
Abstract
The assessment of genetic diversity and karyotypes can help to identify new species and taxonomic units and contribute to species conservation. Ecological niche modeling can be used to predict species distribution and to estimate the impact of climate changes on its survival. The Alouatta guariba group is one of the most threatened primates in the Americas, with two endemic Atlantic Forest subspecies: A. g. guariba and A. g. clamitans, referred to as Northern and Southern populations, respectively. Although extensively studied, the distribution and the genetic diversity of these subspecies remains unclear. We assessed species relationships and genetic diversity in the A. guariba group and evaluated the potential future distribution of populations. We sequenced the mitochondrial cytochrome b gene of 14 captive and 108 free-living individuals to generate a maximum likelihood phylogenetic tree and median-joining haplotype network and to estimate the nucleotide diversity between the taxa. We conducted neutrality tests to test for population expansion. We also linked ten new karyotypes, obtained from cell cultures from blood samples of captive individuals, with the positions of the clades in the gene tree. Finally, we used ecological niche modeling to predict the potential distribution of the Northern and Southern populations. We found three lineages within the A. guariba group: North A and North B associated with A. g. guariba, and South C associated with A. g. clamitans. The results showed that A. guariba group is a species complex, with reduced haplotype and nucleotide diversity within each lineage, compared with other Alouatta species. The neutrality tests and median-joining analysis suggest a recent diversification and expansion of each lineage, and the geographical data, based on the localities of the individuals that we analyzed genetically, increase the geographic distribution of A. g. guariba, expanding the distribution farther south in southeastern Brazil than previously described. Our findings show the distribution of the A. guariba group and genetic diversity of recently expanding populations, contributing to the assessment of this Critically Endangered species. We also suggest that environmental losses and climate change are major factors in the drastic population decline of the A. guariba group.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Primates exhibit a considerable level of genetic diversity within genera, and related species may show different levels of genetic diversity (Osada, 2015). Legislative definitions around the world include taxonomic levels of species, subspecies, varieties, and genetically distinct populations, allowing the protection of genetic diversity and evolutionary processes at different levels (Coates et al., 2018). Identifying lineages, which may be endemic, rare, or endangered, helps to implement private or public policies to conserve specific areas (Winters et al., 2012). Furthermore, the maintenance of genetic diversity is an essential condition for populations to evolve and adapt to environmental change, strengthening the ability of species and populations to survive (Hoban et al., 2021). In particular, human-induced climate change presents numerous potential threats to nonhuman primate species, many of which are already threatened by human activities, such as deforestation, hunting, and trade in exotic animals (Chapman et al., 2006; Ribeiro et al., 2009; Wiederholt & Post, 2010).
Platyrrhines are abundant and very diverse, with high species richness, including around 171 species mainly inhabiting the Amazonia and Atlantic Forest (Estrada et al., 2017). Despite being well studied, their genetic diversity is still insufficiently understood, and several new species and subspecies have been described or revalidated in the past 10 years (Boubli et al., 2016; Costa-Araújo et al., 2021; Dalponte et al., 2014; Gualda-Barros et al., 2012; Marsh, 2014). The main threats to primates are loss of habitat due to agriculture, followed by logging and wood harvesting, with the Atlantic Forest retaining the largest area of agricultural production (Estrada et al., 2017).
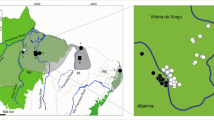
The family Atelidae encompasses the largest-sized platyrrhine genera, including howler monkeys of the genus Alouatta Lacépède, 1799. Howler monkeys are well-adapted to several morphoclimatic domains, with a wide distribution through southern Mexico to northern Argentina (Rylands, 2000), and the number of recognized species ranges from nine to 15 (Cortés-Ortiz et al., 2015a; Gregorin, 2006; Rylands & Mittermeier, 2009). The Alouatta guariba group encompasses two subspecies endemic to the Atlantic Forest: Alouatta guariba clamitans Cabrera, 1940 and A. g. guariba (Humboldt, 1812). The exact geographical distribution of these two subspecies is not well established (Fig. 1). Based on the IUCN Red List distribution, A. g. clamitans, also called the Southern Atlantic Forest population, occurs in the southern Atlantic Forest from Rio Grande do Sul (RS) to the south of Bahia (BA) (Buss et al., 2021), and A. g. guariba, also called the Northern Atlantic Forest population, occurs from the south of Bahia state to northeastern Minas Gerais state (Neves et al., 2021). However, the last taxonomic revision of the Brazilian species of Alouatta (Gregorin, 2006), based on morphological features, extended the distribution of A. g. guariba to Espírito Santo and northern Rio de Janeiro states, and narrowed the distribution of A. g. clamitans in the southeast region (Fig. 1). Although Gregorin (2006) elevated both subspecies to the level of species, most studies and the IUCN Red List (Buss et al., 2021; Neves et al., 2021), continue to refer to them as subspecies.

Maps with localities of the samples used in analyses and the geographic distribution of A. guariba group based on IUCN (Top right, Buss et al., 2021; Neves et al., 2021) and morphological data (Bottom right, Gregorin, 2006). In the top and bottom right maps, the area delimited in grey shows the distribution of A. guariba guariba, and white the distribution of A. guariba clamitans. Numbers refer to localities listed in Table S1. Brazilian states are Espírito Santo (ES), Rio de Janeiro (RJ), Minas Gerais (MG), São Paulo (SP), Paraná (PR), Santa Catarina (SC), and Rio Grande do Sul (RS), Goiás (GO) and Mato Grosso do Sul (MS), and Federal District (DF). The samples used from ES, RJ, MG, SP, and RS states were obtained in 2013, 2014, 2017, 2018, and 2019.
Previous studies hypothesized that A. guariba is a species complex based on karyotypes (de Oliveira et al., 1998; Steinberg et al., 2017), molecular markers (de Mello Martins et al., 2011; Harris et al., 2005), and morphological data (Gregorin, 2006), without a clear geographic distribution of each taxon. Like some other Alouatta species, A. guariba males have one fewer chromosome than females due to the translocation of the Y chromosome to an autosome (Koiffmann & Saldanha, 1974). The A. guariba group has two karyotypes, with a diploid number (2n) of 45 for males and 46 for females in the Southern population (de Oliveira et al., 2000; Steinberg et al., 2017), and 49 for males and 50 for females in the Northern population (de Oliveira et al., 1998, 2000). These differences in karyotype have led some authors to assume that the populations are reproductively isolated (de Oliveira et al., 2000; Steinberg et al., 2017). Studies based on phylogenetic analyses of mitochondrial markers proposed that the two clades obtained for the A. guariba group are linked to A. guariba guariba and A. guariba clamitans (Harris et al., 2005; de Melo Martins et al., 2011). Among the morphological characters that differentiate A. g guariba from A. g. clamitans, pelage coloration is the most conspicuous, with homogeneous coloration without sexual dichromatism in A. g guariba, and presence of sexual dichromatism in A. g. clamitans (Gregorin, 2006).
Alouatta guariba guariba is considered to be among the 25 most-threatened primates (Mittermeier et al., 2022; Neves et al., 2021) due to habitat loss, hunting, traffic, and deaths during outbreaks of yellow fever, which reduced both populations considerably: the South mainly in 2008/2009 (Almeida et al., 2012; de Almeida et al., 2019; Estrada et al., 2018) and the North in 2017/2021 (Agostini et al., 2017; Sacchetto et al., 2020). The last outbreaks may have moved A. g. guariba from Vulnerable to Endangered (Bicca-Marques et al., 2017; Estrada et al., 2018). The Atlantic Forest is considered a world biodiversity hotspot, suffering from long-term deforestation, and is completely fragmented, with approximately 12% of its original coverage remaining (Hansen et al., 2013; INPE, 2020; Rezende et al., 2018; Ribeiro et al., 2009).
We assessed species relationships and genetic diversity in the A. guariba group and evaluated the potential future distribution of the Northern and Southern populations. To do this, we: (i) assessed the cytochrome b and karyotype diversity in A. guariba group, concentrating on the Northern population, which is less studied and sampled than the Southern population, and (ii) evaluated the potential future distribution of populations from the northern and southern Atlantic Forest to shed light on the long-term conservation prospects for the group.
Methods
Ethical Note
We obtained blood samples from free-living and captive Alouatta in accordance with methods and protocols approved by the Institutional Ethics Committee for Animal Experimentation (Protocol CEUA/IOC‐029/2016, license L‐037/2016), the Brazilian Ministry of the Environment (SISBIO = System of Authorization and Information in Biodiversity, license number 54707–4 to R. Lourenço-de-Oliveira), and Rio de Janeiro’s Environment agency (INEA 012/2016 and 019/2018). Veterinarian staff collected blood samples from captive animals at Centro de Primatologia do Rio de Janeiro (CPRJ) and Zoo Parque de São Paulo (RTM). Sample collection and management of free-living animals are described in two recent publications (Abreu et al., 2019; Teixeira et al., 2022). Live, free-living animals were released at the same location as they were caught. We also obtained muscle tissue from roadkill animals under license SISBIO 1187/2013 and 514/2014 to C. Bueno (deposited in Mammal collection of Museu Nacional), as well as from individuals that died during the yellow-fever outbreak from samples held in the mammalian collection of the Centro de Coleções Taxonômicas of Universidade Federal de Minas Gerais (UFMG). We obtained samples in accordance with the ethical treatment guidelines of Conselho Nacional de Controle de Experimentação Animal (CONCEA), normative resolution n. 28 (Pissinatti et al., 2016).
Sampling
We obtained blood and muscle samples for DNA analysis from 122 howler monkeys, 14 in captivity and 108 free-living, including animals killed by yellow fever and found run over during road monitoring. The samples included 117 animals from the Northern population (Espírito Santo, Rio de Janeiro, Minas Gerais, São Paulo states) and five from the Southern population (Rio Grande do Sul; Fig. 1; Table S1). We stored muscle samples in a freezer at − 20 °C. We stored blood samples in a refrigerator at 8 °C and processed them within 2 days.
We isolated DNA from 21 muscle samples using the phenol–chloroform protocol (Sambrook & Russel, 2001). We isolated DNA from 101 blood samples with a saline protocol (Miller et al., 1988), Promega® Wizard Genomic DNA Purification Kit®, or QIamp® DNA Blood mini kit according to the kit or protocol available when we obtained the sample.
We karyotyped ten of the animals from the Northern population: seven from Rio de Janeiro state: (a) Parque Estadual da Serra dos Órgãos – CPRJ3622, (b) Cabo Frio -CPRJ2799 (locality 35 in Fig. 1), (c) Niterói – CPRJ3723 (locality 36), (d) Macaé – CPRJ3636, (e) Parque Estadual dos Três Picos – CPRJ2443 (locality 37), (f) Itaocara – CPRJ3216 (locality 23), (a-f) housed in captivity in Centro de Primatologia do Rio de Janeiro/CPRJ, (g) Angra dos Reis – CPRJ140 (locality 38), and three from São Paulo state (h) Parque Estadual das Fontes do Ipiranga/PEFI—RTM1518, RTM1526, RTM1542 (locality 33), housed in Zoo Parque de São Paulo.
Molecular and Statistical Analyses
We amplified the MT-CYB (ca.1140 bp) of 122 samples using polymerase chain reactions with primers L14725 (Irwin et al., 1991) and Cytb rev 5 ‘-GAATATCAGCTTTGGGTGTTGRTG-3’. Nomenclature of the mitochondrial cytochrome b protein-coding gene followed the HGNC Database, HUGO Gene Nomenclature Committee for the use of the MT-CYB abbreviation.
We used the same primers plus the internal primers CB-in1 5’-GAATATCAGCTTTGGGTGTTGRTG-3’ and CB-in2 5`-TGAGGACAAATATCATTYTGAG-3` for sequencing. The amplification reaction mixture contained 1.0 µl of DNA template, 2.5 µl of 10 × buffer, 2.0 µl of MgCl2 (25 mM), 1.0 µl of forward primer 1 (10 mM), 1.0 µl of reverse primer 2 (10 mM), 0.4 µl of dNTPs (10 mM), 0.3 µl of Platinum® Taq DNA Polymerase (Invitrogen), and Ultrapure water for a total volume of 25 µl. PCR amplification conditions consisted of an initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 54 °C for 30 s, extension at 72 °C for 90 s, and the final extension at 72 °C for 5 min. We obtained amplicons using a ProFlex™ 3 thermal cycler × 32-well Sample Block by Life Technologies, and purified them with PureLink TM Quick Gel Extraction & PCR Purification Combo kit by Thermo Fisher Scientific following the manufacturer’s recommendations. We performed the sequencing in the Applied Biosystems 3730xl DNA Analyzer platform using the reagents of BigDye™ Terminator v3.1 Cycle Sequencing Kit. The sequencing reaction mixture in each well contained 2–4 μl of DNA, 1 µl of Ready Reaction kit, 1.5 µl of BigDye sequencing buffer (5x), 1 µl of oligonucleotide, and ultrapure water to a volume of 10 µl. The sequencing conditions consisted of an initial denaturation at 96 °C for 1 min, followed by 20–40 cycles of denaturation at 96 °C for 15 s, annealing at 50 °C for 15 s, and extension at 60 °C for 4 min. We assembled MT-CYB in sequence contigs using Chromas PRO version 2 (TechnelysiumPtyLtd) and aligned the sequences using Muscle in Mega X (Kumar et al., 2018). We checked for possible nuclear mitochondrial DNA pseudogenes (NUMTs) by translating the nucleotide sequence into protein and verifying the length and the presence of stop-codons in the wrong position. We did not identify any sequences as potential NUMTs. The phylogenetic positions in the analyses were as expected, suggesting that the inclusion of NUMTs was not an issue.
In addition to the sequences that we obtained, we added sequences available in GenBank with at least 800 base pairs (bp) identified as A. guariba clamitans (AF289986-987, AY065898-899, HQ385483-494, HQ385496-501, DQ679770-775, DQ679777-784, KY202428, KR528422), A. belzebul (DQ387044), A. caraya (DQ350637), A. seniculus (EU232713), A. macconnelli (AJ489759), A. sara (AY065887), corresponding to the South American Alouatta clade, and A. pigra (AY065885) and A. palliata (AY065880) from the Mesoamerican clade (Cortés-Ortiz et al., 2003). As outgroups, we used available sequences of Ateles geoffroyi (KR902382) and Ateles belzebuth (KR202369). To check the phylogenetic relationship and identify geographic structure within the samples obtained for the A. guariba group, we performed a maximum likelihood (ML) gene tree estimation using Iqtree (Nguyen et al., 2015) based on MT-CYB. The best-fit model of nucleotide substitution was TN + F + G4, selected with ModelFinder (Kalyaanamoorthy et al., 2017). We estimated branch support using SH-LRT (Shimodaira-Hasegawa-like procedure; Guindon et al., 2010) and UFBoot (ultrafast bootstrap approximation; Hoang et al., 2018).
To confirm the grouping of the samples obtained in the ML tree and understand the mutational steps separating each lineage, we produced a haplotype network. First, we identified the haplotypes with DNASP program version 6 (Rozas et al., 2017) and drew the median-joining (MJ) network analyses using Network program version 10 (fluxus-engineering.com; Bandelt et al., 1999). To identify haplotypes, we excluded sites with gaps and missing data to avoid biasing the haplotype distribution pattern, because missing data may represent poorly sequenced nucleotides (Joly et al., 2007). We used 690 sites to generate the haplotype network. MJ analysis is commonly used to infer an unrooted branching diagram from intraspecific data and to visualize the relationships among DNA sequences within populations or species, where alternative branching may represent potential ancestral sequences. We also used DNASP to calculate indices of nucleotide (π) and haplotype diversity to estimate the level of genetic diversity.
We carried out neutrality tests of Tajima’s D and Fu’s FS based on the null hypothesis of constant population size and neutral mutations with 1,000 replications (Table I) using Arlequin version 3.5 (Excoffier & Lischer, 2010).
Karyotype Preparation
We cultured each of ten peripheral samples from captive individuals attributed to the northern Atlantic forest (CPRJ3622, CPRJ2799, CPRJ3723, CPRJ3636, CPRJ2443, CPRJ3216, RTM1518, RTM1526, RTM1542, RJ140) in 8 ml of RPMI 1640, 2 ml of fetal calf serum, 20 µl of lectin, 0.5 ml of L-Glutamine, and 60 µl of heparin for 70 h. Then, we added 0.5 ml of colchicine 10−5 M for 2 additional hours. We added a solution containing KCl (0.075 M) for hypotonic shock, and a Carnoy solution (3 methanol: 1 acetic acid) for fixation. After culturing the lymphocytes, we performed conventional staining with Giemsa 2%, producing visible karyotypes with condensed chromosomes. We photographed metaphases with a QImaging Retiga™ 3000 camera coupled to a Zeiss Axio Imager. We also performed C-banding (Sumner, 1972), a technique used to show heterochromatic bands, which are areas of chromosomes that typically remain condensed during interphase and are typically found as blocks surrounding centromeres (Sumner, 1972). However, only one sample (CPRJ3636) showed a suitable result. We assembled the karyotypes in descending order, based on chromosome size and number of arms (de Oliveira et al., 1998).
Predicted Potential Distribution
We used occurrences available at Global Biodiversity Information Facility (GBIF, 2021) and in a dataset of occurrences of primates in the Atlantic Forests (Culot et al., 2019) for the Alouatta guariba group, which included localities in nine Brazilian states (Rio de Janeiro, Minas Gerais, Espírito Santo, São Paulo, Bahia, Santa Catarina, Paraná, Rio Grande do Sul and Mato Grosso do Sul), and ten sites from the province of Misiones, Argentina. We did not include individual records from São Paulo that we could not allocate to the Northern or Southern populations with certainty. We also included records available in studies that identified specimens from Northern and Southern population using morphology, karyotype, or molecular analysis (Cardoso Coimbra, 2015; de Oliveira et al., 2000; Gifalli-Iughetti, 2008; Gregorin, 2006; Machado, 2011; Steinberg et al., 2017).
We gathered the geo-referenced occurrences for Northern (n = 207; Table S2) and Southern (n = 259; Table S3) populations, defined South America as the background extent for species distribution models (SDM), and removed duplicate records of each grid cell for analysis. We used Maxent (version 3.4.4) to generate SDM. Maxent uses presence data based on georeferenced points of occurrence and environmental variables to estimate the occurrence of a species realized niche and model their potential distribution (Phillips et al., 2006). We conducted all SDM analyses in R (version 4.0.4), using the “raster” (Hijmans et al., 2015), “dismo” (Hijmans et al., 2017), and “rJava” (Urbanek, 2021) packages. We used the following parameters for all models: logistic output format (habitat suitability measured on a scale of 0–1), 80% of records used as training data and 20% for testing, maximum 500 iterations, 5 replicates, 10,000 background points (Phillips et al., 2006). We defined the threshold for the distribution of suitable habitat as the tenth percentile training presence (TPTP) threshold superimposed on the model training area (Pearson et al., 2007).
We modeled potential species distribution using 19 bioclimatic variables available at WorldClim 2.1 (Fick & Hijmans, 2017; Hijmans et al., 2005), which represent recent (years 1970 to 2000) and future conditions (years 2081 to 2100). We also included current altitude data that was used to produce WorldClim 2.1 as a variable in the present and future models. We used anthropogenic emissions quantification data for two main future scenarios (SSP2-4.5 and SSP5-8.5) of greenhouse gas concentration. We used the mean projections from eight global climate models (GCMs): BCC-CSM2-MR, CNRM-CM6-1, CNRM-ESM2-1, CanESM5, IPSL-CM6A-LR, MIROC-ES2L, MIROC6, and MRI-ESM2-0, available at WorldClim 2.1 (Fick & Hijmans, 2017; Hijmans et al., 2005). We projected all scenarios using a spatial resolution of 2.5 arc-minutes (~ 4.5 km; Fick & Hijmans, 2017; Hijmans et al., 2005). We assessed the proportional contributions (%) of each environmental variable to each model by using jackknife tests (Phillips et al., 2006).
To estimate the predictive accuracy of the potential habitat models, we used area under the curve (AUC) metrics, based on the area under the receiver operating characteristic (ROC) curve (Elith et al., 2006; Swets, 1988). AUC assesses how well the model’s predictions discriminate between places where observations are present and absent and is one of the most widely used threshold independent evaluators of the model’s discriminatory power. AUC values above 0.9 indicate high model performance, whereas AUC values close to 0.5 indicate models equal to or worse than random (Peterson et al., 2011). For comparative purposes, we reclassified the images resulting from each model (with continuous values from 0 to 1) into four classes of environmental suitability: unsuitable (UNS; 0, value TPTP), moderately suitable (MOS; 0.5, value TPTP), highly suitable (HIS; 0.5–0.75), and very highly suitable (VHS; 0.75–1). We plotted a final map with the occurrences and the final distribution model in ArcGIS. For more details of the methods, localities of occurrences, results of the AUC, and Jackknife test, see Appendix S1.
Results
Phylogenetics, Genetics, and Median-Joining Analyses
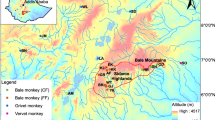
The maximum likelihood analysis divided the clade containing the A. guariba group into two major clades and three lineages: lineages A and B (Northern Clade) (94.1 SH-aLRT, 90 UFBoot); and lineage C (Southern Clade) (88.8 SH-aLRT, 91 UFBoot) (Fig. 2).

Left, maximum likelihood topology of the Alouatta guariba group, with haplotypes, locality numbers (corresponding to map in Fig. 1 and Supplementary Table 1) in parentheses for each haplotype. Values close to the nodes are SH-aLRT and UFBoot supports. Brazilian states are Bahia (BA), Espírito Santo (ES), Goiás (GO), Minas Gerais (MG), Paraná (PR), Rio de Janeiro (RJ), Rio Grande do Sul (RS), São Paulo (SP), and Santa Catarina (SC). Top right, map with sampling localities according to the color of the clades and haplogroups found in the maximum likelihood and median-joining network (MJ) analyses, gray circles represent lineage A (Northern Clade), black circles the lineage B (Northern Clade), and white circles the lineage C (Southern Clade). Bottom right, the MJ network where circles are haplotypes with dimensions proportional to the number of shared sequences, numbers in the lines connecting the circles are nucleotide substitutions and diamond the median vector. The samples used here from ES, RJ, MG, SP, and RS states were obtained in 2013, 2014, 2017, 2018, and 2019.
The MJ network analysis (Fig. 2) with 37 variable sites and 161 sequences showed 26 haplotypes (h), and haplotype diversity (Hd) of 0.8359. The MJ MJ network analysis showed star-like haplogroups corresponding to lineages A, B, and C. Haplogroups A and B were separated by at least six mutations and haplogroups B and C by at least seven mutations. The nucleotide diversity (π), estimated for the dataset according to the haplotypes and haplogroups recovered in ML and MJ, was 0.00367 for lineage A (Northern Clade), 0.00309 for lineage B (Northern Clade), and 0.00306 for lineage C (Southern Clade).
Karyotypes
Karyotypes of lineage A (Northern Clade) from Rio de Janeiro (CPRJ3636 and RJ140) and São Paulo (RTM1526 and RTM1542) are all males with 2n = 49 (Fig. 3). The autosome complement of these four males is composed of seven two-armed pairs, two large, one medium, and four small, and 16 medium to small one-armed pairs. The male sexual chromosome is translocated to an autosome, and the X is a medium biarmed chromosome. Pair numbers 18 and 20 showed a secondary constriction in these four samples, and the karyotype of sample CPRJ3636 also presented a heteromorphic 14th pair (Fig. 3).

Karyotypes of the Northern Clade: lineage A (Northern Clade) with C-banding pattern (RTM1526 from São Paulo state), and Giemsa coloration (CPRJ3636 and RJ140 from Rio de Janeiro state, and RTM1542 from São Paulo state); lineage B (Northern Clade) with Giemsa coloration (females CPRJ2799, CPRJ3622, CPRJ3723, and CPRJ3216 from Rio de Janeiro and males CPRJ2443 from Rio de Janeiro state and RTM1518 from São Paulo state). X and Y are sexual chromosomes. Arrows indicate chromosome pairs with secondary constrictions. The samples from Rio de Janeiro state were obtained in 2018 and the samples from São Paulo in 2019, Brazil.
Karyotypes of males from lineage B (Northern Clade) from Rio de Janeiro (male CPRJ2443) and São Paulo (male RTM1518) also showed 2n = 49 and a chromosome morphology similar to males of lineage A (Northern Clade). The CPRJ2443 karyotype presented a heteromorphic 20th pair, and the CPRJ3723 karyotype presented a heteromorphic 15th pair (Fig. 3).
The karyotypes of females from lineage B (Northern Clade) from Rio de Janeiro (females CPRJ2799, CPRJ3622, CPRJ3723, CPRJ3216) showed 2n = 50 (Fig. 3). The autosome complement is composed of eight two-armed pairs, two large, one medium, and five small, and 16 medium to small pairs with one arm. The X sexual chromosome is a medium biarmed chromosome. Females also showed two pairs of chromosomes with secondary constrictions.
Predicted Potential Distribution
After removing duplicates from each grid cell, we used 249 presence records to model the potential distribution of the Southern Clade and 197 to model that of Northern Clade. The mean training AUC for the replicated runs was 0.982 (SD = 0.001) for the Southern population habitat suitability model and 0.985 (SD = 0.001) for the Northern population habitat suitability model, meaning that the model performance was high (Figure S1). The jackknife test of variable importance for the Southern and Northern population habitat suitability models showed that the environmental variable that best explained the population distributions when used alone is BIO4 (Temperature Seasonality), which is a measure of the change in temperature throughout the year (Figure S2). For the Southern population, the second environmental variable that best explained population distribution is BIO11 (Mean Temperature of Coldest Quarter). This index measures mean temperatures during the three coldest months of the year. The second variable that best explained the distribution of the Northern population is BIO3 (Isothermality), which quantifies how much day-to-night temperatures oscillated relative to annual oscillations.
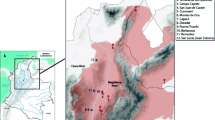
Based on the TPTP cutoff criteria, SDM identified regions in Brazil suitable for the occurrence of Southern and Northern populations (Fig. 4), mainly in Atlantic Forest areas. In the assessment of the recent potential distribution (years 1970 to 2000) for the two species, the geographic range of the Southern population had an area of 464,912.37 km and the Northern population had an area of 492,811.88 km. When assessing the potential distribution for two greenhouse gas emission scenarios (a lower-emission scenario (SSP245) and a worse scenario (SSP585)) in 60 to 79 years (2081–2100), the ideal climatic conditions for the two populations were greatly reduced in geographic coverage. The Southern population had a reduction in geographic coverage of 52% (227,420.43 km) in the most optimistic scenario, and 81% (94,104.18 km) in the worst scenario. The Northern population had a reduction in geographic coverage of 72% (141,025.01 km) in the most optimistic scenario, and 92% (46,944.69 km) in the worst scenario (Fig. 4).

Southern (S) and Northern (N) population habitat suitability model of the A. guariba group as predicted by MaxEnt: (N1 and S1) presence records, (N2 and S2) current distribution suitability, years 1970 to 2000, and two future distribution suitability in 2081–2100, (N3 and S3) in the better scenario SSP2-4.5, and (N4 and S4) in the worse scenario SSP5-8.5.
Discussion
Genetic Diversity of the A. guariba Group Lineages
Our MT-CYB gene tree divided the A. guariba group into two major clades, one with individuals from the South and another with individuals from the North, similar to previous studies (de Mello Martins et al., 2011; Harris et al., 2005; Machado, 2011; Steinberg et al., 2014). Our findings reinforce the association of the Southern Clade with A. g. clamitans and the Northern Clade with A. g. guariba. The Northern Clade was further divided into two clades, named lineage A and lineage B, and we named the Southern Clade, named lineage C. Lineage A (Northern Clade) occurs mostly in the state of Rio de Janeiro, and Lineage B (Northern Clade) in Minas Gerais state, but we found areas of overlap between lineages A and B, and A, B, and C. Although mitochondrial DNA evolves faster than single-copy nuclear DNA and can be used to clarify taxonomic ambiguities and evolutionarily significant units, it still shows some of the limitations associated with using data from a single locus to infer phylogenetic history. Mitochondrial DNA only represents the maternal side and records scant evidence of recent events. As a result, it does not accurately represent the species tree (Wan et al., 2004). Although biparental markers should be taken into consideration for future research with the Alouatta guariba group, mitochondrial DNA is still a powerful tool in conservation genetics, because its shows a strong phylogenetic signal at specific and population level.
In the MJ network and MT-CYB analyses, Southern and Northern populations showed a low number of haplotypes compared to the number of sequences used, and a low nucleotide diversity estimate, even when we analyzed lineages A and B (Northern Clade) separately, a pattern similar to previous studies based on MT-CYB (Bonvicino et al., 2015; Machado, 2011). The greater sample size and the number of sites that we analyzed for samples in the Northern Clade allowed us to better understand the geographic limits of these populations and find instances of sympatry between lineages A and B in the states of Minas Gerais and Rio de Janeiro. These areas of overlap represent an expansion of lineage B (Northern Clade) as shown by neutrality tests, for which the negative value deviated significantly from neutrality, suggesting an excess of rare nucleotide site variants. It is likely that lineages A and B separated recently and accumulated nucleotide differences, allowing us to observe these two isolated lineages. Evidence of expansion was found in the Southern population ca. 5,000 years ago (Machado, 2011), right after a postglacial period (Nascimento et al., 2005), in which the populations emerged from a state of isolation to start expanding. Based on the observed star-like pattern in each lineage, we suggest that the three lineages were likely recently isolated and are expanding. The areas of overlap between lineages A and B and between the three lineages in São Paulo may be associated with the expansion of these three lineages.
A recent study based on mitochondrial marker MT-COI (Petit-Marty et al., 2021) estimated that the nucleotide diversity (π) of threatened mammalian species can be much lower, ranging π = 0.0044–0.0102, than that of nonthreatened mammalian species, ranging π = 0.0128–0.0165. Compared with other Alouatta species, such as Amazon and Atlantic Forest populations of A. belzebul, analyzed by using the same mitochondrial marker (Nascimento et al., 2005, 2007; Povill et al., 2022), the A. guariba group had fewer haplotypes and low nucleotide diversity (Table S4). Alouatta caraya showed low MT-CYB nucleotide diversity than A. guariba group, but more haplotypes (Ascunce et al., 2007; Nascimento et al., 2007). This finding may be related to the large distribution of A. caraya, the strong geographic structuring of the populations, and its limited dispersal, which occurs only along forest corridors (Nascimento et al., 2007). Threatened species, such as A. pigra and A. palliata mexicana, that inhabit areas of continuous forest reduction (Dunn et al., 2014; James et al., 1997; Melo-Carrillo et al., 2020) showed very low mitochondrial nucleotide diversity (Dunn et al., 2014). A. palliata mexicana showed lower genetic diversity than any other Alouatta species. Some species of Alouatta from South America or the Cis-Andean species, such as A. palliata, have low nucleotide diversity (Table S4). A. caraya, A. belzebul (Amazon population), A. palliata mexicana, and the A. guariba group occur in very different environments. Both A. guariba group and A. palliata are species complexes that inhabit small patches, in which the genetic diversity was lower than the other species. The low genetic nucleotide diversity observed in the A. guariba group may result from genetic drift in small and isolated populations, or habitat fragmentation and climatic variation.
Karyotype Analyses Support the Hypothesis that Northern and Southern Populations are Distinct Taxa
We attributed the Northern Clade to A. guariba guariba and the Southern Clade to A. guariba clamitans, based on previous studies and our data for karyotype and phylogenetic relationships, and the geographic distribution of A. guariba guariba (Gregorin, 2006; Kinzey, 1982). However, the type locality of A. guariba guariba in its original description was defined as Brazil by Humboldt in 1812 and later was restricted to Rio Paraguaçu, Bahia state (Cabrera, 1958), a contested action not considered in later publications (Gregorin, 2006) due to the lack of plausible explanations for this restriction. The type locality of A. guariba clamitans was referred to as São Sebastião in São Paulo state, based on the lectotype (Gregorin, 2006).
We attribute the karyotype 2n = 49–50 to A. guariba guariba (Northern Clade) based on previous publications (de Oliveira et al., 2000; Steinberg et al., 2014) and our karyotype results. We attribute the karyotype 2n = 45–46 to A. guariba clamitans (Southern Clade) based on previous karyological (de Oliveira et al., 2000; Steinberg et al., 2014, 2017) and molecular (de Mello Martins et al., 2011; Harris et al., 2005) studies. Analysis of a possible hybrid with karyotype 2n = 48 using fluorescence in situ hybridization showed the presence of chromosomes from lineages from Southern Clade (2n = 45–46) and Northern Clade (2n = 49–50) (Cardoso Coimbra, 2015). The single specimen with 2n = 48, among roughly 65 specimens analyzed with 2n = 45–46 or 2n = 49–50, and the absence of offspring from this hybrid suggests a postzygotic barrier between the lineages from the north and south (Cardoso Coimbra, 2015; Gifalli-Iughetti, 2008) that causes hybrid sterility due to cumulative negative interactions between loci. Evidence of hybridization also has been observed between two Mesoamerican species of Alouatta: A. pigra and A. palliata (Cortés-Ortiz et al., 2015b; Kelaita & Cortés-Ortiz, 2013). The differences in diploid number among Northern and Southern clades of A. guariba group and the differences we found in the number of chromosomal arms present in the autosomal complement of a somatic cell in the Northern Clade support the hypothesis that the A. guariba group is in a process of chromosomal differentiation or speciation (Gifalli-Iughetti, 2008).
Predicted Potential Distributions
In the current potential distribution areas, the two populations are sympatric in a small area in the southeastern part of São Paulo state, and this overlap is lost in the SDM worst scenario for the future. Temperature seasonality (BIO4) was the most important environmental variable to explain the distribution of the northern and southern populations. For the southern population, the second variable (BIO11), related to the coldest quarter in the year, appears to affect the distribution of this lineage, whereas for the northern population, the variable Isothermality (BIO3), related to temperature fluctuations during the year, could influence its distribution (O’Donnell & Ignizio, 2012).
According to the models, the northern and southern populations will lose a large percentage of their suitable distribution areas in the future (2081–2100) due to changes in climate, in both the most optimistic or pessimistic scenarios. The SDM results showed fluctuations in temperature throughout the year and the colder months as the most influential variables in the Northern and Southern A. guariba population distributions. Another factor reinforcing the SDM scenario is a likely reduction in the Atlantic Forest territory, as a result of forest being replaced by savanna (Salazar et al., 2007), caused mainly by the direct effect of the change in land use (Ribeiro et al., 2009). Other studies of the potential distribution of Alouatta suggest that populations are affected by both deforestation and climate changes (Freire Filho & Palmeirim, 2020; Holzmann et al., 2015), supporting the scenario that we suggest, that fluctuations in temperature and the decrease in the Atlantic Forest territory are major threats to the Alouatta populations.
Variations in effective population size may be caused by climate change, as shown in the potential distribution model of the recent past and the future scenarios for the Northern and Southern clades. Another factor affecting population size is pandemic diseases (Harding et al., 2002), which are strong enough to remove the effect of species abundance on mtDNA diversity in mammals (Nabholz et al., 2008). Alouatta guariba lineages are endemic and mostly adapted to the Atlantic Forest, a very fragmented morphoclimatic domain where outbreaks of yellow fever are increasingly frequent. These outbreaks are considered the most important threat to this species in Misiones province, Argentina (Agostini et al., 2017), and are responsible for population declines in other platyrrhine primates from the Atlantic Forest, such as titi monkeys (Berthet et al., 2021). The recent devastating yellow fever epizootics are one of the reasons that the A. guariba group was considered one of the most threatened primates of the world (Estrada et al., 2018; Mittermeier et al., 2022). Outbreaks of yellow fever are more frequent as a result of climate change and forest alterations that also influenced the resilience of Northern and Southern populations of A. guariba. Yellow fever and environmental modifications together influence their future predicted distributions, indicating very pessimistic outcomes.
Conclusions
Our results add to our understanding of the current and future species distribution of the Alouatta guariba group and reveal low genetic diversity in both Northern and Southern populations. The distribution limits for A. guariba guariba and A. guariba clamitans, based on differences between the Northern (A. guariba guariba) and Southern (A. guariba clamitans) populations in karyotype, morphology, and phylogenetics, differ from those established by IUCN. The status of A. guariba guariba and A. guariba clamitans are well supported by molecular, karyotype, and morphologic analyses. Despite being widely distributed, the Northern and Southern clades, designated here as A. g. guariba and A. g. clamitans, respectively, are restricted to small patches in the southeast. To better understand the genetic diversity of A. guariba group, studies should assess their nuclear DNA diversity, such as levels of heterozygosity. The low MT-CYB nucleotide diversity is an additional concern for these populations, and the high fragmentation of Atlantic Forest and recurrent outbreaks of yellow fever represent potential threats to these taxa. These data reinforce the IUCN Red List categories of A. guariba clamitans as Vulnerable (Buss et al., 2021) and A. guariba guariba as Critically Endangered (Neves et al., 2021).
Data Availability
The datasets generated and analyzed during the present study were uploaded to NCBI (Accession OP985658—OP985779).
Change history
01 July 2023
A minor correction has been made in the article title.
References
Abreu, F. V. S., Dos Santos, E., Gomes, M. Q., Vargas, W. P., de Oliveira Passos, P. H., Nunes e Silva, C., Araújo, P. C., Pires, J. R., Romano, A. P. M., Teixeira, D. S., & Lourenço-de-Oliveira, R. (2019). Capture of Alouatta guariba clamitans for the surveillance of sylvatic yellow fever and zoonotic malaria: Which is the best strategy in the tropical Atlantic Forest? American Journal of Primatology, 81(6), e23000. https://doi.org/10.1002/ajp.23000
Agostini, I., Holzmann, I., & Di Bitetti, M. (2017). The brown howler monkey (Alouatta guariba clamitans) in Argentina: Current status, threats and conservation challenges. In M. Kowalewski & L. I. Oklander (Eds.), Primatology in Argentina (pp. 475–480). Sociedad Argentina para Estudio de los Mamíferos SAREM.
Almeida, M. A. B., Santos, E., Cardoso, J. C., Fonseca, D. F., Noll, C. A., Silveira, V. R., Maeda, A. Y., de Souza, R. P., Kanamura, C., & Brasil, R. A. (2012). Yellow fever outbreak affecting Alouatta populations in Southern Brazil (Rio Grande do Sul State), 2008–2009. American Journal of Primatology, 74(1), 68–76. https://doi.org/10.1002/ajp.21010
Ascunce, M. S., Hasson, E., Mulligan, C. J., & Mudry, M. D. (2007). Mitochondrial sequence diversity of the southernmost extant New World monkey, Alouatta Caraya. Molecular Phylogenetics and Evolution, 43(1), 202–215. https://doi.org/10.1016/j.ympev.2006.10.004
Bandelt, H. J., Forster, P., & Röhl, A. (1999). A Median-joining network for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16, 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Berthet, M., Mesbahi, G., Duvot, G., Zuberbühler, K., Cäsar, C., & Bicca-Marques, J. C. (2021). Dramatic decline in a titi monkey population after the 2016–2018 sylvatic yellow fever outbreak in Brazil. American Journal of Primatology, 83(12), e23335. https://doi.org/10.1002/ajp.23335
Bicca-Marques, J. C., Calegaro-Marques, C., Rylands, A. B., Strier, K. B., Mittermeier, R. A., De Almeida, M. A. B., De Castro, P. H. G., Chaves, Ó. M., Ferraz, L. P., Fortes, V. B., Hirano, Z. M. B., Jerusalinsky, L., Kowalewski, M., Martins, W. P., De Melo, F. R., Mendes, S. L., Neves, L. G., Passos, F. C., Port-Carvalho, M., … Texeira, D. S. (2017). Yellow fever threatens Atlantic forest primates. Science Advances, 3, e1600946.
Bonvicino, C. R., Viana, M. C., Bueno, C., Santos, H. S., & Geise, L. (2015). Genetic diversity of Alouatta (Primates) from Brazilian Atlantic Forest. Journal of Primatology, 4(131), 2. https://doi.org/10.4172/2167-6801.1000131
Boubli, J. P., Byrne, H., Silva, M. N. F., José Silva-Júnior, J. S., Araújo, R. C., Bertuol, F., Gonçalves, J., Melo, F. R., Rylands, A. B., Mittermeier, R. A., Silva, F. E., Nash, S. D., Canale, G., Alencar, R. M., Rossi, R. V., Carneiro, J., Sampaio, I., Farias, I. P., Scheneider, H., & Hrbek, T. (2016). On a new species of titi monkey (Primates: Plecturocebus), from Alta Floresta, southern Amazon, Brazil. Molecular Phylogenetics and Evolution, 132, 117–137. https://doi.org/10.1016/j.ympev.2018.11.012
Buss, G., Bicca-Marques, J. C., Alves, S. L., Ingberman, B., Fries, B. G., Alonso, A. C., da Cunha, R. G. T., Miranda, J. M. D., de Melo, F. R., Jerusalinsky, L., Mittermeier, R. A., Cortés-Ortiz, L., & Talebi, M. (2021). Alouatta guariba ssp. clamitans (amended version of 2020 assessment). The IUCN Red List of Threatened Species, 2021, e.T39918A190419216. https://doi.org/10.2305/IUCN.UK.2021-1.RLTS.T39918A190419216.en
Cabrera, A. (1958). Catálogo de los mamíferos de América del Sur. Revista del Museo Argentino de Ciencias Naturales “Bernardino Rivadavia”. 4(1), 1–307.
Cardoso Coimbra, A. P. (2015). Cytogenetic studies in Alouatta clamitans (Primates, Platyrrhini): analysis of chromosomal variability of howler monkeys from South and Southeast regions of Brazil [Master’s dissertation], University of São Paulo.
Chapman, C. A., Lawes, M. J., & Eeley, H. A. C. (2006). What hope for African primate diversity? African Journal of Ecology, 44, 116–133. https://doi.org/10.1111/j.1365-2028.2006.00636.x
Coates, D. J., Byrne, M., & Moritz, C. (2018). Genetic diversity and conservation units: Dealing with the species-population continuum in the age of genomics. Frontiers in Ecology and Evolution, 6, 165. https://doi.org/10.3389/fevo.2018.00165
Collins, M., Knutti, R., Arblaster, J., Dufresne, J.-L., Fichefet, T., Friedlingstein, P., Gao, X., Gutowski, W. J., Johns, T., Krinner, G., Shongwe, M., Tebaldi, C., Weaver, A. J., Wehner, M. F., Allen, M. R., Andrews, T., Beyerle, U., Bitz, C. M., Bony, S., & Booth, B. B. B. (2013). Long-term climate change: Projections, commitments and irreversibility. In T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, & P. M. Midgley (Eds.), Climate change 2013: the physical science basis. Contribution of working group I to the fifth assessment report of the intergovernmental panel on climate change. Cambridge University Press.
Cortés-Ortiz, L., Bermingham, E., Rico, C., Rodríguez-Luna, E., Sampaio, I., & Ruiz-Garcia, M. (2003). Molecular systematics and biogeography of the Neotropical monkey genus, Alouatta. Molecular Phylogenetics and Evolution, 26(1), 64–81. https://doi.org/10.1016/S1055-7903(02)00308-1
Cortés-Ortiz, L., Rylands, A. B., & Mittermeier, R. A. (2015a). The taxonomy of howler monkeys: integrating old and new knowledge from morphological and genetic studies. In M. Kowalewski, P. Garber, L. Cortés-Ortiz, B. Urbani, & D. Youlatos (Eds.), Howler monkeys (pp. 55–84). Developments in primatology: progress and prospects. Springer. https://doi.org/10.1007/978-1-4939-1957-4_3
Cortés-Ortiz, L., Agostini, I., Aguiar, L. M., Kelaita, M., Silva, F. E., & Bicca-Marques, J. C. (2015b). Hybridization in howler monkeys: Current understanding and future directions. In M. Kowalewski, P. Garber, L. Cortés-Ortiz, B. Urbani, & D. Youlatos (Eds.), Howler monkeys (pp. 107–131). Developments in primatology: progress and prospects. Springer. https://doi.org/10.1007/978-1-4939-1957-4_5
Costa-Araújo, R., Silva, J. S., Boubli, J. P., Rossi, R. V., Canale, G. R., Melo, F. R., Bertuol, F., Silva, F. E., Silva, D. A., Nash, S. D., Sampaio, I., Farias, I. P., & Hrbek, T. (2021). An integrative analysis uncovers a new, pseudo-cryptic species of Amazonian marmoset (Primates: Callitrichidae: Mico) from the arc of deforestation. Scientific Reports, 11, 15665. https://doi.org/10.1038/s41598-021-93943-w
Culot, L., Pereira, L. A., Agostini, I., de Almeida, M. A. B., Alves, R. S. C., Aximoff, I., Bager, A., Baldovino, M. C., Bella, T. R., Bicca-Marques, J. C., Braga, C., Brocardo, C. R., Campelo, A. K. N., Canale, G. R., Cardoso, J. C., Carrano, E., Casanova, D. C., Cassano, C. R., Castro, E., & Galetti, M. (2019). Atlantic-primates: a dataset of communities and occurrences of primates in the Atlantic Forests of South America. Ecology, 100(1), e02525. https://doi.org/10.1002/ecy.2525
Dalponte, J. C., Silva, F. E., & Silva, J. S., Jr. (2014). New species of titi monkey, genus Callicebus Thomas, 1903 (Primates, Pitheciidae), from Southern Amazonia, Brazil. Papeis Avulsos De Zoologia, 54(32), 457–472. https://doi.org/10.1590/0031-1049.2014.54.32
de Almeida, M. A., Dos Santos, E., Cardoso, J. D. C., da Silva, L. G., Rabelo, R. M., & Bicca-Marques, J. C. (2019). Predicting yellow fever through species distribution modeling of virus, vector, and monkeys. EcoHealth, 16(1), 95–108. https://doi.org/10.1007/s10393-018-1388-4
de Mello Martins, F., Gifalli-Iughetti, C., Koiffman, C. P., & Harris, E. E. (2011). Coalescent analysis of mtDNA indicates Pleistocene divergence among three species of howler monkey (Alouatta spp.) and population subdivision within the Atlantic Coastal Forest species, A. guariba. Primates, 52(1), 77–87. https://doi.org/10.1007/s10329-010-0226-2
de Oliveira, E. H. C., de Lima, M. C., Sbalqueiro, I. J., & Pissinati, A. (1998). The karyotype of Alouatta fusca clamitans from Rio de Janeiro, Brazil: Evidence for a y-autosome translocation. Genetics and Molecular Biology, 21(3). https://doi.org/10.1590/S1415-47571998000300012
de Oliveira, E. H. C., Suemitsu, E., da Silva, A. F., & Sbalqueiro, I. J. (2000). Geographical variation of chromosomal number in Alouatta fusca clamitans (Primates, Atelidae). Caryologia, 53(2), 163–168. https://doi.org/10.1080/00087114.2000.10589192
Dunn, J. C., Shedden-González, A., Cristóbal-Azkarate, J., Cortés-Ortiz, L., Rodríguez-Luna, E., & Knapp, L. A. (2014). Limited genetic diversity in the critically endangered Mexican howler monkey (Alouatta palliata mexicana) in the Selva Zoque. Mexico. Primates, 55(2), 155–160. https://doi.org/10.1007/s10329-013-0399-6
Elith, J., Graham, C. H., Anderson, R. P., Dudík, M., Ferrier, S., Guisan, A., Hijmans, R. J., Huettmann, F., Leathwick, J. R., Lehmann, A., Li, J., Lohmann, L. G., Loiselle, B. A., Manion, G., Moritz, C., Nakamura, M., Nakazawa, Y., Overton, J. M. C., Peterson, A. T., … Zimmermann, N. E. (2006). Novel methods improve prediction of species’ distributions from occurrence data. Ecography, 29, 129–151. https://doi.org/10.1111/j.2006.0906-7590.04596.x
Estrada, A., Garber, P. A., Rylands, A. B., Roos, C., Fernandez-Duque, E., Di Fiore, A., Nekaris, K. A., Nijman, V., Heymann, E. W., Lambert, J. E., Rovero, F., Barelli, C., Setchell, J. M., Gillespie, T. R., Mittermeier, R. A., Arregoitia, L. V., Guinea, M., Gouveia, S., Dobrovolski, R., … Li, B. (2017). Impending extinction crisis of the world’s primates: Why primates matter. Science Advances, 3(1), e1600946. https://doi.org/10.1126/sciadv.1600946
Estrada, A., Garber, P. A., Mittermeier, R. A., Wich, S., Gouveia, S., Dobrovolski, R., Nekaris, K. A. I., Nijman, V., Rylands, A. B., Maisels, F., Williamson, E. A., Bicca-Marques, J., Fuentes, Jerusalinsky, L., Johnson, S., Melo, F. R., Oliveira, L., Schitzer, C., Ross, C., … Setiawan, A. (2018). Primates in peril: the significance of Brazil, Madagascar, Indonesia and the Democratic Republic of the Congo for global primate conservation. PeerJ, 6, e4869. https://doi.org/10.7717/peerj.4869
Excoffier, L., & Lischer, H. E. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fick, S. E., & Hijmans, R. J. (2017). WorldClim 2: New 1km spatial resolution climate surfaces for global land areas. International Journal of Climatology, 37(12), 4302–4315. https://doi.org/10.1002/joc.5086
Frankham, R., Ballou, S. E. J. D., Briscoe, D. A., & Ballou, J. D. (2002). Introduction to conservation genetics. Cambridge University Press.
Freire Filho, R., & Palmeirim, J. M. (2020). Potential distribution of and priority conservation areas for the endangered Caatinga howler monkey Alouatta ululata in north-eastern Brazil. Oryx, 54(6), 794–802. https://doi.org/10.1017/S0030605318001084
GBIF.org (2021). GBIF Home Page. Available from: https://www.gbif.org. Accessed 17 March 2021.
Gifalli-Iughetti, C. (2008). Evolução cromossômica: Estudo da variabilidade cariotípica em Platyrrhini e das homeologias e sintenias com cromossomos humanos [unpublished doctoral dissertation]. Universidade de São Paulo.
Gregorin, R. (2006). Taxonomia e variação geográfica das espécies do gênero Alouatta Lacépède (Primates, Atelidae) no Brasil. Revista Brasileira De Zoologia, 23(1), 64–144. https://doi.org/10.1590/S0101-81752006000100005
Gualda-Barros, J., Nascimento, F. O., & Amaral, M. K. (2012). A new species of Callicebus Thomas, 1903 (Primates, Pitheciidae) from the states of Mato Grosso and Pará, Brazil. Papeis Avulsos De Zoologia, 52(23), 261–279. https://doi.org/10.1590/S0031-10492012002300001
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., & Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3), 307–321. https://doi.org/10.1093/sysbio/syq010
Hansen, M. C., Potapov, P. V., Moore, R., Hancher, M., Turubanova, S. A., Tyukavina, A., Thau, D., Stehman, S. V., Goetz, S. J., Loveland, T. R., Kommareddy, A., Egorov, A., Chini, L., Justice, C. O., & Townshend, J. R. G. (2013). High-resolution global maps of 21st-Century forest cover change. Science, 342(6160), 850–853. https://doi.org/10.1126/science.1244693
Harding, K. C., Härkönen, T., & Caswell, H. (2002). The 2002 European seal plague: Epidemiology and population consequences. Ecology Letters, 5(6), 696–704. https://doi.org/10.1046/j.1461-0248.2002.00390.x
Harris, E., Gifalli-Iughetti, C., Hirano, Z., & Koiffmann, C. (2005). Cytochrome b sequences show subdivision between populations of the brown howler monkey (Alouatta guariba) from Rio de Janeiro and Santa Catarina. Brazil. Neotropical Primates, 13(2), 16–21. https://doi.org/10.1896/1413-4705.13.2.16
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G., & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology: A Journal of the Royal Meteorological Society, 25(15), 1965–1978. https://doi.org/10.1002/joc.1276
Hijmans, R. J., Phillips, S., Leathwick, J., Elith, J., & Hijmans, M. R. J. (2017). Package ‘dismo.’ Circles, 9(1), 1–68.
Hijmans, R. J., Van Etten, J., Cheng, J., Mattiuzzi, M., Sumner, M., Greenberg, J. A., Lamigueiro, O. P., Bevan, A., Racine, E. B., Shortridge, A., & Hijmans, M. R. J. (2015). Package ‘raster’. R package, 734.
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., & Vinh, L. S. (2018). UFBoot2: Improving the ultrafast bootstrap approximation. Molecular Biology and Evolution, 35(2), 518–522. https://doi.org/10.1093/molbev/msx281
Hoban, S., Campbell, C. D., da Silva, J. M., Ekblom, R., Funk, W. C., Garner, B. A., Godoy, J. A., Kershaw, F., MacDonald, A. J., Mergeay, J., Minter, M., O’Brien, D., Vinas, I. P., Pearson, S. K., Pérez-Espona, S., Potter, K. M., Russo, I.-R.M., Segelbacher, G., Vernesi, C., & Hunter, M. E. (2021). Genetic diversity is considered important but interpreted narrowly in country reports to the convention on biological diversity: Current actions and indicators are insufficient. Biological Conservation, 261, 109233. https://doi.org/10.1016/j.biocon.2021.109233
Holzmann, I., Agostini, I., DeMatteo, K., Areta, J. I., Merino, M. L., & Di Bitetti, M. S. (2015). Using species distribution modeling to assess factors that determine the distribution of two parapatric howlers (Alouatta spp.) in South America. International Journal of Primatology, 36(1), 18–32. https://doi.org/10.1007/s10764-014-9805-1
INPE. (2020). Instituto Nacional de Pesquisas Espaciais. Available from: http://www.inpe.br/noticias/noticia.php?Cod_Noticia=5450. Accessed 18 Jan 2022.
Irwin, D. M., Kocher, T. D., & Wilson, A. C. (1991). Evolution of the cytochrome b gene of mammals. Journal of Molecular Evolution, 32(2), 128–144. https://doi.org/10.1007/BF02515385
James, R. A., Leberg, P. L., Quattro, J. M., & Vrijenhoek, R. C. (1997). Genetic diversity in black howler monkeys (Alouatta pigra) from Belize. American Journal of Physical Anthropology, 102(3), 329–336. https://doi.org/10.1002/(SICI)1096-8644(199703)102:3%3c329::AID-AJPA3%3e3.0.CO;2-Q
Joly, S., Stevens, M. I., & van Vuuren, B. J. (2007). Haplotype networks can be misleading in the presence of missing data. Systematic Biology, 56(5), 857–862. https://doi.org/10.1080/10635150701633153
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., Von Haeseler, A., & Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nature Methods, 14(6), 587–589. https://doi.org/10.1038/nmeth.4285
Kelaita, M. A., & Cortés-Ortiz, L. (2013). Morphological variation of genetically confirmed Alouatta pigra× A. palliata hybrids from a natural hybrid zone in Tabasco. Mexico. American Journal of Physical Anthropology, 150(2), 223–234. https://doi.org/10.1002/ajpa.22196
Kinzey, W. G. (1982). Distribution of primates and forest refuges. In G. T. Prance (Ed.), Biological diversification in the tropics (pp. 455–482). Columbia Univ. Press.
Koiffmann, C. P., & Saldanha, P. H. (1974). Cytogenetics of Brazilian monkeys. Journal of Human Evolution, 3(4), 275–282. https://doi.org/10.1016/0047-2484(74)90021-9
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). Mega X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. https://doi.org/10.1093/molbev/msy096
Machado, S. (2011). Filogeografia do Bugio Ruivo, Alouatta guariba (Primates, Atelidae). [unpublished Master’s dissertation], Pontifícia Universidade Católica do Rio Grande do Sul.
Marsh, L. K. (2014). A taxonomic revision of the saki monkeys, Pithecia Desmarest, 1804. Neotropical Primates, 21(1), 1–163. https://doi.org/10.1896/044.021.0101
Melo-Carrillo, A., Dunn, J. C., & Cortés-Ortiz, L. (2020). Low genetic diversity and limited genetic structure across the range of the critically endangered Mexican howler monkey (Alouatta palliata mexicana). American Journal of Primatology, 82(8), e23160. https://doi.org/10.1002/ajp.23160
Miller, S. A., Dykes, D. D., & Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 16(3), 1215. https://doi.org/10.1093/nar/16.3.1215
Mittermeier, R. A., Reuter, K. E., Rylands, A. B., Jerusalinsky, L., Schwitzer, C., Strier, K. B., Ratsimbazafy, J., & Humle, T. (Eds.). (2022). Primates in peril: The world’s 25 most endangered primates 2022–2023. IUCN SSC Primate Specialist Group.
Nabholz, B., Mauffrey, J. F., Bazin, E., Galtier, N., & Glemin, S. (2008). Determination of mitochondrial genetic diversity in mammals. Genetics, 178(1), 351–361. https://doi.org/10.1534/genetics.107.073346
Nascimento, F. D., Bonvicino, C. R., Da Silva, F. C. D., Schneider, M. P. C., & Seuánez, H. N. (2005). Cytochrome b polymorphisms and population structure of two species of Alouatta (Primates). Cytogenetic and Genome Research, 108(1–3), 106–111. https://doi.org/10.1159/000080808
Nascimento, F. F., Bonvicino, C. R., & Seuánez, H. N. (2007). Population genetic studies of Alouatta caraya (Alouattinae, Primates): Inferences on geographic distribution and ecology. American Journal of Primatology, 69(10), 1093–1104. https://doi.org/10.1002/ajp.20423
Nascimento, F. F., Bonvicino, C. R., de Oliveira, M. M., Schneider, M. P. C., & Seuánez, H. N. (2008). Population genetic studies of Alouatta belzebul from the Amazonian and Atlantic Forests. American Journal of Primatology, 70(5), 423–431. https://doi.org/10.1002/ajp.20507
Neves, L. G., Jerusalinsky, L., Talebi, M., Mittermeier, R. A., Cortés-Ortiz, L., & de Melo, F. R. (2021). Alouatta guariba ssp. guariba (amended version of 2020 assessment). The IUCN Red List of Threatened Species 2021: e.T39917A190420483. Downloaded on 05 April 2021.
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., & Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274. https://doi.org/10.1093/molbev/msu300
O’Donnell, M. S., & Ignizio, D. A. (2012). Bioclimatic predictors for supporting ecological applications in the conterminous United States. US Geological Survey Data Series, 691(10), 4–9.
Osada, N. (2015). Genetic diversity in humans and non-human primates and its evolutionary consequences. Genes & Genetic Systems, 90(3), 133–145. https://doi.org/10.1266/ggs.90.133
Pearson, R. G., Raxworthy, C. J., Nakamura, M., & Townsend Peterson, A. (2007). Predicting species distributions from small numbers of occurrence records: A test case using cryptic geckos in Madagascar. Journal of Biogeography, 34(1), 102–117. https://doi.org/10.1111/j.1365-2699.2006.01594.x
Peterson, A. T., Soberon, J., Pearson, R. G., Anderson, R. P., Martinez-Meyer, E., Nakamura, M., & Araujo, M. B. (2011). Ecological niches and geographic distributions. Princeton University Press.
Petit-Marty, N., Vázquez-Luis, M., & Hendriks, I. E. (2021). Use of the nucleotide diversity in COI mitochondrial gene as an early diagnostic of conservation status of animal species. Conservation Letters, 14(1), e12756. https://doi.org/10.1111/conl.12756
Phillips, S. J., Anderson, R. P., & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190, 231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026
Pissinatti, A., Muniz, J. A. P. C., da Silva, K. S. M., & Andrade, M. C. R. (2016). Primatas não humanos mantidos em instalações de instituições de ensino e pesquisa científica. Resolução Normativa nº28 in CONCEA (Ed.), Guia brasileiro de produção, manutenção ou utilização de animais em atividades de ensino ou pesquisa científica (pp. 40–93). Ministério da Ciência, Tecnologia e Inovação.
Povill, C., de Assis Passos Oliveira, M., de Melo, F. R., & Bonvicino, C. R. (2022). Phylogenetic relationships, population demography, and species delimitation of the Alouatta belzebul species complex (Atelidae: Alouattinae). Primates, 63, 65–78.https://doi.org/10.1007/s10329-021-00959-x
Reed, D. H., & Frankham, R. (2003). Correlation between fitness and genetic diversity. Conservation Biology, 17, 230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Rezende, C. L., Scarano, F. R., Assad, E. D., Joly, C. A., Metzger, J. P., Strassburg, B. B. N., Tabarelli, M., Fonseca, G. A., & Mittermeier, R. A. (2018). From hotspot to hopespot: An opportunity for the Brazilian Atlantic Forest. Perspectives in Ecology and Conservation, 16, 208–214. https://doi.org/10.1016/j.pecon.2018.10.002
Ribeiro, M. C., Metzger, J. P., Martensen, A. C., Ponzoni, F. J., & Hirota, M. M. (2009). The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implications for conservation. Biological Conservation, 142, 1141–1153. https://doi.org/10.1016/j.biocon.2009.02.021
Ricklefs, R. E. (2004). A comprehensive framework for global patterns in biodiversity. Ecology Letters, 7, 1–15. https://doi.org/10.1046/j.1461-0248.2003.00554.x
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., & Sánchez-Gracia, A. (2017). DnaSP 6: DNA sequence polymorphism analysis of large datasets. Molecular Biology and Evolution, 34(12), 3299–3302. https://doi.org/10.1093/molbev/msx248
Rylands, A. B. (2000). An assessment of the diversity of New World primates. Neotropical Primates, 8, 61–93.
Rylands, A. B., & Mittermeier, R. A. (2009). The diversity of the New World primates (Platyrrhini): an annotated taxonomy. In P. A. Garber, A. Estrada, J, C. Bicca-Marques, E. W. Heymann, & K. B. Strier (Eds.), South American Primates (pp. 23–54). Springer. https://doi.org/10.1007/978-0-387-78705-3_2
Sacchetto, L., Silva, N. I. O., Rezende, I. M. D., Arruda, M. S., Costa, T. A., de Mello, É. M., Oliveira, G. F. G., Alves, P. A., de Mendonça, V. E., Stumpp, R. G. A. V., Prado, A. I. A., Paglia, A. P., Perini, F. A., Nogueira, M. L., Kroon, E. G., de Thoisy, B., Trindade, G. S., & Drumond, B. P. (2020). Neighbor danger: Yellow fever virus epizootics in urban and urban-rural transition areas of Minas Gerais state, during 2017–2018 yellow fever outbreaks in Brazil. PLoS Neglected Tropical Diseases, 14(10), e0008658. https://doi.org/10.1371/journal.pntd.0008658
Salazar, L. F., Nobre, C. A., & Oyama, M. D. (2007). Climate change consequences on the biome distribution in tropical South America. Geophysical Research Letters, 34(9), L09708. https://doi.org/10.1029/2007GL029695
Sambrook, J., & Russell, D. W. (2001). Molecular cloning: a laboratory manual (3rd ed.). Cold Spring Harbor Laboratory Press.
Steinberg, E. R., Nieves, M., & Mudry, M. D. (2014). Multiple sex chromosome systems in howler monkeys (Platyrrhini, Alouatta). Comparative Cytogenetics, 8(1), 43. https://doi.org/10.3897/CompCytogen.v8i1.6716
Steinberg, E. R., Fortes, V. B., Rossi, L. F., Murer, L., Lovato, M., Merani, M. S., & Mudry, M. D. (2017). Cytogenetic characterization of brown howler monkeys, Alouatta guariba clamitans (Atelidae, Platyrrhini): Meiotic confirmation of an X1X1X2X2X3X3/X1X2X3Y1Y2 sex chromosome system. Cytogenetic and Genome Research, 151(3), 131–140. https://doi.org/10.1159/000464375
Sumner, A. T. (1972). A simple technique for demonstrating centromeric heterochromatin. Experimental Cell Research, 75, 304–306.
Swets, J. A. (1988). Measuring the accuracy of diagnostic systems. Science, 240(4857), 1285–1293. https://doi.org/10.1126/science.3287615
Teixeira, D. S., Bernal-Valle, S., Ramos, A. V. V., Santos, L. K. N. S. S., dos Abreu, F. V. S., Santos, E., Bandeira, J. C., & de Almeida, R. M. (2022). Capture and collection of biological samples from free-living Neotropical Primates. Primate Conservation, 36, 01–12.
Urbanek, S. (2021). Package ‘rJava’. R package, 48.
Vincent, L. A., Peterson, T. C., Barros, V. R., Marino, M. B., Rusticucci, M., Carrasco, Ramirez, E., Alves, L. M., Ambrizzi, T., Berlato, M. A., Grimm, A. M., Marengo, J. A., Molion, L., Moncunill, D. F., Rebello, E., Anunciação, Y. M. T., Quintana, J., Santoso, J. L., Baez, J., … Karoly, D. (2005). Observed trends in indices of daily temperature extremes in South America 1960–2000. Journal of Climate, 18(23), 5011–5023. https://doi.org/10.1175/JCLI3589.1
Wan, Q. H., Wu, H., Fujihara, T., & Fang, S. G. (2004). Which genetic marker for which conservation genetics issue? Electrophoresis, 25(14), 2165–2176. https://doi.org/10.1002/elps.200305922
Wiederholt, R., & Post, E. (2010). Tropical warming and the dynamics of endangered primates. Biology Letters, 6(2), 257–260. https://doi.org/10.1098/rsbl.2009.0710
Winters, M., Devictor, V., & Schweiger, O. (2012). Phylogenetic diversity and nature conservation: Where are we? Trends in Ecology and Evolution, 28, 199–204.
Acknowledgements
The authors are grateful to Dr. Julio Cesar Bicca-Marques and anonymous reviewers for the comments and suggestions in a previous version of this manuscript. Thanks to the veterinarian Silvia Bahadian Moreira for samples collection of Alouatta in captivity from Centro de Primatologia do Rio de Janeiro; to Department of Applied Research of Fundação Parque Zoológico de São Paulo for samples from Parque Estadual das Fontes do Ipiranga (PEFI) and the veterinarian staff; and to Danilo Simonini Teixeira and Marcelo Quintela Gomes for their support during the fieldwork in Rio de Janeiro. This study was developed thanks to scholarship awarded in part by the Coordenação de Aperfeiçoamento de Nível Superior (CAPES-88882.331370/2019-01) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ-E-26/200.742/2019) to Cintia Povill, and from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq- 304498/2014-9 and 312446/2018-7) and FAPERJ (E26/201.200/2014 and E-26/210.047/2014) to Cibele R. Bonvicino and R. Lourenço-de-Oliveira, respectively, and Marcione B. Oliveira thanks to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, 162273/2018-5) for the PhD scholarship.
Author information
Authors and Affiliations
Contributions
CP conceived (lead), designed (lead), performed the experiments (lead), analyzed the data (lead), writing, review and editing (lead). CRB conceived (equal), funding acquisition (lead), writing review & editing (equal). MBO analyzed and wrote spatial distribution (lead), writing review and editing (equal). FVSA performed the experiments (equal). FVSA, RLO, ES, FAP, CB, CM, AP, CRB got the samples, writing review and editing (supporting), contributed to the final version (supporting).
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Handling Editor: Julio Cesar Bicca-Marques
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Povill, C., de Oliveira, M.B., de Abreu, F.V.S. et al. Genetic Diversity and Insights into the Distribution of Brown Howler Monkeys (Alouatta guariba Group) (Atelidae, Alouattinae). Int J Primatol 44, 517–539 (2023). https://doi.org/10.1007/s10764-023-00352-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10764-023-00352-z