were each divided again into two peaks due to the influence of another H on a different carbon. This explains the appearance of an ABAB peak, corresponding to the absorption peak #2 with chemical shifts of 2.59, 2.55, 2.42 and 2.38 ppm [34]. Figure 6b is obtained by adding 0.15 mol L−1 CuSO4·5H2O to the solution presented in Fig. 6a. The complexation of Cu and C6H8O7 in the solution caused –CH2– to not rotate freely, and the absorption peak became broad and less intense. The slower the frequency of the molecule itself, the blunter is the peak. Figure 6c is obtained by adding 0.08 mol L−1 Na2SnO3·3H2O to the solution used in Fig. 6a. The complexation of Sn and C6H8O7 hardly occurred. Figure 6d is obtained by adding 0.15 mol L−1 CuSO4·5H2O to the solution used in Fig. 6c. The absorption peak was significantly broadened, and the complexation of copper ions with C6H8O7 was further confirmed. At the same time, compared with Fig. 6b, after adding Na2SnO3·3H2O into the electroplating solution of Fig. 6b, sodium stannate reduced the degree of complexation of Cu ions and citric acid, causing the citric acid to dissociate to some extent. Therefore, some of the –CH2– can be freely rotated to increase the intensity of the absorption peak. Figure 6e is obtained by adding 3.0 mol L−1 H3PO4 to the solution used in Fig. 6d. Figure 6f is obtained by adding 0.6 mL L−1 H2O2 to the solution used in Fig. 6e. As shown in Table 2, the chemical shifts and peak shapes of peak #2 in Fig. 6e and f hardly changed.
3.6 Reaction mechanism of electroplating
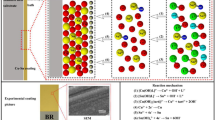
When the pH of the electroplating solutions is greater than 7.5, citric acid (abbreviated as H3Cit) is mainly present in the form of Cit3−. When the pH of the solution was greater than 8, the complex ions obtained by the copper ions and citric acid were mainly in the form of Cu2Cit2H-24−, and the cathodic electrochemical reactions of copper ions and their complex ions in this system were as follows: [13, 35,36,37,38,39]
Amongst them, the reaction rate constant of the reaction (2) was 1.55 × 10−16 mol·m−2 s−1 [35]. When the pH of the plating solution was greater than 8.0, the complex ions were mainly present in the form of SnO32−. SnO32− is hydrolysed in the strong base to form Sn(OH)62−, and the cathodic electrochemical reactions of the complex ions were as follows:
According to the analysis of the above complex deposition mechanism, when the pH of the electroplating solution was 9.5, the copper ions and citric acid formed a complex, and the complex ions were mainly in the form of Cu2Cit2H-24−. Sodium stannate and NaOH complexed in the form of Sn(OH)62−. This conclusion was consistent with the previous NMR results.
The content of H3PO4 or H2O2 had an effect on the dissolution of anode material by the result of electrochemical analysis, and H3PO4 or H2O2 will affect the uniformity and particle size of the coating by SEM results.
4 Conclusion
This experiment explored the process of electroplating Cu–Sn alloy in a citric acid system. The composition of the electroplating solution was 0.15 mol L−1 CuSO4·5H2O, 0.08 mol L−1 Na2SnO3·3H2O, 0.83 mol L−1 C6H8O7·H2O and 3.0 mol L−1 H3PO4 at pH 9.5. The process conditions were as follows: the temperature was set to 293–303 K, the cathode current density was 0.4 A/dm2 and the plating time was 5 min. The electroplated layer obtained by the BR electroplating solution was a golden yellow Cu–Sn alloy, which was composed of 86.28% Cu and 13.72% Sn. The layer was dense and had no large particles attached to the surface. The binary alloy layer consisted of Cu, [Cu, Sn], Cu6Sn5 and Cu10Sn3 phases. At the same time, the cathode material only had a deposition peak at − 1.3 V vs. Hg|HgO. At pH 9.5, the copper ions and citric acid formed a complex, and the complex ions were mainly in the form of Cu2Cit2H-24−. Sodium stannate and NaOH complexed in the form of Sn(OH)62−. Therefore, the formed chelate can be reduced together to deposit Cu and Cu–Sn alloy at the same voltage. The main salt, complexing agent, H3PO4 and H2O2 and their amounts in the plating bath had no effect on the electrode reaction, and the pH had a great influence on the plating layer.
References
Piccinini N, Ruggiero GN, Baldi G, Robotto A (2000) J Hazard Mater 71:395–407
Orhan G, Gezgin GG (2011) Metall Mater Trans A 42:771–782
Survila A, Mockus Z, Kanapeckaitė S, Jasulaitienė V, Juškėnas R (2010) J Electroanal Chem 647:123–127
Ding LF, Liu F, Cheng J, Niu YL (2018) J Appl Electrochem 48:175–185
Barbano EP, Oliveira GMD, Carvalho MFD, Carlos IA (2014) Surf Coat Technol 240:14–22
Barbosa LL, Finazzi GA, Tulio PC, Carlos IA (2008) J Appl Electrochem 38:115–125
Rubin W, Oliveira EMD, Carlos IA (2012) J Appl Electrochem 42:11–20
Pewnim N, Roy S (2013) Electrochim Acta 90:498–506
Zdemir RO, Karahan IH, Karabulut O (2016) Metall Mater Trans A 11:5609–5617
Meng GZ, Sun FL, Wang SJ, Shao YW, Zhang T, Wang FH (2010) Electrochim Acta 55:2238–2245
Almeida MRHD, Barbano EP, Zacarin MG, Brito MMD, Tulio PC, Carlos IA (2015) Appl Surf Sci 333:13–22
Volov I, Sun X, Gadikota G, Shi P, West AC (2013) Electrochim Acta 89:792–797
Slupska M, Ozga P (2014) Electrochim Acta 141:149–160
Saeki I, Seguchi T, Kourakata Y, Hayashi Y (2013) Electrochim Acta 114:827–831
Walsh FC, Low CTJ (2016) Surf Coat Technol 288:79–94
Ashworth MA, Wilcox GD, Higginson RL (2014) J Electron Mater 43:1005–1016
Heidari G, Khoie SMM, Abrishami ME, Javanbakht M (2015) J Mater Sci Mater Electron 26:1969–1976
Gougaud C, Rai D, Delbos S, Elisabeth C, Daniel L (2013) J Electrochem Soc 160:485–494
Meudre C, Ricq L, Hihn JY, Moutarlier V, Monnin A, Heintz O (2014) Surf Coat Technol 252:93–101
Darban AK, Aazami M, Meléndez AM, Abdollahy M, Gonzalez I (2011) Hydrometallurgy 105:296–303
Yin KB, Xia YD, Chan CY, Zhang WQ, Wang QJ (2008) Scr Mater 58:65–68
Shin S, Park C, Kim C, Kim Y, Park S, Lee JH (2016) Curr Appl Phys 16:207–210
Im B, Kim S (2014) Electrochim Acta 130:52–59
Ballesteros JC, Chaînet E, Ozil P, Trejo G, Meas Y (2010) J Electroanal Chem 645:94–102
Kulyk N, Cherevko S, Chung CH (2012) Electrochim Acta 59:179–185
Jung M, Lee G, Choi J (2017) Electrochim Acta 241:229–236
Xue L, Fu Z, Yu Y, Tao H, Yu A (2010) Electrochim Acta 55:7310–7314
Orhan G, Gezgin GG (2011) Metall Mater Trans B 42:771–782
Murase K, Ito A, Ichii T, Sugimura H (2011) J Electrochem Soc 158:335–341
Ilczyszyn M, Ratajczak H (1996) J Mol Struct 375:23–35
Yang G, Hu Q, Yang J, Huang Z, Yin J (2002) Anal Bioanal Chem 374:1325–1329
Schah-Mohammedi P, Shenderovich IG, Detering C, Limbach HH, Tolstoy PM, Smirnov S, Denisov S, Golubev S (2013) J Am Chem Soc 122:12878–12879
Wong WS, Ng CF, Kuck D, Chow HF (2017) Angew Chem 129:12528–12532
Palanki S, Akiyama H, Campochiaro P, Cao J, Chow CP (2008) J Med Chem 51:1546–1559
Rode S, Henninot C, Vallières C, Matlosz M (2004) J Electrochem Soc 151:405–411
Oaki Y, Nakamura K, Imai H (2012) Chem Eur J 18:2825–2831
El-Ashram T, Canché-Canul L, Oskam G (2011) Electrochim Acta 56:9391–9396
Zhang ZJ, Liu AM, Ren XF, Zhang JQ, Yang PX, An MZ (2014) Rsc Adv 4:38012–38026
Salhi Y, Cherrouf S, Cherkaoui M, Abdelouahdi K (2016) Appl Surf Sci 367:64–69
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. NSFC51604180), Cultivate Scientific Research Excellence Programs of Higher Education Institutions in Shanxi (Grant No. CSREP2019KJ038), General Project of Shanxi Province Key R & D Plan (Social Development Field, 201903D321068) and Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (Grant No. 2019L0235).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ding, L., Chen, C., Li, Q. et al. Process and theoretical research on electroplating Cu–Sn alloys of low Sn. J Appl Electrochem 51, 1287–1299 (2021). https://doi.org/10.1007/s10800-021-01584-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-021-01584-8