
Abstract
Germline mutations in BRCA1 and BRCA2 cause hereditary breast and ovarian cancer. Molecular screening of these two genes in patients with a family history of breast or ovarian cancer has revealed pathogenic variants as well as genetic variants of unknown significance (VUS). These VUS may cause a challenge in the genetic counseling process regarding clinical management of the patient and the family. We investigated 32 variants previously detected in 33 samples from patients with a family history of breast or ovarian cancer. cDNA was analyzed for alternative transcripts and selected missense variants located in the BRCT domains of BRCA1 were assessed for their trans-activation ability. Although an extensive cDNA analysis was done, only three of the 32 variants appeared to affect the splice-process (BRCA1 c.213-5T>A, BRCA1 c.5434C>G and BRCA2 c.68-7T>A). In addition, two variants located in the BRCT domains of BRCA1 (c.5075A>C p.Asp1692Ala and c.5513T>G p.Val1838Gly) were shown to abolish the BRCT domain trans-activation ability, whereas BRCA1 c.5125G>A p.Gly1709Arg exhibited equal trans-activation capability as the WT domain. These functional studies may offer further insights into the pathogenicity of certain identified variants; however, this assay is only applicable for a subset of missense variants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
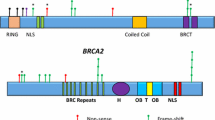
The BRCA1 gene consists of 23 exons and encodes a 208 kDa protein encompassing 1863 amino acids (aa) [1]. N-terminally, BRCA1 has a RING-domain (aa 8–96) and two nuclear localization signals (aa 200–300) [2]. It also contains a phosphorylation site for Checkpoint Kinase 2 (CHEK2) protein at Ser988, a coiled coil domain (aa 1364–1437), followed by several phosphorylation sites for Ataxia Telangiectasia Mutated protein (ATM) (between aa 1280–1524) and two trans-activating BRCT-domains (aa 1646–1859) [2]. BRCA1 has several interactions partners, for instance BRCA1 associated RING domain 1 (BARD1) protein, which interacts with the RING-domain during homologous recombination repair (HRR) [2].
The BRCA2 gene consists of 27 exons and encodes a 384 kDa protein encompassing 3418 aa [1]. BRCA2 has eight BRC-repeats spaced evenly from aa 1009–2083, a helical domain, three oligonucleotide binding folds and a tower domain [2]. C-terminally, BRCA2 has two nuclear localization signals and a Cyclin Dependent Kinase 2 (CDK2) phosphorylation site at Ser3291 [2]. N-terminally, BRCA2 has the ability to interact with Partner And Localizer of BRCA2 (PALB2) at aa 21–39, overlapping with exon 3 (aa 23–106) [3]. The physical connection between BRCA2 and PALB2 is important because PALB2 links BRCA2 and BRCA1 during HRR, at the coiled coil domain of BRCA1 [2].
Together, mutated BRCA1 and BRCA2 are responsible for about 15–25 % of familial breast and ovarian cancer cases [4, 5]. Pathogenic variants in BRCA1 and BRCA2 are estimated to give a 40–87 % risk of breast cancer and a 11–68 % risk of ovarian cancer by age 70 [6]. Since the identification of BRCA1 and BRCA2, many pathogenic variants have been reported in these two genes. The Breast cancer information core (BIC) database includes over 1700 distinct variants in BRCA1 and approximately 2000 in BRCA2 (https://research.nhgri.nih.gov/projects/bic/). However, many of these variants are classified as variants of unknown significance (VUS) and include synonymous, missense, intronic and in-frame deletions/insertions. Missense mutations have the capacity to affect protein function; additionally they may also disturb mRNA splicing. Similarly, synonymous variants, intronic variants outside the consensus splice sites (ss) and deletions/insertions may also cause aberrant splicing. This has been reported for several genes including BRCA1 and BRCA2 [7–9].
Several normal alternative transcripts have been reported both for BRCA1 and BRCA2 [10–13]. The Evidence based Network for the Interpretation of Germline Mutation Alleles (ENIGMA) consortium reported 63 splicing events in BRCA1 and 24 in BRCA2 [11, 13]. Ten of the 63 BRCA1 alternative splicing events and four of the BRCA2 alternative splicing events were considered major splicing events, thus complicating the investigation of aberrant splicing [11, 13]. In this study we assessed the consequences of some of the variants detected in a Norwegian breast and ovarian cancer cohort, by performing cDNA analysis and evaluating the functional consequences of variants located in the BRCA1 C-Terminal (BRCT) domains (aa 1646–1859) using a trans-activation assay [14, 15].
Materials and methods
Patients and samples
Thirty-three whole-blood samples collected in RNA preserving tubes (PAXgene tubes) were obtained from the University Hospital of Oslo, Norway. The samples were collected from unrelated patients who were carriers of sequence variants in BRCA1 or BRCA2 (Table 1). All patients had a family history of breast or ovarian cancer. Complete sequencing of the coding regions, corresponding exon–intron borders and parts of the 5′ and 3′ untranslated regions in BRCA1 and BRCA2 as well as multiplex ligation-dependent probe amplification (MLPA) were previously performed for all patients. In total, these patients carried 18 variants in BRCA1 and 14 variants in BRCA2 (Table 1). As controls, samples from individuals without a family history of breast and ovarian cancer were used.
RNA isolation and cDNA synthesis
RNA was isolated from the PAXgene tubes using the PAXgene Blood RNA Kit (PreAnalytiX, Hombrechtikon, Switzerland) according to the manufacturer’s protocol. cDNA was synthesized using the SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen, Waltham, MA USA).
Nomenclature
Variants were named following recommendations from the Human Genome Variation Society (HGVS) [16]. Reference sequences for BRCA1 and BRCA2 were NM_007294.3 and NM_000059.3, respectively. Custom numbering was used for BRCA1.
Bioinformatic tools
Primers were designed using the Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0/) [17, 18]. In silico evaluation of the variants was done with Alamut Visual version 2.7 (Interactive Biosoftware, Rouen, France), which includes the missense prediction programs Align GVGD, SIFT, MutationTaster and PolyPhen-2 and the the splice prediction tools SpliceSiteFinder-like (SSF), MaxEntScan (MES), NNSPLICE, GeneSplicer (GS) and Human Splicing Finder (HSF). Thresholds were set to zero for all splice prediction tools. The Alamut Visual software also provides results and/or links to the following databases the Exome Aggregation Consortium (ExAC), the Exome Variant Server (EVS), the Single Nucleotide Polymorphism Database (dbSNP), ClinVar and Human Gene Mutation Database (HGMD). In addition, information from the Breast Cancer Information Core (BIC database was utilized.
cDNA analysis
The variants were investigated for their effect on splicing. Primers were positioned in flanking exons, preferentially so PCR-products covered at least one exon on either side of the exon containing the variant of interest (Table 2). Due to the size of the large exons 11 of BRCA1 and BRCA2, alternative strategies were used. For these exons, the corresponding PCR-products did not contain the entire exon 11, as one of the primers in each set was located in exon 11 (Table 2). The PCR-products were visualized on agarose gels, sequenced using Sanger sequencing and evaluated in Sequencher® version 5.3 (Gene Codes Inc. [19]). All exonic variants were used as markers for biallelic expression. All PCR-reactions were repeated using a second cDNA preparation as template (prepared from the same RNA sample).
Trans-activation (TA) assay
Plasmids, mutagenesis and transformation
A fusion construct containing GAL4 DBD:BRCA1 (amino acids 1396–1863) WT and the known neutral variant c.4837A>G (p.Ser1613Gly) sub-cloned into pcDNA3 were kindly provided by Alvaro N. A. Monteiro [15]. As an internal transfection control, the phRG-TK vector was used. The phRG-TK contains a Renilla-luciferase gene under the control of a constitutive TK-promoter. The pGAL4-e1b-Luc containing the Firefly-luciferase gene was used as a reporter for measuring the trans-activating ability (Fig. 2a). Variants c.5075A>C (p.Asp1692Ala), c.5125G>A (p.Gly1709Arg), c.5513T>G (p.Val1838Gly), and the pathogenic control c.5324T>G (p.Met1775Arg) [15], were introduced in pcDNA3 GAL4 DBD:BRCA1 (amino acid 1396–1863) WT using the QuikChange XL Site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA USA) according to the manufacturer’s protocol. Mutant plasmids were transformed into XL-10 Gold or Top10 competent cells and successful mutagenesis was confirmed by Sanger sequencing.
Transfection and harvesting
Both BHK-21 and HEK293 cells (ATCC, www.atcc.org) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Waltham, MA USA) with 10 % Fetal Bovine Serum (Life Technologies) and 60 U/ml Penicillin–Streptomycin (Life Technologies). Approximately 150,000 BHK-21 and 300,000 HEK293 cells were transferred to each well of a 6-well plate and grown overnight before transfection. One µg of pcDNA3 GAL4 DBD:BRCA1 was co-transfected with one µg of pGAL4-e1b-Luc and 100 ng phRG-TK (internal transfection control). Fugene® HD Transfection Reagent (Promega, Madison, WI USA) was used as transfecting agent according to the protocol recommended by the supplier. Untransfected cells, cells transfected exclusively with the reporter plasmids (pGAL4-e1b-Luc and phRG-TK) and cells transfected with the plasmid containing the BRCA1 WT, the p.Ser1613Gly (neutral) and p.Met1775Arg (pathogenic) variants, were used as controls. Cells were harvested 24 h post-transfection. The transfection experiments were repeated three times.
Luciferase measurements
The Dual-Luciferase Assay System (Promega) was used to measure the trans-activation activity. In short, 50 µl Luciferase Assay Reagent II (LARII) was injected into wells containing 20 µl cell lysate. The amount of light produced was measured and subsequently 50 µl Stop & Glo Reagent was injected. A CLARIOstar (BMG LABTECH, Ortenberg, Germany) was used for injections and recordings. For each lysate, both Renilla- and Firefly-luciferase activities were measured in triplicates. The data are presented as ratios of Firefly- to Renilla-excitation values. The activity-ratios obtained from cells transfected with only the reporter plasmid were defined as background and thus subtracted from the activity-ratios obtained from the BRCT-containing plasmids. For each WT lysate/triplicates, the average was calculated. All luciferase measurements within the same transfection set-up were then calculated as the percentage of the corresponding WT average. Values were combined, before the average and standard deviations were calculated.
Western blot
Lysates from one of the HEK293 transfections and one of the BHK-21 transfections were used for western blot analysis to confirm the presence of fusion proteins. The amount of light produced by the internal transfection control (Renilla luciferase) was used for normalization of samples. Samples were loaded on NuPAGE 4–12 % Bis–Tris pre cast gels (Life Technologies) and the proteins were separated for 1.5 h at 200 V and 120 mA. Proteins were subsequently transferred to polyvinylidene difluoride (PVDF) membranes (Life Technologies) (1.5 h at 25 V and 160 mA), blocked for 1 h in phosphate buffered saline (PBS) with 5 % nonfat dried milk powder (PanReac AppliChem, Darmstadt, Germany) and incubated overnight with 1:200 dilution of BRCA1 (C-20) primary antibodies (Santa Cruz Biotechnology, Dallas, Texas USA). Membranes were incubated for 1 h with HRP-Chicken anti-rabbit secondary antibodies (1:50,000) (Santa Cruz Biotechnology) followed by treatment with Signal® West Dura Extended Duration Substrate (Thermo Scientific, Waltham, MA USA). The ImageQuant Las4000 (GE Healthcare Life Sciences, Buckinghamshire, UK) was used to capture images.
Results
cDNA analysis
Eighteen BRCA1 variants, comprising three intronic and 15 exonic variants, and 14 BRCA2 variants, comprising one intronic and 13 exonic variants were investigated (Tables 1, 3). All variants, except BRCA1 c.3418A>G and BRCA2 c.4068G>A (which were earlier identified as benign variants [20, 21]), were screened for their effect on splicing. In addition, all exonic variants (including BRCA1 c.3418A>G and BRCA2 c. 4068G>A) were used as markers to investigate biallelic expression.
In the performed cDNA analysis, three variants appeared to cause alterations in the normal splicing. BRCA1 c.213-5T>A (intron 5) resulted in inclusion of 59 nucleotides of the 3′-end of intron 5, leading to a frame-shift introducing an early stop-codon (r.212_213ins213-59_213-1 p.Arg71Serfs*11) (Fig. 1a). BRCA1 c.5434C>G (exon 23) induced skipping of exon 23, also leading to a frame-shift and subsequently an early stop-codon (r.5407_5467del p.Gly1803Glnfs*11) (Fig. 1b). BRCA2 c.68-7T>A (intron 2) appeared to increase skipping of exon 3 (Fig. 1c). Skipping of exon 3 is an in-frame deletion (r.68_316del p.Asp23_Leu105del) which was also detected in controls. Splice site predictions for these three variants are shown in Table 4.

cDNA analysis. At the top of each image the wild type (WT) sequence is shown, followed by the alternative sequences observed in the patient samples. At the bottom the electropherograms are displayed. a BRCA1 c.213-5T>A resulted in an inclusion of 59 nucleotides from the 3′ end of intron 5 (r.212_213ins213-59_213-1 p.Arg71Serfs*11). b BRCA1 c.5434C>G resulted in skipping of exon 23 (r.5407_5467del p.Gly1803Glnfs*11). Electropherogram displayed with sequences from the reverse primer. c BRCA2 c.68-7T>A resulted in increased skipping of exon 3 (r.68_316del p.Asp23_Leu105del), which is a normal alternative splicing event
Heterozygous positions identified in gDNA that appear homozygous when cDNA is investigated suggest the loss of expression from one of the alleles or alternative splicing in the investigated region. The majority of patients with an exonic variant were confirmed to have both alleles transcribed (exception marked in Table 1).
Trans-activation assay
Seven patients were carriers of variants in the BRCT domains of BRCA1 (c.5075A>C, c.5096G>A, c.5117G>C, c.5123C>T, c.5125G>A, c.5434C>G and c.5513T>G). Of these, three variants were novel (c.5075A>C p.Asp1692Ala, c.5125G>A p.Gly1709Arg and c.5513T>G p.Val1838Gly). These three variants were further investigated for their trans-activating ability. For the remaining variants c.5434C>G, c.5096G>A, c.5117G>C and c.5123C>T, we were able to confirm that the sequence variant c.5434C>G caused aberrant splicing, hence this variant was not included in the TA assay. Variants c.5096G>A (p.Arg1699Gln), c.5117G>C (p.Gly1706Ala) and c.5123C>T (p.Ala1708Val) have previously been evaluated by trans-activation assays and were accordingly not included in the TA assay [22–24].
BRCA1 p.Asp1692Ala and p.Val1838Gly were unable to induce transcription of the firefly luciferase, equal to the known pathogenic variant p.Met1775Arg, which was apparent in both BHK-21 and HEK293 cells (Fig. 2b). BRCA1 p.Gly1709Arg however, showed trans-activation activity similar to the WT and the known benign variant p.Ser1613Gly (Fig. 2b).

Trans-activation assay. a A simplified view of the assay set-up. Plasmids with constructs encoding a DNA binding domain (DBD) and the C-terminal of BRCA1 (amino acids 1396–1863) were co-transfected into HEK293 and BHK-21 cells with a reporter plasmid containing firefly luciferase. If the plasmids with the C-terminal part of BRCA1 have trans-activation activity, they will activate transcription of firefly luciferase, luciferase activity is then measured and quantitated. b The dual luciferase reporter assay (Promega) was used to evaluate the trans-activation activity of BRCA1 BRCT variants in BHK-21 cells and HEK293 cells. The first three columns represent controls: wild type (WT) BRCA1, a neutral polymorphism (p.Ser1613Gly) and a pathogenic variant (p.Met1775Arg), respectively. p.Asp1692Ala (BRCA1 c.5075A>C) and p.Val1838Gly (BRCA1 c.5513T>G) had no trans-activation activity, whereas p.Gly1709Arg (BRCA1 c.5125G>A) showed normal activity. c Western blot results from proteins isolated from one of the transfections in BHK-21 cells and HEK293 cells. Samples were normalized according to renilla expression measured by CLARIOstar (BMG LABTECH)
Western blot results indicated an equal expression of the plasmid constructs in the BHK-21 cells, but showed some variation in HEK293 cells despite adjusting the protein concentrations according to the transfection control, Renilla luciferase (Fig. 2c). However, the BRCT mutants were expressed in both cell types, indicating that the reduced values were due to reduced trans-activation ability and not due to variations in expression/stability.
Discussion
Prophylactic mastectomy and salphingo-oophorectomy are potent, but invasive risk reducing managements for carriers of pathogenic BRCA1/2 variants. Accordingly, identifying a VUS pose a considerable challenge for genetic counsellors and medical geneticists in advising clinical management. In this study, we characterized some of the variants detected in a Norwegian breast and ovarian cancer cohort, both by cDNA analysis and analysis of the trans-activation ability of variants located in the BRCT domains.
cDNA analysis
Alternative splicing allows for a more diverse expression of genes, and can regulate localization, enzymatic properties and different interaction properties of proteins [25]. The majority of variants located in the consensus ss (GT-AG in position ± 1, 2) lead to abnormal splicing [26], but the effects of variants at positions further away from the exon–intron border are more difficult to predict. In addition, both missense variants and silent exonic variants might affect splicing [27], either by creating cryptic ss, remove binding sites for exonic splicing enhancers (ESE) or create binding sites for exonic splicing silencers (ESS). However, normal alternative splicing can counteract the effect of some variants leading to aberrant splicing [28]. de la Hoya et al. [28] recently reported a variant causing an out-of-frame deletion of BRCA1 exon 10. The potential effect of this variant, however, was counteracted by a normal in-frame alternative splice event deleting exons 9–10 from the transcript [28].
In the current study, three of the 32 variants had a consequence on pre-mRNA splicing
BRCA1 c.213-5T>A, a novel variant located in intron 5, resulted in usage of a cryptic ss 59 nucleotides upstream of the original site. Three splice prediction tools, SSF, MES and HSF anticipated a 3′ss at the original position. The variant led to reduced predictions of the original ss (Table 4) and the cryptic ss 59 bases upstream was strongly predicted by all prediction programs (also in the WT sequence). Inclusion of 59 nucleotides caused a frame-shift and introduced a premature stop-codon after 75 codons. Another variant in this region, BRCA1 c.213-11T>G, has previously been shown to lead to the use of the same cryptic ss [8]. The presence of a premature stop-codon likely activates the nonsense-mediated mRNA decay pathway [29]. However, variants in BRCA1, which introduce a stop-codon before position c.297, are presumed to allow re-initiation of translation at the AUG at this position [30]. A re-initiation at c.297 would lead to BRCA1 proteins lacking the RING-finger motif located at the N-termini (amino acids 8–96) [14]. Binding of the BRCA1 RING-domain to BARD1 protein seems to be essential for tumor suppression [31], accordingly, variants lacking this domain are expected to be of clinical importance.
BRCA1 c.5434C>G in exon 23 was the only exonic sequence variant introducing exon skipping in our cancer cohort. This variant was previously reported by Gaildrat et al. [7] to cause skipping of exon 23, possibly by affecting a splice regulatory element (SRE), by removing an ESE or by introducing an ESS [7]. This demonstrates the importance of experimentally assessing the effect of exonic variants on splicing.
BRCA2 c.68-7T>A in intron 2 had previously been reported by Vreeswijk et al. [32] and Sanz et al. [33], who performed mini-gene assays that revealed partial skipping of exon 3 (p.Asp23_Leu105del). Prediction programs suggested a reduced strength of the downstream original 3′ss in the presence of the variant (Table 4), and cDNA analysis indicated that the variant led to increased exon 3 skipping. However, the skipping of exon 3 resulted in an in-frame alternative transcript, also present in normal controls (albeit at lower levels). Exon 3 in BRCA2 encodes the part of BRCA2 that interacts with PALB2 [34], however, the consequence (if any) of reduced interaction with PALB2 is currently unknown. Santos and colleagues have shown that in two families, BRCA2 c.68-7T>A did not segregate with the disease, suggesting that the variant is neutral [34].
Recently, Hoya et al. [28] suggested that variants in BRCA1 not leading to more than 70–80 % loss of functional transcripts from one of the alleles still can show tumor suppressor haplosufficiency, implicating the importance of knowing normal alternative splicing events in the genes investigated.
Splice predictions as cDNA analysis inclusion criteria
In 2012, Houdayer et al. introduced specific criteria for selection of variants which should be tested for splicing [36]. They concluded that as long as the original splice site in BRCA1 or BRCA2 has a prediction value over three for the MES prediction tool and over 60 for the SSF prediction tool, a reduction of 15 and 5 %, respectively, was sufficient to include variants for cDNA analysis. Both BRCA1 c.213-5T>A and BRCA2 c.68-7T>A would have been included using these criteria. However, BRCA1 c.5434C>G would have been omitted from cDNA analysis, since this variant most likely affects an SRE. In summary, although prediction programs can indicate that some variants can cause aberrant splicing, the true outcome can only be identified experimentally.
Trans-activation assay
We investigated three novel BRCA1 variants for their effect on BRCA1′s trans-activation activity (Table 1). Two of the three variants (BRCA1 c.5075A>C p.Asp1692Ala and c.5513T>G p.Val1838Gly) showed a clear loss of activity (Fig. 2b). BRCA1 p.Asp1692Ala exchanging the highly conserved aspartate to an alanine and BRCA1 p.Val1838Gly, substituting the highly conserved valine to a glycine, are both predicted to be pathogenic by the missense prediction tools Align GVGD, SIFT and MutationTaster. However, PolyPhen-2 only predicts p.Val1838Gly to be damaging. Both these variants result in changes in the BRCT domains and our functional study indicated their pathogenicity by loss of trans-activation activity (Fig. 2b). Other variants changing aspartate at position 1692 and valine at position 1838 (p.Asp1692His, p.Asp1692Asn, p.Asp1692Tyr and p.Val1838Glu), which have all previously been shown to have a functional impact using the TA-assay, indicating the importance of the conserved amino acids at these positions [37, 38]. BRCA1 c.5125G>A p.Gly1709Arg however, substituting the highly conserved glycine with arginine, is predicted differently by Align GVGD, SIFT, Mutation taster and PolyPhen-2 (Table 3). Even though some of the prediction programs indicated pathogenicity, p.Gly1709Arg displayed normal trans-activation activity.
Although the in vitro trans-activation studies suggest the pathogenicity of BRCA1 c.5075A>C and c.5513T>G, we only investigated a limited part of the BRCA1 protein. Further assessment including segregation studies in families with these variants are needed to establish their classification.
Several BRCA1 variants in our cohort are classified as either likely pathogenic, likely benign or benign based on cDNA analysis, functional studies, segregation analysis, frequency in control populations, among others (Tables 1, 3). However, some remain classified as VUS. Two variants identified in our cohort (BRCA1 c.734A>T and c.1419C>T) have not been previously reported in the literature and both are reported with a low frequency in the ExAC database [39], accordingly, their clinical significance is uncertain (Table 1). BRCA1 c.3708T>G and c.5123C>T were previously reported in both the literature and with low frequencies in databases (Table 3).
In BRCA2 none of the variants identified in our cohort were classified as likely pathogenic. One variant (c.4068G>A) was classified as benign and five variants (c.750G>A, c.2680 G>A, c.3568C>T, c.6100C>T and c.6821G>T) were classified as likely benign (Table 1). Eight variants remained classified as VUS; The BRCA2 variant c.40A>G has not been previously reported in the investigated databases nor in the literature (Table 3) and c.8323A>G has not earlier been reported in the literature but is reported with low frequency in the ExAC database (Table 3). The five remaining variants, c.4828G>A, c.5272_5274delAAT, c.7301A>C, c.8177A>G and c.9116C>T, have been reported in the literature and all except c.8177A>G are reported with low frequencies in the investigated databases (Table 3). Our current study was unable to disclose new variants located in regulatory sequences, potentially affecting the expression of one of the alleles.
Conclusion
In the current study, we identified three variants leading to abnormal splicing of pre-mRNA; Two variants located intronically, BRCA1 c.213-5T>A and BRCA2 c.68-7T>A, and one exonic variant, BRCA1 c.5434C>G. In addition, functional studies assessing the trans-activation activity of the BRCT domains resulted in identification of two variants, c.5075A>C p.Asp1692Ala and c.5513T>G p.Val1838Gly, which lacked trans-activation activity. The use of partial proteins can lead to further understanding of how variants may affect protein function, however, the use of full-length proteins would be preferable in functional studies.
References
Safran M, Dalah I, Alexander J, et al (2010) GeneCards Version 3: the human gene integrator. Database (Oxford) 2010: baq020 DOI 10.1093/database/baq020
Roy R, Chun J, Powell SN (2012) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12(1):68–78. doi:10.1038/nrc3181
Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH (2009) Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep 10(9):990–996. doi:10.1038/embor.2009.126
Kast K, Rhiem K, Wappenschmidt B et al (2016) Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet 53(7):465–471. doi:10.1136/jmedgenet-2015-103672
Frank TS, Deffenbaugh AM, Reid JE, et al (2002) Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 20(6):1480–1490
Barnes DR, Antoniou AC (2012) Unravelling modifiers of breast and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers: update on genetic modifiers. J Intern Med 271(4):331–343. doi:10.1111/j.1365-2796.2011.02502.x
Gaildrat P, Krieger S, Thery JC et al (2010) The BRCA1 c.5434C → G (p. Pro1812Ala) variant induces a deleterious exon 23 skipping by affecting exonic splicing regulatory elements. J Med Genet 47(6):398–403. doi:10.1136/jmg.2009.074047
Friedman LS, Ostermeyer EA, Szabo CI et al (1994) Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet 8(4):399–404. doi:10.1038/ng1294-399
Hoffman JD, Hallam SE, Venne VL, Lyon E, Ward K (1998) Implications of a novel cryptic splice site in the BRCA1 gene. Am J Med Genet 80(2):140–144
Fetzer S, Tworek HA, Piver MS, Dicioccio RA (1998) An alternative splice site junction in exon 1a of the BRCA1 gene. Cancer Genet Cytogenet 105(1):90–92
Colombo M, Blok MJ, Whiley P et al (2014) Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: a report from the ENIGMA consortium. Hum Mol Genet 23(14):3666–3680. doi:10.1093/hmg/ddu075
Jakubowska A, Gorski B, Byrski T et al (2001) Detection of germline mutations in the BRCA1 gene by RNA-based sequencing. Hum Mutat 18(2):149–156. doi:10.1002/humu.1164
Fackenthal JD, Yoshimatsu T, Zhang B et al (2016) Naturally occurring BRCA2 alternative mRNA splicing events in clinically relevant samples. J Med Genet. doi:10.1136/jmedgenet-2015-103570
Narod SA, Foulkes WD (2004) BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 4(9):665–676. doi:10.1038/nrc1431
Carvalho MA, Marsillac SM, Karchin R et al (2007) Determination of cancer risk associated with germ line BRCA1 missense variants by functional analysis. Cancer Res 67(4):1494–1501. doi:10.1158/0008-5472.CAN-06-3297
den Dunnen JT, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15(1):7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7:AID-HUMU4>3.0.CO;2-N
Untergasser A, Cutcutache I, Koressaar T et al (2012) Primer3—new capabilities and interfaces. Nucl Acids Res 40(15):e115. doi:10.1093/nar/gks596
Koressaar T, Remm M (2007) Enhancements and modifications of primer design program Primer3. Bioinformatics 23(10):1289–1291. doi:10.1093/bioinformatics/btm091
Sequencher® version 5.3 sequence analysis software. Gene Codes Corporation, Ann Arbor, MI USA. http://www.genecodes.com
Lindor NM, Guidugli L, Wang X et al (2012) A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat 33(1):8–21. doi:10.1002/humu.21627
Cherbal F, Salhi N, Bakour R, Adane S, Boualga K, Maillet P (2012) BRCA1 and BRCA2 unclassified variants and missense polymorphisms in Algerian breast/ovarian cancer families. Dis Markers 32(6):343–353. doi:10.3233/DMA-2012-0893
Spurdle AB, Whiley PJ, Thompson B et al (2012) BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J Med Genet 49(8):525–532. doi:10.1136/jmedgenet-2012-101037
Bouwman P, van der Gulden H, van der Heijden I et al (2013) A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov 3(10):1142–1155. doi:10.1158/2159-8290.CD-13-0094
Lovelock PK, Spurdle AB, Mok MT et al (2007) Identification of BRCA1 missense substitutions that confer partial functional activity: Potential moderate risk variants? Breast Cancer Res 9(6):R82. doi:10.1186/bcr1826
Kelemen O, Convertini P, Zhang Z et al (2013) Function of alternative splicing. Gene 514(1):1–30. doi:10.1016/j.gene.2012.07.083
Easton DF, Deffenbaugh AM, Pruss D et al (2007) A systematic genetic assessment of 1433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet 81(5):873–883. doi:10.1086/521032
Cooper TA, Mattox W (1997) The regulation of splice-site selection, and its role in human disease. Am J Hum Genet 61(2):259–266. doi:10.1086/514856
Hoya M, Soukarieh O, Lopez-Perolio I et al (2016) Combined genetic and splicing analysis of BRCA1 c.[594-2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum Mol Genet. doi:10.1093/hmg/ddw094
Palacios IM (2013) Nonsense-mediated mRNA decay: from mechanistic insights to impacts on human health. Brief Funct Genom 12(1):25–36. doi:10.1093/bfgp/els051
Buisson M, Anczukow O, Zetoune AB, Ware MD, Mazoyer S (2006) The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum Mutat 27(10):1024–1029. doi:10.1002/humu.20384
Shakya R, Reid LJ, Reczek CR et al (2011) BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334(6055):525–528. doi:10.1126/science.1209909
Vreeswijk MP, Kraan JN, van der Klift HM et al (2009) Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum Mutat 30(1):107–114. doi:10.1002/humu.20811
Sanz DJ, Acedo A, Infante M et al (2010) A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in breast/ovarian cancer patients. Clin Cancer Res 16(6):1957–1967. doi:10.1158/1078-0432.CCR-09-2564
Xia B, Sheng Q, Nakanishi K et al (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22(6):719–729. doi:10.1016/j.molcel.2006.05.022
Santos C, Peixoto A, Rocha P et al (2014) Pathogenicity evaluation of BRCA1 and BRCA2 unclassified variants identified in Portuguese breast/ovarian cancer families. J Mol Diagn 16(3):324–334. doi:10.1016/j.jmoldx.2014.01.005
Houdayer C, Caux-Moncoutier V, Krieger S et al (2012) Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat 33(8):1228–1238. doi:10.1002/humu.22101
Lee MS, Green R, Marsillac SM et al (2010) Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res 70(12):4880–4890. doi:10.1158/0008-5472.CAN-09-4563
Jhuraney A, Velkova A, Johnson RC et al (2015) BRCA1 Circos: a visualisation resource for functional analysis of missense variants. J Med Genet 52(4):224–230. doi:10.1136/jmedgenet-2014-102766
Lek M, Karczewski K, Minikel E, et al (2015) Analysis of protein-coding genetic variation in 60,706 humans. Biorxiv. doi:10.1101/030338
Simard J, Dumont M, Moisan AM et al (2007) Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J Med Genet 44(2):107–121. doi:10.1136/jmg.2006.044388
Castera L, Krieger S, Rousselin A et al (2014) Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet 22(11):1305–1313. doi:10.1038/ejhg.2014.16
Millot GA, Berger A, Lejour V et al (2011) Assessment of human Nter and Cter BRCA1 mutations using growth and localization assays in yeast. Hum Mutat 32(12):1470–1480. doi:10.1002/humu.21608
Scottish/Northern Irish BBC (2003) BRCA1 and BRCA2 mutations in Scotland and Northern Ireland. Br J Cancer 88(8):1256–1262. doi:10.1038/sj.bjc.6600840
Bonnet C, Krieger S, Vezain M et al (2008) Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J Med Genet 45(7):438–446. doi:10.1136/jmg.2007.056895
Schoumacher F, Glaus A, Mueller H, Eppenberger U, Bolliger B, Senn HJ (2001) BRCA1/2 mutations in Swiss patients with familial or early-onset breast and ovarian cancer. Swiss Med Wkly 131(15–16):223–226
Barker DF, Almeida ER, Casey G et al (1996) BRCA1 R841 W: a strong candidate for a common mutation with moderate phenotype. Genet Epidemiol 13(6):595–604. doi:10.1002/(SICI)1098-2272(1996)13:6<595:AID-GEPI5>3.0.CO;2-#
Durocher F, Shattuck-Eidens D, McClure M et al (1996) Comparison of BRCA1 polymorphisms, rare sequence variants and/or missense mutations in unaffected and breast/ovarian cancer populations. Hum Mol Genet 5(6):835–842
Goldgar DE, Easton DF, Deffenbaugh AM et al (2004) Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet 75(4):535–544. doi:10.1086/424388
Panguluri RC, Brody LC, Modali R et al (1999) BRCA1 mutations in African Americans. Hum Genet 105(1–2):28–31
van Orsouw NJ, Dhanda RK, Elhaji Y et al (1999) A highly accurate, low cost test for BRCA1 mutations. J Med Genet 36(10):747–753
Vallon-Christersson J, Cayanan C, Haraldsson K et al (2001) Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum Mol Genet 10(4):353–360
Scott CL, Jenkins MA, Southey MC et al (2003) Average age-specific cumulative risk of breast cancer according to type and site of germline mutations in BRCA1 and BRCA2 estimated from multiple-case breast cancer families attending Australian family cancer clinics. Hum Genet 112(5–6):542–551. doi:10.1007/s00439-003-0908-6
Laraqui A, Uhrhammer N, Lahlou-Amine I et al (2013) Mutation screening of the BRCA1 gene in early onset and familial breast/ovarian cancer in Moroccan population. Int J Med Sci 10(1):60–67. doi:10.7150/ijms.5014
Chenevix-Trench G, Healey S, Lakhani S et al (2006) Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res 66(4):2019–2027. doi:10.1158/0008-5472.CAN-05-3546
Martinez-Ferrandis JI, Vega A, Chirivella I et al (2003) Mutational analysis of BRCA1 and BRCA2 in Mediterranean Spanish women with early-onset breast cancer: identification of three novel pathogenic mutations. Hum Mutat 22(5):417–418. doi:10.1002/humu.9188
Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE (2002) Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst 94(18):1396–1406
Thery JC, Krieger S, Gaildrat P et al (2011) Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur J Hum Genet 19(10):1052–1058. doi:10.1038/ejhg.2011.100
Guidugli L, Carreira A, Caputo SM et al (2014) Functional assays for analysis of variants of uncertain significance in BRCA2. Hum Mutat 35(2):151–164. doi:10.1002/humu.22478
Stegel V, Krajc M, Zgajnar J et al (2011) The occurrence of germline BRCA1 and BRCA2 sequence alterations in Slovenian population. BMC Med Genet 12:9. doi:10.1186/1471-2350-12-9
Wagner TM, Hirtenlehner K, Shen P et al (1999) Global sequence diversity of BRCA2: analysis of 71 breast cancer families and 95 control individuals of worldwide populations. Hum Mol Genet 8(3):413–423
Kanchi KL, Johnson KJ, Lu C et al (2014) Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun 5:3156. doi:10.1038/ncomms4156
Karchin R, Agarwal M, Sali A, Couch F, Beattie MS (2008) Classifying Variants of Undetermined Significance in BRCA2 with protein likelihood ratios. Cancer Inform 6:203–216
Guidugli L, Pankratz VS, Singh N et al (2013) A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res 73(1):265–275. doi:10.1158/0008-5472.CAN-12-2081
Llort G, Munoz CY, Tuser MP et al (2002) Low frequency of recurrent BRCA1 and BRCA2 mutations in Spain. Hum Mutat 19(3):307. doi:10.1002/humu.9014
Menendez M, Castellsague J, Mirete M et al (2012) Assessing the RNA effect of 26 DNA variants in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat 132(3):979–992. doi:10.1007/s10549-011-1661-5
Acknowledgments
We thank Alvaro N. A. Monteiro for kindly providing us with the BRCT containing plasmids necessary for the trans-activation assay. We also thank “Helse Nord” for providing the necessary funding for this study (Grant # SFP1161-14).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
All participants gave written informed consent for diagnostic testing. The project was submitted to the appropriate regional ethics committee, however, since the samples were tested diagnostically the regional ethical committee waved the need for ethical approval based on the Norwegian regional health organization law § 2 and § 9 and the Norwegian research ethical law § 4.
Rights and permissions
About this article

Cite this article
Jarhelle, E., Riise Stensland, H.M.F., Mæhle, L. et al. Characterization of BRCA1 and BRCA2 variants found in a Norwegian breast or ovarian cancer cohort. Familial Cancer 16, 1–16 (2017). https://doi.org/10.1007/s10689-016-9916-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-016-9916-2